Ursolic Acid Attenuates Hepatic Steatosis, Fibrosis, and Insulin Resistance by Modulating the Circadian Rhythm Pathway in Diet-Induced Obese Mice

Abstract
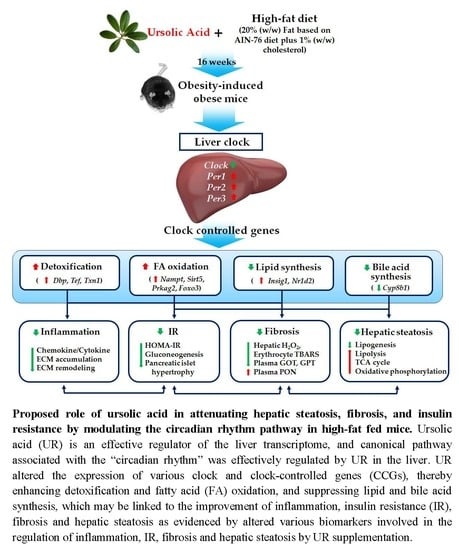
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Blood Glucose, Intraperitoneal Glucose Tolerance Test (IPGTT), Homeostasis Model Assessment-Estimated IR (HOMA-IR) Index, and Plasma Biomarkers
2.3. Analyses of Plasma and Hepatic Lipids
2.4. Morphology of Liver, Fat, and Pancreatic Tissues
2.5. Hepatic Enzyme Activities and Glycogen Concentrations
2.6. Hepatic H2O2 and Erythrocyte Thiobarbituric Acid Reactive Substance (TBARS) Concentrations, and Plasma Paraoxonase (PON) Activity
2.7. RNA Preparation and Quality Control
2.8. Quantitative Real-Time PCR
2.9. Microarray Analysis
2.10. Statistical Analysis
3. Results
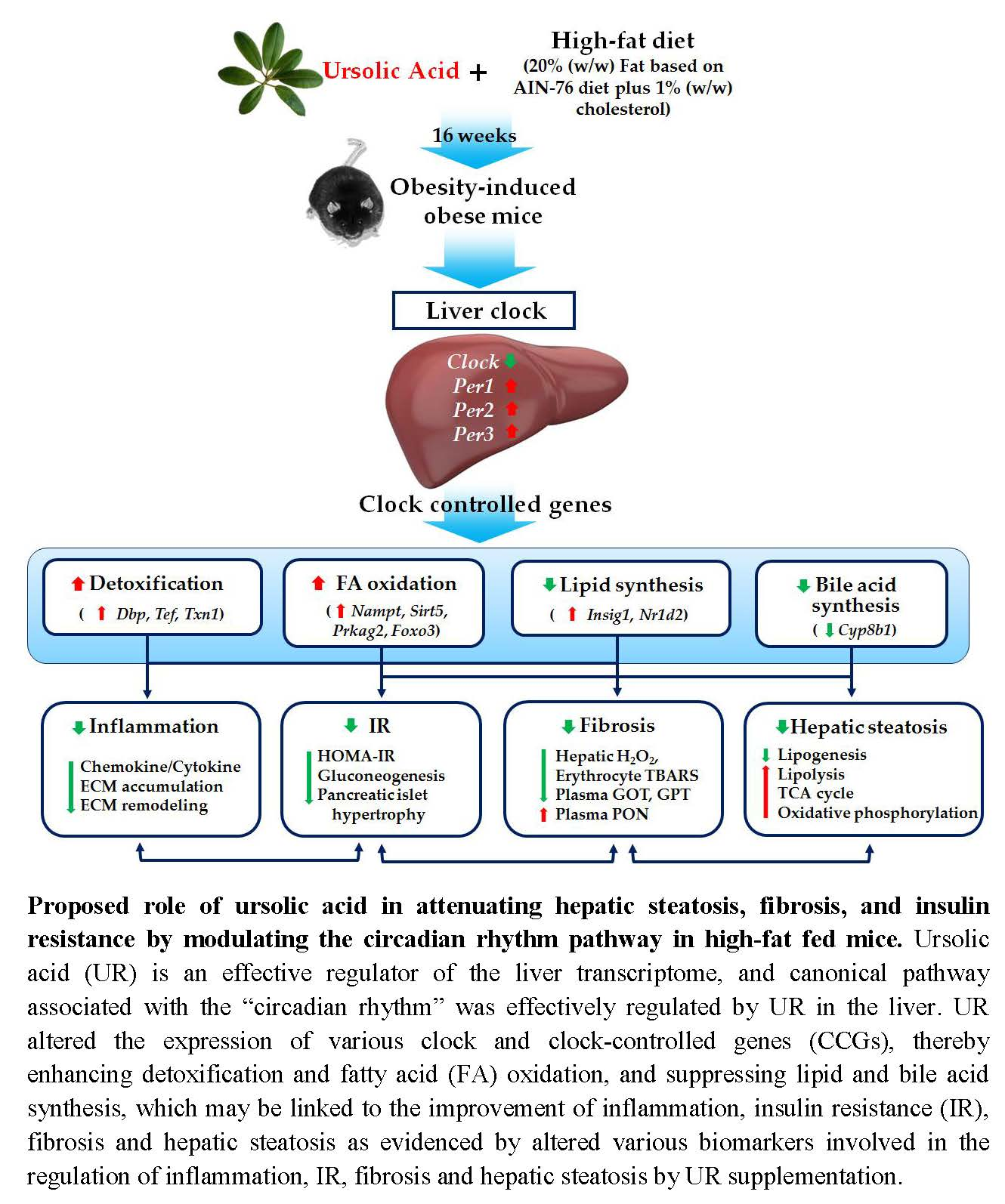
3.1. UR Decreased Adiposity and Plasma Lipids Levels
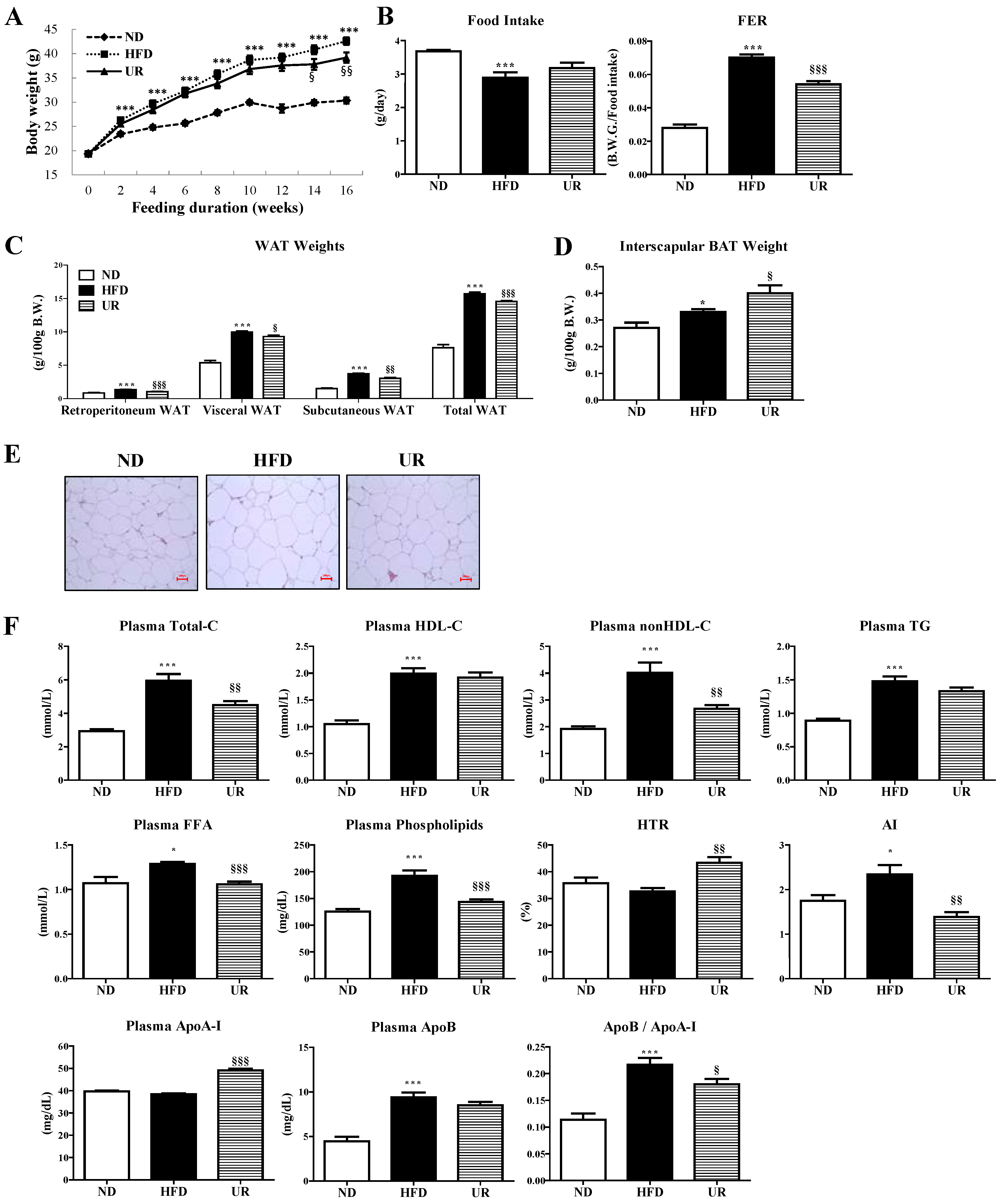
3.2. UR Abated HFD-Induced Hepatic Steatosis by Modulating Hepatic Lipid-Regulating Enzyme Activities
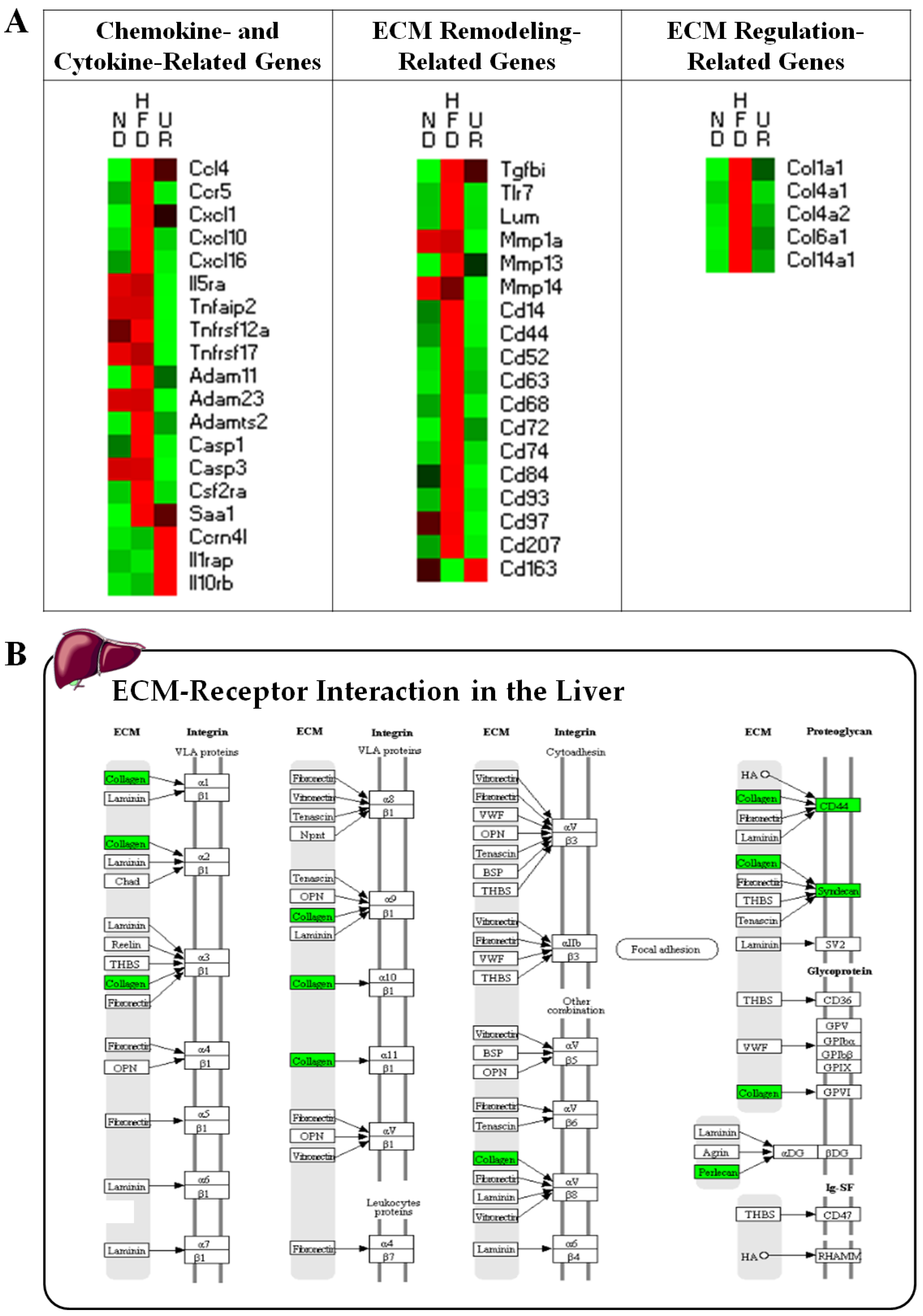
3.3. UR Ameliorated Tissue Fibrosis in Mice with DIO
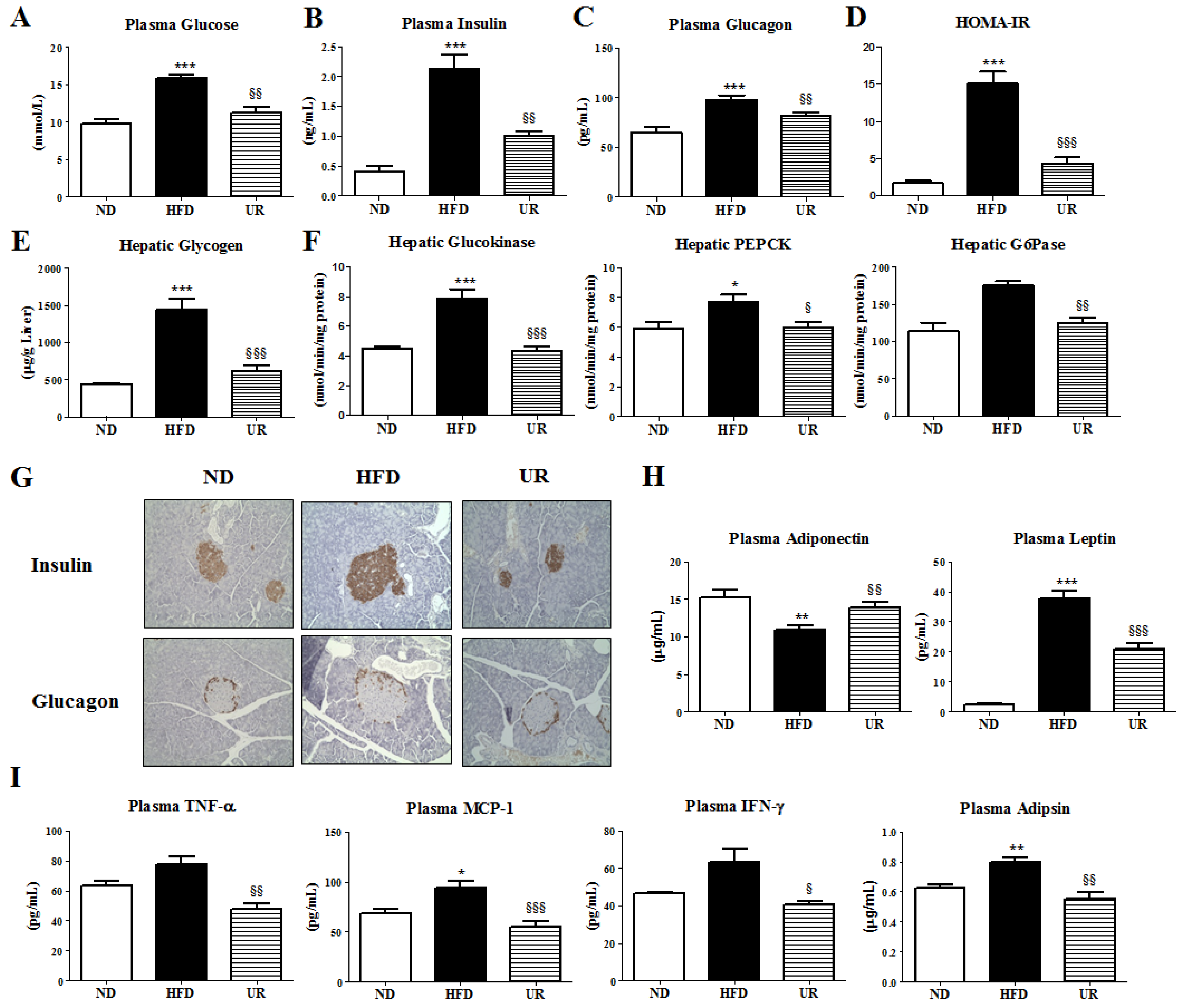
3.4. UR Prevented HFD-Induced IR and Pancreatic Islet Hypertrophy
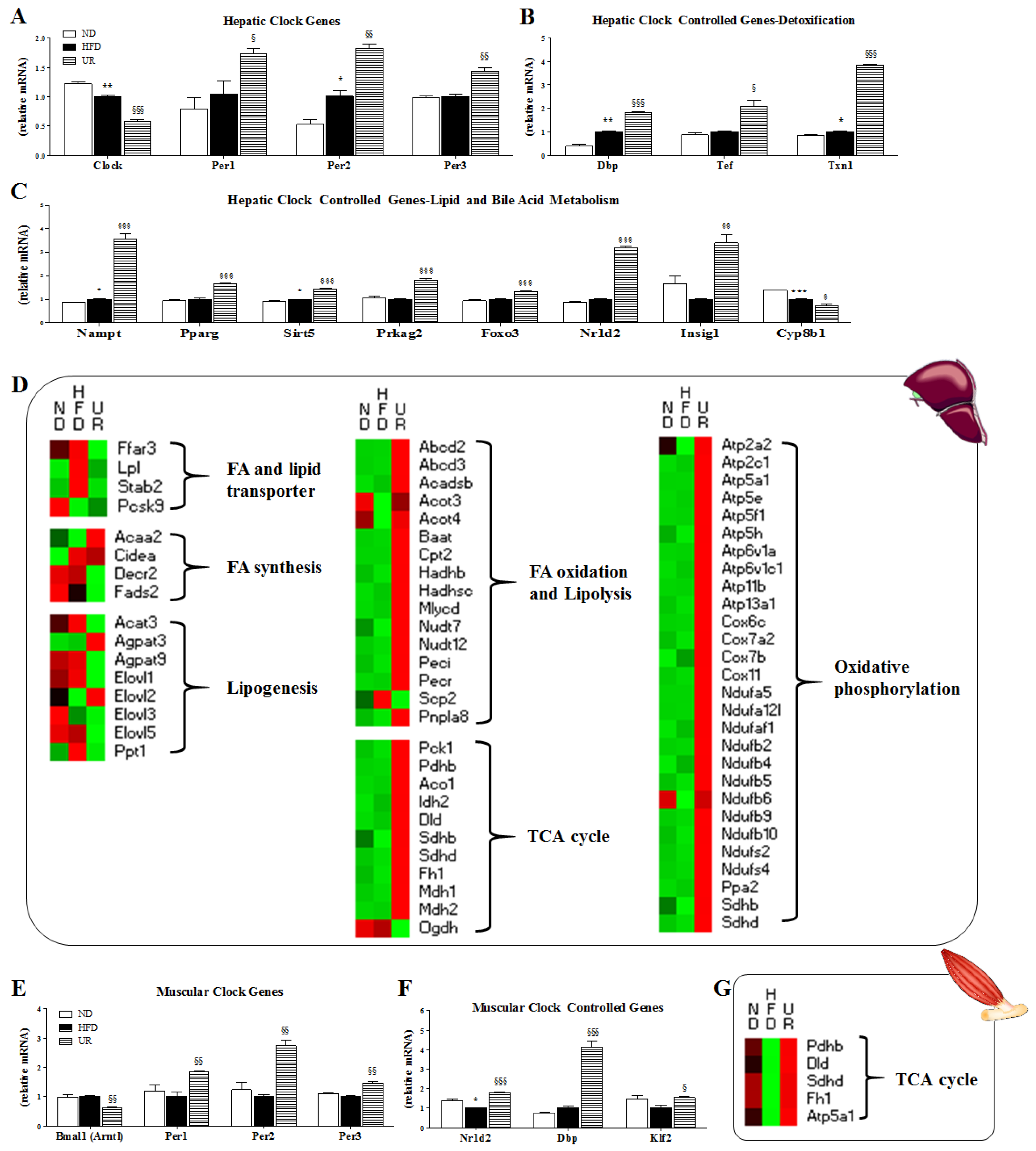
3.5. UR Altered the Transcriptional Responses Involved in Circadian Clock and ECM in the Liver and Muscle Tissue of DIO Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guh, D.P.; Zhang, W.; Bansback, N.; Amarsi, Z.; Birmingham, C.L.; Anis, A.H. The incidence of co-morbidities related to obesity and overweight: A systematic review and meta-analysis. BMC Public Health 2009, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Eng, F.J.; Friedman, S.L. Fibrogenesis I. New insights into hepatic stellate cell activation: The simple becomes complex. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G7–G11. [Google Scholar] [CrossRef] [PubMed]
- Carmiel-Haggai, M.; Cederbaum, A.I.; Nieto, N. A high-fat diet leads to the progression of non-alcoholic fatty liver disease in obese rats. FASEB J. 2005, 19, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Ferrell, J.M.; Chiang, J.Y. Circadian rhythms in liver metabolism and disease. Acta Pharm. Sin. B 2015, 5, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, R.A.; Reddy, A.B.; Maywood, E.S.; Clayton, J.D.; King, V.M.; Smith, A.G.; Gant, T.W.; Hastings, M.H.; Kyriacou, C.P. Circadian cycling of the mouse liver transcriptome, as revealed by cDNA microarray, is driven by the suprachiasmatic nucleus. Curr. Biol. 2002, 12, 540–550. [Google Scholar] [CrossRef]
- Ohta, Y.; Taguchi, A.; Matsumura, T.; Nakabayashi, H.; Akiyama, M.; Yamamoto, K.; Fujimoto, R.; Suetomi, R.; Yanai, A.; Shinoda, K.; et al. Clock gene dysregulation induced by chronic er stress disrupts β-cell function. EBioMedicine 2017, 18, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Turek, F.W.; Joshu, C.; Kohsaka, A.; Lin, E.; Ivanova, G.; McDearmon, E.; Laposky, A.; Losee-Olson, S.; Easton, A.; Jensen, D.R.; et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 2005, 308, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Pekovic-Vaughan, V.; Gibbs, J.; Yoshitane, H.; Yang, N.; Pathiranage, D.; Guo, B.; Sagami, A.; Taguchi, K.; Bechtold, D.; Loudon, A.; et al. The circadian clock regulates rhythmic activation of the NRF2/glutathione-mediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes Dev. 2014, 28, 548–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Liao, X.; Meng, F.; Wang, Y.; Sun, Z.; Guo, F.; Li, X.; Meng, M.; Li, Y.; Sun, C. Therapeutic role of ursolic acid on ameliorating hepatic steatosis and improving metabolic disorders in high-fat diet-induced non-alcoholic fatty liver disease rats. PLoS ONE 2014, 9, e86724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Song, C.; Li, H.; Hou, J.; Li, D. Ursolic acid prevents augmented peripheral inflammation and inflammatory hyperalgesia in high-fat diet-induced obese rats by restoring downregulated spinal PPARα. Mol. Med. Rep. 2016, 13, 5309–5316. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; He, X.; Shi, Z.; Li, C.; Guo, F.; Li, S.; Li, Y.; Na, L.; Sun, C. Ursolic acid increases energy expenditure through enhancing free fatty acid uptake and β-oxidation via an UCP3/AMPK-dependent pathway in skeletal muscle. Mol. Nutr. Food Res. 2015, 59, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.S.; de Melo, C.L.; Queiroz, M.G.; Lemos, T.L.; Menezes, D.B.; Melo, T.S.; Santos, F.A. Ursolic acid, a pentacyclic triterpene from Sambucus australis, prevents abdominal adiposity in mice fed a high-fat diet. J. Med. Food 2011, 14, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Shishodia, S.; Majumdar, S.; Banerjee, S.; Aggarwal, B.B. Ursolic acid inhibits nuclear factor-kappaB activation induced by carcinogenic agents through suppression of IkappaBalpha kinase and p65 phosphorylation: Correlation with down-regulation of cyclooxygenase 2, matrix metalloproteinase 9, and cyclin D1. Cancer Res. 2003, 63, 4375–4383. [Google Scholar] [PubMed]
- Kunkel, S.D.; Elmore, C.J.; Bongers, K.S.; Ebert, S.M.; Fox, D.K.; Dyle, M.C.; Bullard, S.A.; Adams, C.M. Ursolic acid increases skeletal muscle and brown fat and decreases diet-induced obesity, glucose intolerance and fatty liver disease. PLoS ONE 2012, 7, e39332. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Shao, R.; Ring, S.C.; Tarloff, J.B. Coincubation of rat renal proximal tubules with hepatic subcellular fractions potentiates the effects of para-aminophenol. Fundam. Appl. Toxicol. 1997, 39, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Nepokroeff, C.M.; Lakshmanan, M.R.; Porter, J.W. Fatty-acid synthase from rat liver. Methods Enzymol. 1975, 35, 37–44. [Google Scholar] [PubMed]
- Walton, P.A.; Possmayer, F. Mg2+-dependent phosphatidate phosphohydrolase of rat lung: Development of an assay employing a defined chemical substrate which reflects the phosphohydrolase activity measured using membrane-bound substrate. Anal. Biochem. 1985, 151, 479–486. [Google Scholar] [CrossRef]
- Markwell, M.A.; McGroarty, E.J.; Bieber, L.L.; Tolbert, N.E. The subcellular distribution of carnitine acyltransferases in mammalian liver and kidney. A new peroxisomal enzyme. J. Biol. Chem. 1973, 248, 3426–3432. [Google Scholar] [PubMed]
- Lazarow, P. Assay of peroxisomal b-oxidation of fatty acids. Methods Enzymol. 1981, 72, 315–319. [Google Scholar] [PubMed]
- Shapiro, D.J.; Nordstrom, J.L.; Mitschelen, J.J.; Rodwell, V.W.; Schimke, R.T. Micro assay for 3-hydroxy-3-methylglutaryl-CoA reductase in rat liver and in L-cell fibroblasts. Biochim. Biophys. Acta 1974, 370, 369–377. [Google Scholar] [CrossRef]
- Gillies, P.J.; Rathgeb, K.A.; Perri, M.A.; Robinson, C.S. Regulation of acyl-CoA:cholesterol acyltransferase activity in normal and atherosclerotic rabbit aortas: Role of a cholesterol substrate pool. Exp. Mol. Pathol. 1986, 44, 329–339. [Google Scholar] [CrossRef]
- Alegre, M.; Ciudad, C.J.; Fillat, C.; Guinovart, J.J. Determination of glucose-6-phosphatase activity using the glucose dehydrogenase-coupled reaction. Anal. Biochem. 1988, 173, 185–189. [Google Scholar] [CrossRef]
- Davidson, A.L.; Arion, W.J. Factors underlying significant underestimations of glucokinase activity in crude liver extracts: Physiological implications of higher cellular activity. Arch. Biochem. Biophys. 1987, 253, 156–167. [Google Scholar] [CrossRef]
- Bentle, L.A.; Lardy, H.A. Interaction of anions and divalent metal ions with phosphoenolpyruvate carboxykinase. J. Biol. Chem. 1976, 251, 2916–2921. [Google Scholar] [PubMed]
- Seifter, S.; Dayton, S.; Muntwyler, N.; Muntwyler, E. The estimation of glycogen with the anthrone reagent. Arch. Biochem. 1950, 25, 191–200. [Google Scholar] [PubMed]
- Wolff, S.P. Ferrous ion oxidation in presence of ferric ion indicator xylenol orange for measurement of hydroperoxides. Methods Enzymol. 1994, 233, 182–189. [Google Scholar]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Mackness, M.I.; Harty, D.; Bhatnagar, D.; Winocour, P.H.; Arrol, S.; Ishola, M.; Durrington, P.N. Serum paraoxonase activity in familial hypercholesterolaemia and insulin-dependent diabetes mellitus. Atherosclerosis 1991, 86, 193–199. [Google Scholar] [CrossRef]
- Do, G.M.; Kwon, E.Y.; Kim, E.; Kim, H.S.; Choi, M.S. Hepatic transcription response to high-fat treatment in mice: Microarray comparison of individual versus. pooled RNA samples. Biotechnol. J. 2010, 5, 970–973. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Stangherlin, A.; Reddy, A.B. Regulation of circadian clocks by redox homeostasis. J. Biol. Chem. 2013, 288, 26505–26511. [Google Scholar] [CrossRef] [PubMed]
- Gachon, F.; Olela, F.F.; Schaad, O.; Descombes, P.; Schibler, U. The circadian PAR-domain basic leucine zipper transcription factors DBP, TEF, and HLF modulate basal and inducible xenobiotic detoxification. Cell Metab. 2006, 4, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, E.M.; Abdelrahman, S.A.; Hussein, S.; Shalaby, S.M.; Mosaad, H.; Awad, A.M. Effect of human umbilical cord blood mesenchymal stem cells administered by intravenous or intravitreal routes on cryo-induced retinal injury. IUBMB Life 2017, 69, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Dahl, T.B.; Haukeland, J.W.; Yndestad, A.; Ranheim, T.; Gladhaug, I.P.; Damås, J.K.; Haaland, T.; Løberg, E.M.; Arntsen, B.; Birkeland, K.; et al. Intracellular nicotinamide phosphoribosyltransferase protects against hepatocyte apoptosis and is down-regulated in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 2010, 95, 3039–3047. [Google Scholar] [CrossRef] [PubMed]
- Revollo, J.R.; Grimm, A.A.; Imai, S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004, 279, 50754–50763. [Google Scholar] [CrossRef] [PubMed]
- Raspé, E.; Duez, H.; Mansén, A.; Fontaine, C.; Fiévet, C.; Fruchart, J.C.; Vennström, B.; Staels, B. Identification of Rev-erbalpha as a physiological repressor of apoC-III gene transcription. J. Lipid Res. 2002, 43, 2172–2179. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, K.; Duplomb, L.; Wang, M.Y.; Li, J.; Unger, R.H. Hepatic insig-1 or -2 overexpression reduces lipogenesis in obese Zucker diabetic fatty rats and in fasted/refed normal rats. Proc. Natl. Acad. Sci. USA 2004, 101, 7106–7111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stromsdorfer, K.L.; Yamaguchi, S.; Yoon, M.J.; Moseley, A.C.; Franczyk, M.P.; Kelly, S.C.; Qi, N.; Imai, S.; Yoshino, J. NAMPT-Mediated NAD(+) Biosynthesis in Adipocytes Regulates Adipose Tissue Function and Multi-organ Insulin Sensitivity in Mice. Cell Rep. 2016, 16, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Ayala, J.E.; Lee-Young, R.S.; Zhang, Z.; James, F.D.; Neufer, P.D.; Pozzi, A.; Zutter, M.M.; Wasserman, D.H. Diet-induced muscle insulin resistance is associated with extracellular matrix remodeling and interaction with integrin alpha2beta1 in mice. Diabetes 2011, 60, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Berria, R.; Wang, L.; Richardson, D.K.; Finlayson, J.; Belfort, R.; Pratipanawatr, T.; De Filippis, E.A.; Kashyap, S.; Mandarino, L.J. Increased collagen content in insulin-resistant skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E560–E565. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, E.-Y.; Shin, S.-K.; Choi, M.-S. Ursolic Acid Attenuates Hepatic Steatosis, Fibrosis, and Insulin Resistance by Modulating the Circadian Rhythm Pathway in Diet-Induced Obese Mice. Nutrients 2018, 10, 1719. https://doi.org/10.3390/nu10111719
Kwon E-Y, Shin S-K, Choi M-S. Ursolic Acid Attenuates Hepatic Steatosis, Fibrosis, and Insulin Resistance by Modulating the Circadian Rhythm Pathway in Diet-Induced Obese Mice. Nutrients. 2018; 10(11):1719. https://doi.org/10.3390/nu10111719
Chicago/Turabian StyleKwon, Eun-Young, Su-Kyung Shin, and Myung-Sook Choi. 2018. "Ursolic Acid Attenuates Hepatic Steatosis, Fibrosis, and Insulin Resistance by Modulating the Circadian Rhythm Pathway in Diet-Induced Obese Mice" Nutrients 10, no. 11: 1719. https://doi.org/10.3390/nu10111719