Induction of Apoptosis and Cytotoxicity by Isothiocyanate Sulforaphene in Human Hepatocarcinoma HepG2 Cells

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
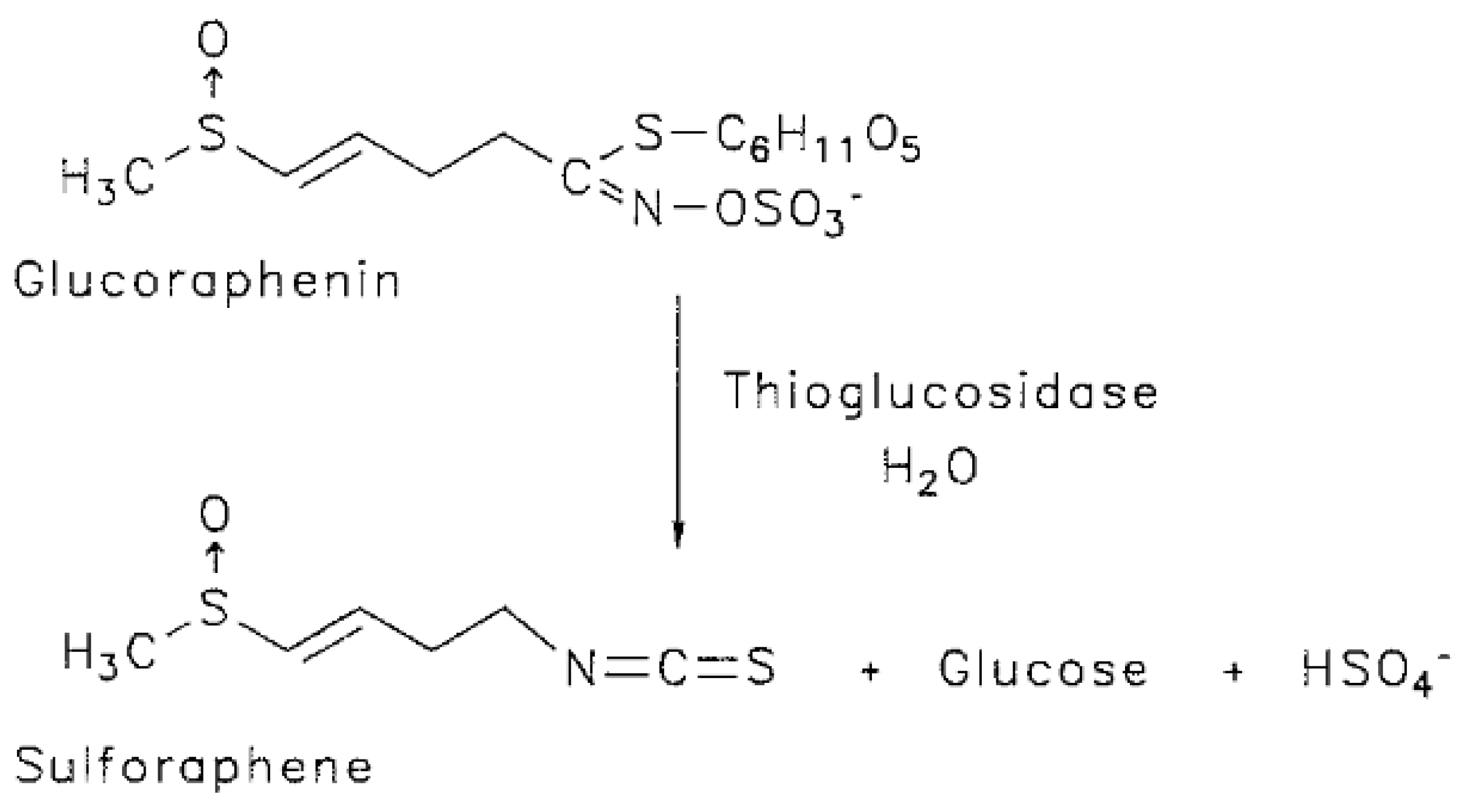
2.1. Isolation of Glucoraphenin (GRE)
2.2. Cell Culture
2.3. Cytotoxicity Assay of GRE, Sulforaphene and Cisplatin
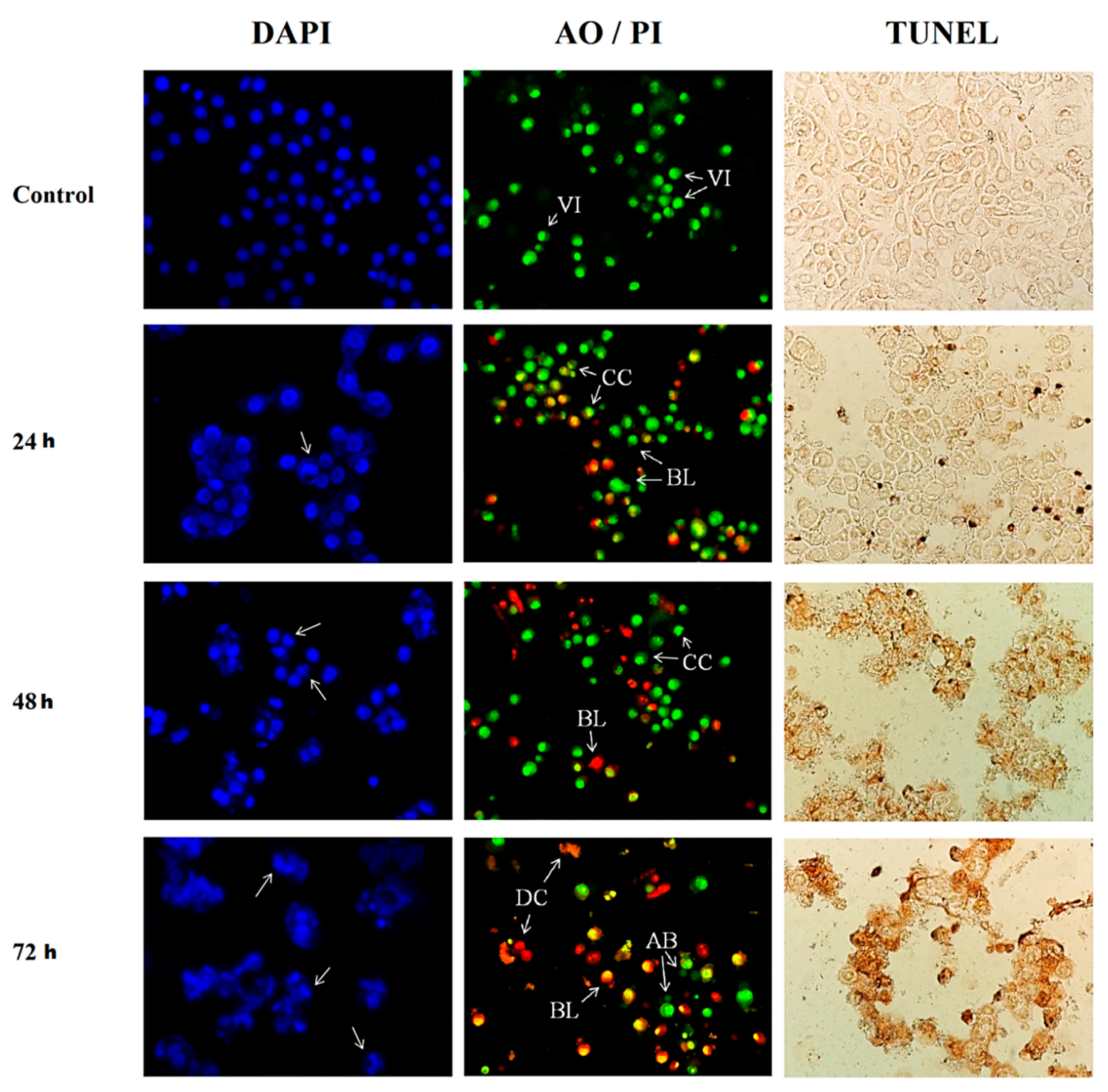
2.4. Morphological Assessment of Apoptotic Cells by TUNEL Assay
2.5. Morphological Assessment of Apoptotic Cells by DAPI Staining
2.6. Morphological Assessment of Apoptotic Cells by Acridine Orange (AO) Propidium Iodide (PI) Double Staining
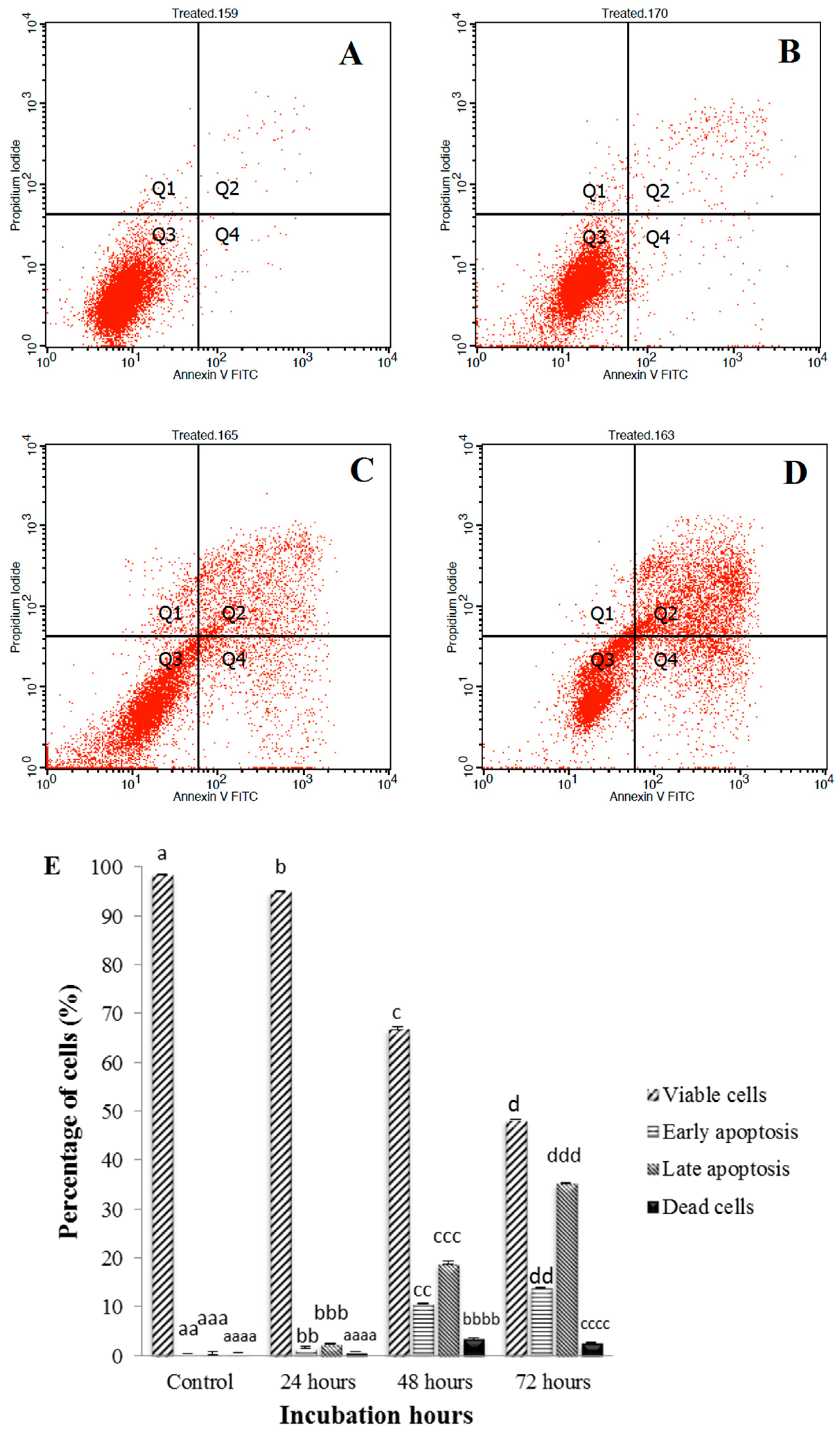
2.7. Annexin V-Fitc Assay
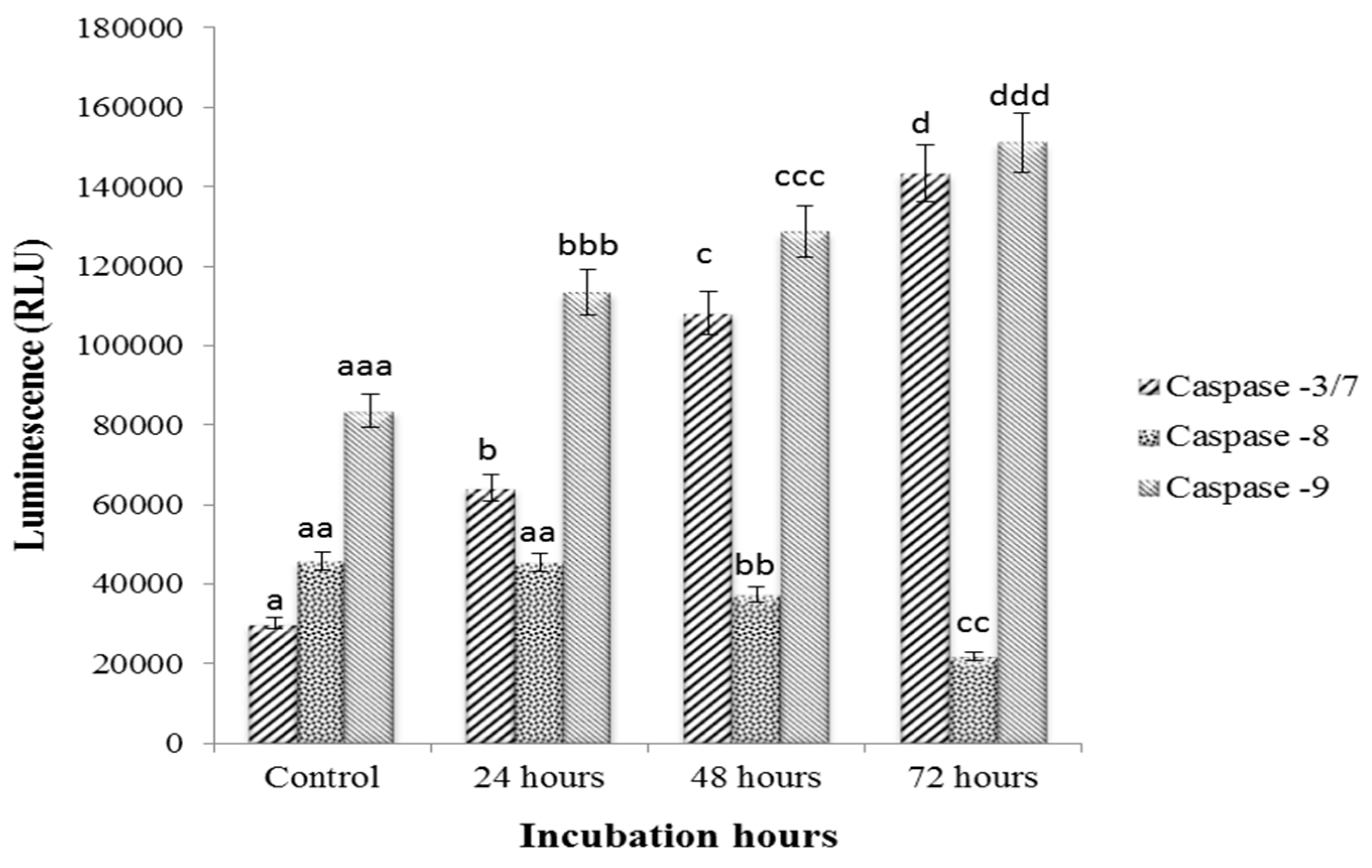
2.8. Caspase-3/7, -8 and -9 Activity Assay
2.9. Cell Cycle Analysis by Flow Cytometry
2.10. Statistical Analysis
3. Results
3.1. Cytotoxicity of GRE, Sulforaphene and Cisplatin
3.2. Morphological Assessment of Apoptotic Cells by DAPI Staining, AO/PI Double Staining and TUNEL Assay
3.3. Annexin V-FITC Assay of Sulforaphene Treated HepG2 Cells
3.4. caspase-3/7, -8 and -9 Activity on Sulforaphene Treated HepG2 Cells
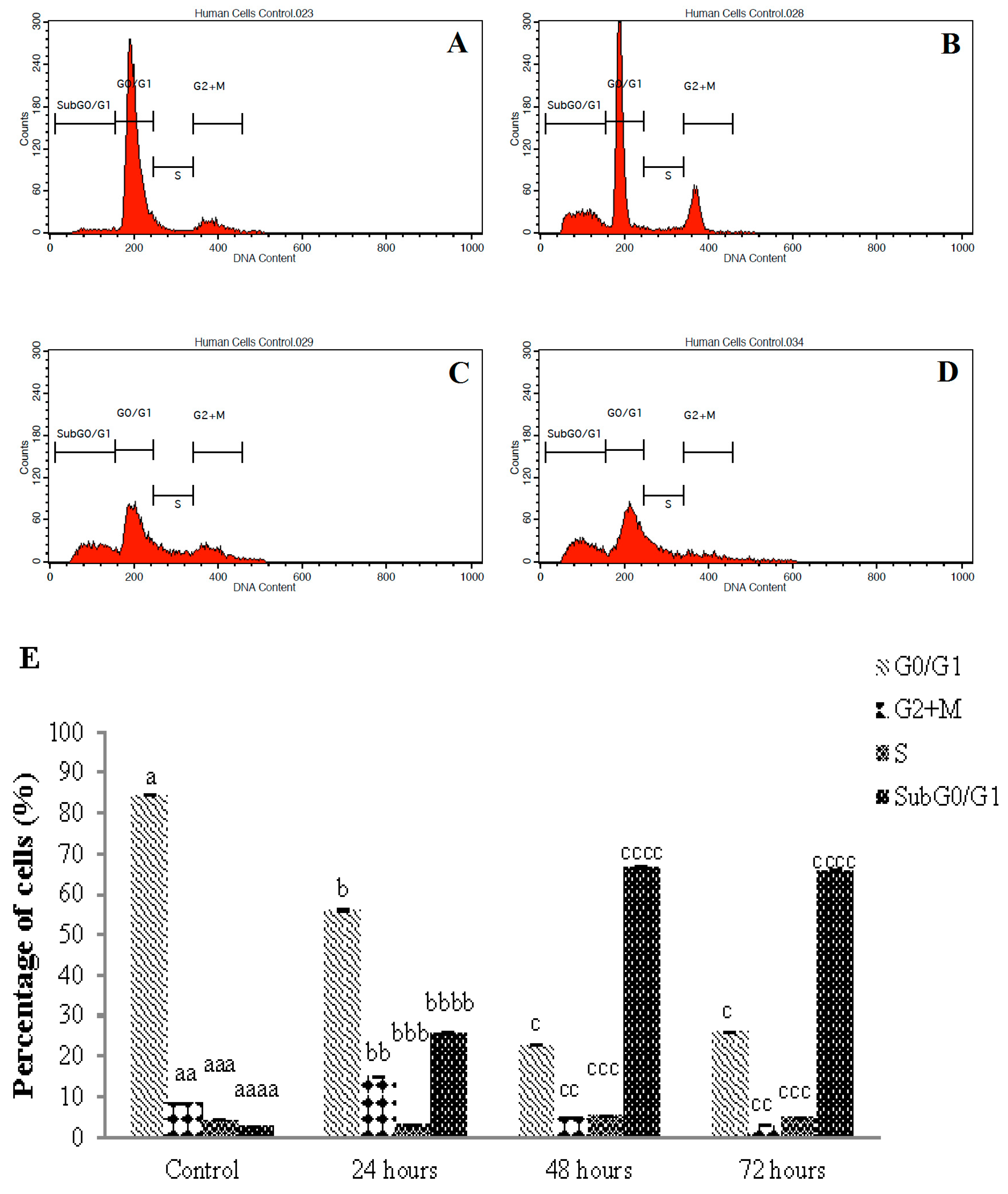
3.5. Cell Cycle Arrest of Sulforaphene Treated HepG2 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ramirez-Tagle, R.; Escobar, C.A.; Romero, V.; Montorfano, I.; Armisen, R.; Borgna, V.; Jeldes, E.; Pizzarro, L.; Simon, F.; Echeverria, C. Chalcone-induced apoptosis through caspase-dependent intrinsic pathways in human hepatocellular carcinoma cells. Int. J. Mol. Sci. 2016, 17, 260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, F.; Wang, J.; Peng, L.; Luo, H. MiR-15b mediates liver cancer cells proliferation through targeting BCL-2. Int. J. Clin. Exp. Pathol. 2015, 8, 15677–15683. [Google Scholar] [PubMed]
- Gu, E.H.; Su’udi, M.; Han, N.; Kwon, B.; Lim, S.; Kim, J. Increase in aliphatic glucosinolates synthesis during early seedling growth and insect herbivory in radish (Raphanus sativus L.) plant. Hortic. Environ. Biotechnol. 2015, 56, 255–262. [Google Scholar] [CrossRef]
- Byun, S.; Shin, S.H.; Park, J.; Lim, S.; Lee, C.; Sung, D.; Farrand, L.; Lee, S.R.; Kim, K.H.; Dong, Z.; et al. Sulforaphene supresses growth of colon cancer-derived tumours via induction of glutathione depletion and microtubule depolymerization. Mol. Nutr. Food Res. 2016, 60, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, M.B.; Lim, S.B. Formation and stabilization of raphasatin and sulforaphene from radish roots by endogenous enzymolysis. Prev. Nutr. Food Sci. 2015, 20, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Beevi, S.S.; Mangamoori, L.N.; Subathra, M.; Edula, J.R. Hexane extract of Raphanus sativus L. roots inhibits cell proliferation and induces apoptosis in human cancer cells by modulating genes related to apoptotic pathway. Plant Foods Hum. Nutr. 2010, 65, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Barillari, J.; Cervellati, R.; Paolini, M.; Tatibouet, A.; Rollin, P.; Iori, R. Isolation of 4-methylthio-3-butenyl glucosinolate from Raphanus sativus sprouts (Kaiware Daikon) and its redox properties. J. Agric. Food Chem. 2005, 53, 9890–9896. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Tyagi, A.K.; Kaur, H. Cancer modulation by glucosinolates: A review. Curr. Sci. 2000, 79, 1665–1671. [Google Scholar]
- Abdull Razis, A.F.; Mohd Noor, N. Sulforaphane is superior to glucoraphanin in modulating carcinogen-metabolising enzymes in HepG2 cells. Asian Pac. J. Cancer Prev. 2013, 14, 4235–4238. [Google Scholar] [CrossRef] [PubMed]
- Higdon, J.V.; Delage, B.; Williams, D.E.; Dashwood, R.H. Cruciferous vegetables and human cancer risk: Epidemiologic evidence and mechanistic basis. Pharmacol. Res. 2007, 55, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Pledgie-Tracy, A.; Sobolewski, M.D.; Davidson, N.E. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol. Cancer Ther. 2007, 6, 1013–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canistro, D.; Croce, C.D.; Iori, R.; Barillari, J.; Bronzetti, G.; Poi, G.; Cini, M.; Caltavuturo, L.; Perocco, P.; Paolini, M. Genetic and metabolic effects of gluconasturtiin, a glucosinolate derived from cruciferae. Mutat. Res. 2004, 545, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Heber, D. Vegetables, fruits and phytoestrogens in the prevention of diseases. J. Postgrad. Med. 2004, 50, 145–149. [Google Scholar] [PubMed]
- Park, S.; Arasu, M.V.; Lee, M.K.; Chun, J.H.; Seo, J.M.; Al-Dhabi, N.A.; Kim, S.J. Analysis and metabolite profiling of glucosinolates, anthocyanins and free amino acids in inbred lines of green and red cabbage (Brassica oleracea L.). LWT Food Sci. Technol. 2014, 58, 203–213. [Google Scholar] [CrossRef]
- Kuang, P.; Song, D.; Yuan, Q.; Lv, X.; Zhao, D.; Liang, H. Preparative separation and purification of sulforaphene from radish seeds by high-speed countercurrent chromatography. Food Chem. 2013, 136, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Ippoushi, K.; Takeuchi, A.; Ito, H.; Horie, H.; Azuma, K. Antioxidative effects of daikon sprout (Raphanus sativus L.) and ginger (Zingiber officinale Roscoe) in rats. Food Chem. 2007, 102, 237–242. [Google Scholar] [CrossRef]
- Brinker, A.M.; Spencer, G.F. Herbicidal activity of sulforaphene from stock (Matthiola incana). J. Chem. Ecol. 1993, 19, 2279–2284. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid calorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Papi, A.; Farabegoli, F.; Iori, R.; Orlandi, M.; Nicola, G.R.D.; Bagatta, M.; Angelino, D.; Gennari, L.; Ninfali, P. Vitexin-2-O-xyloside, raphasatin and (-)-epigallocatechin-3-gallate synergistically affect cell growth and apoptosis of colon cancer cells. Food Chem. 2013, 138, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Arbab, I.A.; Abdul, A.B.; Sukari, M.A.; Abdullah, R.; Syam, S.; Kamalidehghan, B.; Ibrahim, M.Y.; Taha, M.M.E.; Abdelwahab, S.I.; Ali, H.M.; et al. Dentatin isolated from Clausena excavate induces apoptosis in MCF-7 cells through the intrinsic pathway with involvement of NF-kB signalling and G0/G1 cell cycle arrest: A bioassay-guided approach. J. Ethnopharmacol. 2013, 145, 343–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.S.; Kobayashi, C.; Agata, K.; Ikeo, K.; Gojobori, T. Detection of apoptosis during planarian regeneration BYT the expression of apoptosis-related genes and TUNEL assay. Gene 2004, 333, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, J.C. Mechanism of apoptosis. Am. J. Pathol. 2000, 157, 1415–1430. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, L.; Gonzalez, V. Selected isothiocyanates rapidly induce growth inhibition of cancer cells. Mol. Cancer Ther. 2003, 2, 1045–1052. [Google Scholar] [PubMed]
- Tian, G.; Tang, P.; Xie, R.; Cheng, L.; Yuan, Q.; Hu, J. The stability and degradation mechanism of sulforaphene in solvents. Food Chem. 2016, 199, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Sangthong, S.; Weerapreeyakul, N. Simultaneous quantification of sulforaphene and sulforaphane by reverse phase HPLC and their content in Raphanus sativus L. var. caudatus Alef extracts. Food Chem. 2016, 201, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Moon, E.; Kim, S.Y.; Choi, S.U.; Lee, J.H.; Lee, K.R. 4-Methylthio-butanyl derivatives from the seeds of Raphanus sativus and their biological evaluation on anti-inflammatory and antitumor activities. J. Ethnopharmacol. 2014, 151, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Villaluenga, C.; Penas, E.; Ciska, E.; Piskula, M.K.; Kozlowska, H.; Vidal-Valverde, C.; Frias, J. Time dependence of bioactive compounds and antioxidant capacity during germination of different cultivars of broccoli and radish seeds. Food Chem. 2010, 120, 710–716. [Google Scholar] [CrossRef]
- Fahey, J.W.; Talalay, P. Antioxidant functions of sulforaphane: A potent inducer of phase II detoxication enzymes. Food Chem. Toxicol. 1999, 37, 973–979. [Google Scholar] [CrossRef]
- Sangthong, S.; Weerapreeyakul, N.; Barusrux, S. Cell survival inhibitory effect of Raphanus sativus v.caudatus alef extracts against non-resistant HepG2 and resistant HepG2 hepatocellular carcinoma. In Proceedings of the 34th the National Graduate Research Conference, Khon Kaen, Thailand, 27 March 2015. [Google Scholar]
- Suddek, G.M.; Maysara, N.M. A comparative evaluation of cytotoxicity of four different antioxidants: An In Vitro study. Int. J. Med. Pharm. Sci. 2013, 4, 17–24. [Google Scholar]
- Gowtham, K.S.; Ramakrishnan, V.; Madhusudhanan, N.; Thiyagarajan, G.; Langeswaran, V.K.; Balasubramanian, M.P. Apoptosis induced in hepatoma cells (HepG2) by allyl isothiocyanate associated with expression of P53 and activation of caspase-3. Int. J. Biol. Pharm. Res. 2011, 2, 19–26. [Google Scholar]
- Lamy, E.; Hertrampf, A.; Herz, C.; Schuler, J.; Erlacher, M.; Bertele, D.; Bakare, A.; Wagner, M.; Weiland, T.; Lauer, U.; et al. Preclinical evaluation of 4-methylthiobutyl isothiocyanate on liver cancer and cancer stem cells with different p53 status. PLoS ONE 2013, 8, e70846. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.; Kim, G.H. Anticancer and antimicrobial activities of β-phenylethyl isothiocyanate in Brassica Rapa L. Food Sci. Technol. Res. 2008, 14, 377. [Google Scholar] [CrossRef]
- Yeh, C.T.; Yen, G.C. Effect of sulforaphene in metallothionein expression and induction of apoptosis in human hepatoma HepG2 cells. Carcinogenesis 2005, 26, 2138–2148. [Google Scholar] [CrossRef] [PubMed]
- Maiyo, F.C.; Moodley, R.; Singh, M. Cytotoxicity, antioxidant and apoptosis studies of quercetin-3-O glucoside and 4-(β-d-glucopyranosyl-1→4-α-l-rhamnopyranosyloxy)-benzyl isothiocyanate from Moringa oleifera. Anticancer Agents Med. Chem. 2016, 16, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Kissen, R.; Bones, A.M. Nitrile-specifier proteins involved in glucosinolate hydrolysis in Arabidopsis thaliana. J. Biol. Chem. 2009, 284, 12057–12070. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Kelleher, M.O.; Eggleston, I.M. The cancer chemopreventive actions of phytochemicals derived from glucosinolates. Eur. J. Nutr. 2008, 47, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Morra, M.J. Glucosinolate-Containing Seed Meal as a Soil Amendment to Control Plant Pests; Subcontract Report 2000–2002; University of Idaho: Moscow, ID, USA, 2005; pp. 1–199. [Google Scholar]
- Tao, C.; He, B.B. Isolation of intact glucosinolates from mustard seed meal to increase the sustainability of biodiesel utilization. In Proceedings of the ASAE/CSAE Annual International Meeting, Ottawa, ON, Canada, 1–4 August 2004; pp. 6703–6713. [Google Scholar]
- Fimognari, C.; Nusse, M.; Cesari, R.; Iori, R.; Cantelli-Forti, G.; Hrelia, P. Growth inhibition, cell-cycle arrest and apoptosis in human T-cell leukemia by the isothiocyanate sulforaphane. Carcinogenesis 2002, 23, 581–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, I.T. Glucosinolates: Bioavailability and importance to health. Int. J. Vitam. Nutr. Res. 2002, 72, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Charron, C.S.; Kopsell, D.A.; Randle, W.M.; Sams, C.E. Sodium selenite fertilisation increases selenium accumulation and decreases glucosinolate concentration in rapid-cycling Brassica oleracea. J. Sci. Food Agric. 2001, 81, 962–966. [Google Scholar] [CrossRef]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Bottone, M.G.; Santin, G.; Aredia, F.; Bernocchi, G.; Pellicciari, C.; Scovassi, A.I. Morphological features of organelles during apoptosis: An overview. Cells 2013, 2, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Fesik, S.W. Promoting apoptosis as a strategy for cancer drug discovery. Nat. Rev. Cancer 2005, 5, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Abdul, A.B.; Abedlwahab, S.I.; Al-Zubairi, A.S.; Sukari, M.A.; Abdullah, R.; Taha, M.M.E.; Ibrahim, M.Y.; Syam, S. Thyphonium flagelliforme induces apoptosis in CEMss cells via activation of caspase-9, PARP cleavage and cytochrome c release: Its activation coupled with G0/G1 phase cell cycle arrest. J. Ethnopharmacol. 2010, 131, 592–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular mechanism controlling caspase activation and function. Cold Spring Harb. Perspect. Biol. 2013, 5, a008672. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentnall, M.; Rodriguez-Menocal, L.; Guevara, R.B.D.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Mechanism of caspase activation and inhibition during apoptosis. Mol. Cell 2002, 9, 459–470. [Google Scholar] [CrossRef]
- Dickson, M.A.; Schwartz, G.K. Development of cell-cycle inhibitors for cancer therapy. Curr. Oncol. 2009, 16, 36–43. [Google Scholar] [PubMed]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef] [PubMed]
- Angelino, D.; Jeffery, E. Glucosinolate hydrolysis and bioavailability of resulting isothiocyanates: Focus on glucoraphanin. J. Funct. Foods 2014, 7, 67–76. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| Glucoraphenin (GRE) | ND | ND | ND |
| Sulforaphene | 40.1 ± 0.41 | 38.5 ± 0.14 | 33.8 ± 0.52 |
| Cisplatin | 7.5 ± 0.12 | 2.0 ± 0.04 | 0.7 ± 0.00 |
| Compound | IC50 (μM) | ||
|---|---|---|---|
| HepG2 | MFC-7 | HT-29 | |
| Sulforaphene | 40.0 ± 0.08 a | 41.1 ± 0.08 a,b | 42.3 ± 0.05 b |
| ITCs | Incubation Hours | IC50 Value (μM) | References |
|---|---|---|---|
| Glucoraphanin ITC | 48 | 6.13 | [31] |
| Benzyl ITC | 72 | 7.30 | [24] |
| Allyl ITC | 24 | 10.00 | [32] |
| Phenethyl ITC | 72 | 11.20 | [24] |
| Glucoraphanin ITC | 72 | 12.80 | [24] |
| Glucoerucin ITC | 24 | 23.18 | [33] |
| β-phenylethyl ITC | 24 | 24.60 | [34] |
| Sulforaphene | 72 | 33.80 | Current research |
| Allyl ITC | 72 | 35.50 | [24] |
| Glucoraphanin ITC | 24 | 65.20 | [35] |
| Glucoraphenin ITC | 72 | 79.65 * | [30] |
| Glucomoringin ITC | NA | 192.71 * | [36] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kntayya, S.B.; Ibrahim, M.D.; Mohd Ain, N.; Iori, R.; Ioannides, C.; Abdull Razis, A.F. Induction of Apoptosis and Cytotoxicity by Isothiocyanate Sulforaphene in Human Hepatocarcinoma HepG2 Cells. Nutrients 2018, 10, 718. https://doi.org/10.3390/nu10060718
Kntayya SB, Ibrahim MD, Mohd Ain N, Iori R, Ioannides C, Abdull Razis AF. Induction of Apoptosis and Cytotoxicity by Isothiocyanate Sulforaphene in Human Hepatocarcinoma HepG2 Cells. Nutrients. 2018; 10(6):718. https://doi.org/10.3390/nu10060718
Chicago/Turabian StyleKntayya, Saie Brindha, Muhammad Din Ibrahim, Nooraini Mohd Ain, Renato Iori, Costas Ioannides, and Ahmad Faizal Abdull Razis. 2018. "Induction of Apoptosis and Cytotoxicity by Isothiocyanate Sulforaphene in Human Hepatocarcinoma HepG2 Cells" Nutrients 10, no. 6: 718. https://doi.org/10.3390/nu10060718