A Two-Sample Mendelian Randomization Analysis Investigates Associations Between Gut Microbiota and Celiac Disease

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
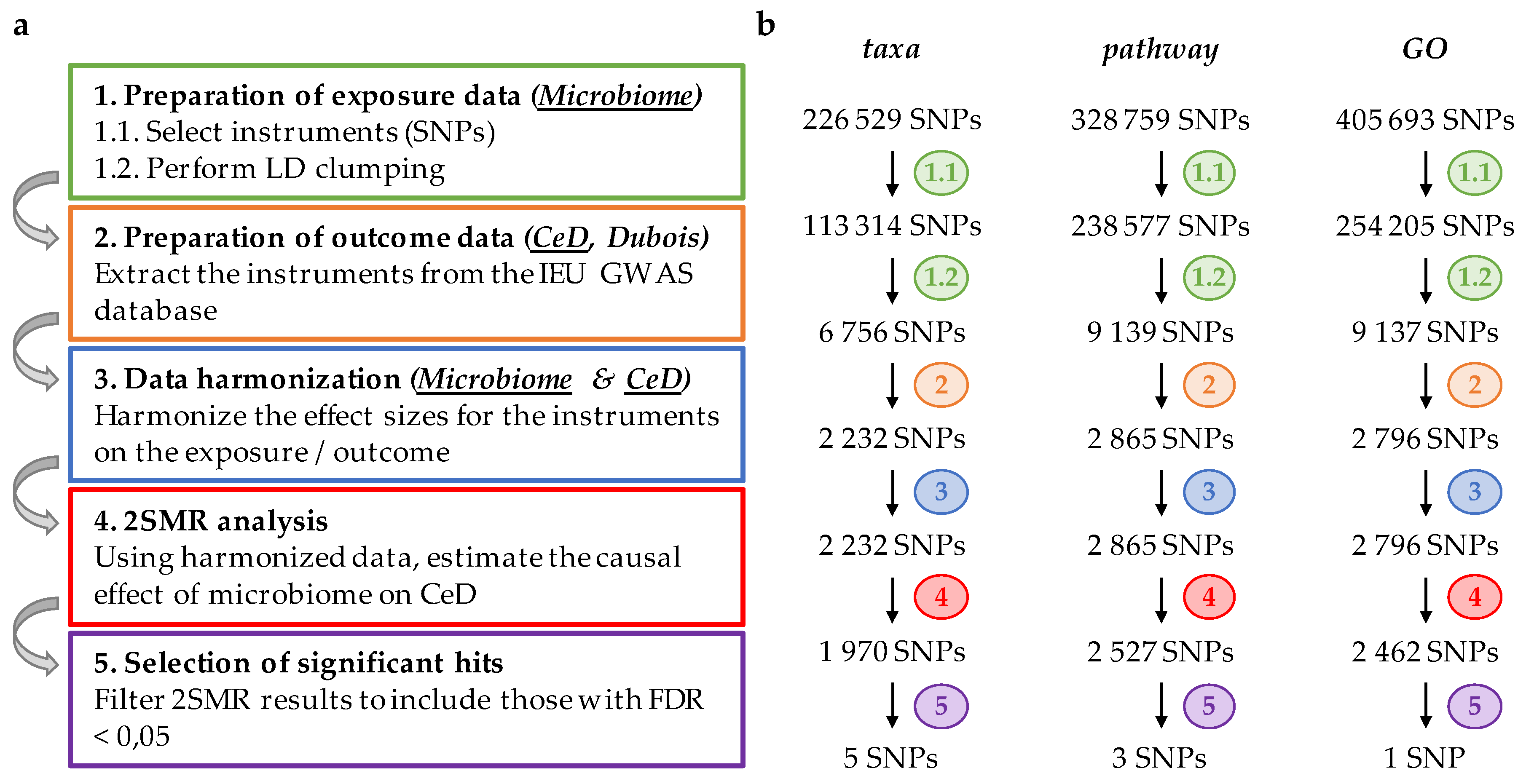
2. Materials and Methods
2.1. Preparation of Exposure Data
2.2. Preparation of Outcome Data
2.3. Harmonization of Exposure and Outcome Data
2.4. Two-Sample Mendelian Randomization (2SMR) and Statistical Analysis
3. Results
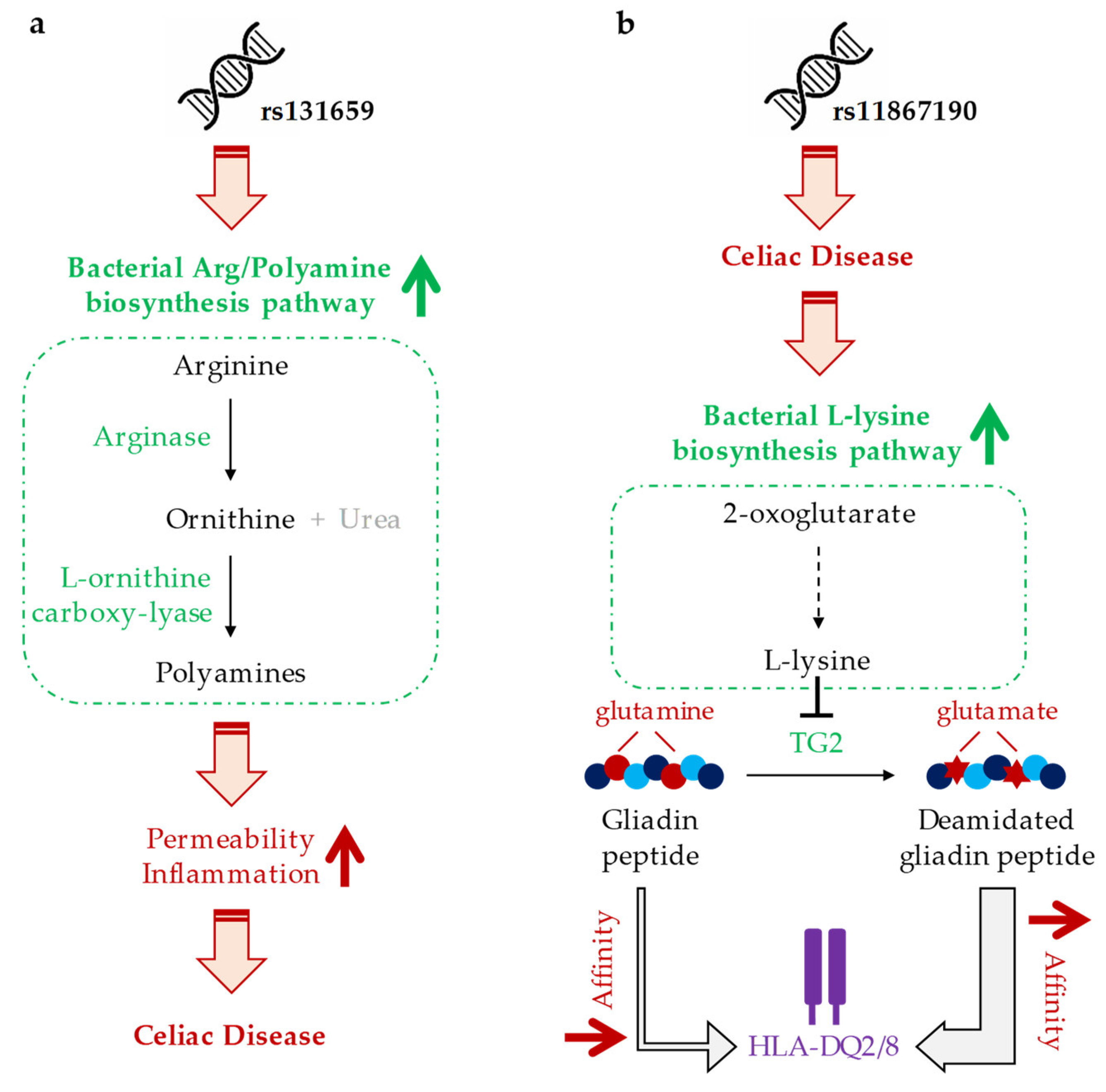
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lindfors, K.; Ciacci, C.; Kurppa, K.; Lundin, K.E.A.; Makharia, G.K.; Mearin, M.L.; Murray, J.A.; Verdu, E.F.; Kaukinen, K. Coeliac disease. Nat. Rev. Dis. Prim. 2019, 5, 1–18. [Google Scholar] [CrossRef]
- Bevan, S.; Popat, S.; Braegger, C.P.; Busch, A.; O’Donoghue, D.; Falth-Magnusson, K.; Ferguson, A.; Godkin, A.; Hogberg, L.; Holmes, G.; et al. Contribution of the MHC region to the familial risk of coeliac disease. J. Med. Genet. 1999, 36, 687–690. [Google Scholar]
- Trynka, G.; Hunt, K.A.; Bockett, N.A.; Romanos, J.; Mistry, V.; Szperl, A.; Bakker, S.F.; Bardella, M.T.; Bhaw-Rosun, L.; Castillejo, G.; et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet. 2011, 43, 1193–1201. [Google Scholar] [CrossRef]
- Dubois, P.C.A.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.R.; Ádány, R.; Aromaa, A.; et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010, 42, 295–302. [Google Scholar] [CrossRef]
- Tamburini, S.; Shen, N.; Wu, H.C.; Clemente, J.C. The microbiome in early life: Implications for health outcomes. Nat. Med. 2016, 22, 713–722. [Google Scholar] [CrossRef]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Wang, J.; Thingholm, L.B.; Skiecevičie, J.; Rausch, P.; Kummen, M.; Hov, J.R.; Degenhardt, F.; Heinsen, F.A.; Rühlemann, M.C.; Szymczak, S.; et al. Genome-wide association analysis identifies variation in Vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 2016, 48, 1396–1406. [Google Scholar] [CrossRef]
- Turpin, W.; Espin-Garcia, O.; Xu, W.; Silverberg, M.S.; Kevans, D.; Smith, M.I.; Guttman, D.S.; Griffiths, A.; Panaccione, R.; Otley, A.; et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat. Genet. 2016, 48, 1413–1417. [Google Scholar] [CrossRef]
- Bonder, M.J.; Kurilshikov, A.; Tigchelaar, E.F.; Mujagic, Z.; Imhann, F.; Vila, A.V.; Deelen, P.; Vatanen, T.; Schirmer, M.; Smeekens, S.P.; et al. The effect of host genetics on the gut microbiome. Nat. Genet. 2016, 48, 1407–1412. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Davenport, E.R.; Beaumont, M.; Jackson, M.A.; Knight, R.; Ober, C.; Spector, T.D.; Bell, J.T.; Clark, A.G.; Ley, R.E. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe 2016, 19, 731–743. [Google Scholar] [CrossRef]
- Jackson, M.A.; Verdi, S.; Maxan, M.E.; Shin, C.M.; Zierer, J.; Bowyer, R.C.E.; Martin, T.; Williams, F.M.K.; Menni, C.; Bell, J.T.; et al. Gut microbiota associations with common diseases and prescription medications in a population-based cohort. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- D’Argenio, V.; Casaburi, G.; Precone, V.; Pagliuca, C.; Colicchio, R.; Sarnataro, D.; Discepolo, V.; Kim, S.M.; Russo, I.; Del Vecchio Blanco, G.; et al. Metagenomics reveals dysbiosis and a potentially pathogenic N. flavescens strain in duodenum of adult celiac patients. Am. J. Gastroenterol. 2016, 111, 879–890. [Google Scholar] [CrossRef]
- Bodkhe, R.; Shetty, S.A.; Dhotre, D.P.; Verma, A.K.; Bhatia, K.; Mishra, A.; Kaur, G.; Pande, P.; Bangarusamy, D.K.; Santosh, B.P.; et al. Comparison of small gut and whole gut microbiota of first-degree relatives with adult celiac disease patients and controls. Front. Microbiol. 2019, 10, 137–140. [Google Scholar] [CrossRef]
- De Palma, G.; Nadal, I.; Medina, M.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Intestinal dysbiosis and reduced immunoglobulin-coated bacteria associated with coeliac disease in children. BMC Microbiol. 2010, 10, 1–7. [Google Scholar] [CrossRef]
- Nadal, I.; Donant, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Imbalance in the composition of the duodenal microbiota of children with coeliac disease. J. Med. Microbiol. 2007, 56, 1669–1674. [Google Scholar] [CrossRef]
- Sellitto, M.; Bai, G.; Serena, G.; Fricke, W.F.; Sturgeon, C.; Gajer, P.; White, J.R.; Koenig, S.S.K.; Sakamoto, J.; Boothe, D.; et al. Proof of concept of microbiome-metabolome analysis and delayed gluten exposure on celiac disease autoimmunity in genetically at-risk infants. PLoS ONE 2012, 7, 33387. [Google Scholar] [CrossRef]
- Sánchez, E.; De Palma, G.; Capilla, A.; Nova, E.; Pozo, T.; Castillejo, G.; Varea, V.; Marcos, A.; Garrote, J.A.; Polanco, I.; et al. Influence of environmental and genetic factors linked to celiac disease risk on infant gut colonization by Bacteroides species. Appl. Environ. Microbiol. 2011, 77, 5316–5323. [Google Scholar] [CrossRef]
- Olivares, M.; Benítez-Páez, A.; de Palma, G.; Capilla, A.; Nova, E.; Castillejo, G.; Varea, V.; Marcos, A.; Garrote, J.A.; Polanco, I.; et al. Increased prevalence of pathogenic bacteria in the gut microbiota of infants at risk of developing celiac disease: The PROFICEL study. Gut Microbes 2018, 9, 551–558. [Google Scholar] [CrossRef]
- Olivares, M.; Walker, A.W.; Capilla, A.; Benítez-Páez, A.; Palau, F.; Parkhill, J.; Castillejo, G.; Sanz, Y. Gut microbiota trajectory in early life may predict development of celiac disease. Microbiome 2018, 6. [Google Scholar] [CrossRef]
- Lawlor, D.A.; Harbord, R.M.; Sterne, J.A.C.; Timpson, N.; Smith, G.D. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat. Med. 2008, 27, 1133–1163. [Google Scholar] [CrossRef]
- Hartwig, F.P.; Davies, N.M.; Hemani, G.; Smith, G.D. Counterfactual causation: Avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int. J. Epidemiol. 2016, 45, 1717–1726. [Google Scholar] [CrossRef]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-base platform supports systematic causal inference across the human phenome. Elife 2018, 7, 1–29. [Google Scholar] [CrossRef]
- Olivares, M.; Neef, A.; Castillejo, G.; De Palma, G.; Varea, V.; Capilla, A.; Palau, F.; Nova, E.; Marcos, A.; Polanco, I.; et al. The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut 2015, 64, 406–417. [Google Scholar] [CrossRef]
- Collado, M.C.; Calabuig, M.; Sanz, Y. Differences between the fecal microbiota of coeliac infants and healthy controls. Curr. Issues Intest. Microbiol. 2007, 8, 9–14. [Google Scholar]
- Ercolini, D.; Francavilla, R.; Vannini, L.; De Filippis, F.; Capriati, T.; Di Cagno, R.; Iacono, G.; De Angelis, M.; Gobbetti, M. From an imbalance to a new imbalance: Italian-style gluten-free diet alters the salivary microbiota and metabolome of African celiac children. Sci. Rep. 2015, 5, 18571. [Google Scholar] [CrossRef]
- Visconti, A.; Le Roy, C.I.; Rosa, F.; Rossi, N.; Martin, T.C.; Mohney, R.P.; Li, W.; de Rinaldis, E.; Bell, J.T.; Venter, J.C.; et al. Interplay between the human gut microbiome and host metabolism. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef]
- Wacklin, P.; Kaukinen, K.; Tuovinen, E.; Collin, P.; Lindfors, K.; Partanen, J.; Mäki, M.; Mättuö, J. The duodenal microbiota composition of adult celiac disease patients is associated with the clinical manifestation of the disease. Inflamm. Bowel Dis. 2013, 19, 934–941. [Google Scholar] [CrossRef]
- Sánchez, E.; Donat, E.; Ribes-Koninckx, C.; Fernández-Murga, M.L.; Sanz, Y. Duodenal-mucosal bacteria associated with celiac disease in children. Appl. Environ. Microbiol. 2013, 79, 5472–5479. [Google Scholar] [CrossRef]
- Verdu, E.F.; Galipeau, H.J.; Jabri, B. Novel players in coeliac disease pathogenesis: Role of the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 497–506. [Google Scholar] [CrossRef]
- Mullaney, J.A.; Stephens, J.E.; Costello, M.E.; Fong, C.; Geeling, B.E.; Gavin, P.G.; Wright, C.M.; Spector, T.D.; Brown, M.A.; Hamilton-Williams, E.E. Type 1 diabetes susceptibility alleles are associated with distinct alterations in the gut microbiota. Microbiome 2018, 6. [Google Scholar]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Barilli, A.; Rotoli, B.M.; Visigalli, R.; Ingoglia, F.; Cirlini, M.; Prandi, B.; Dall’Asta, V. Gliadin-mediated production of polyamines by RAW264.7 macrophages modulates intestinal epithelial permeability in vitro. Biochim. Biophys. Acta—Mol. Basis Dis. 2015, 1852, 1779–1786. [Google Scholar] [CrossRef] [PubMed]
- Barilli, A.; Gaiani, F.; Prandi, B.; Cirlini, M.; Ingoglia, F.; Visigalli, R.; Rotoli, B.M.; De’Angelis, N.; Sforza, S.; De’Angelis, G.L.; et al. Gluten peptides drive healthy and celiac monocytes toward an M2-like polarization. J. Nutr. Biochem. 2018, 54, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Elli, L.; Roncoroni, L.; Hils, M.; Pasternack, R.; Barisani, D.; Terrani, C.; Vaira, V.; Ferrero, S.; Bardella, M.T. Immunological effects of transglutaminase-treated gluten in coeliac disease. Hum. Immunol. 2012, 73, 992–997. [Google Scholar] [CrossRef]
- Wang, J.; Kurilshikov, A.; Radjabzadeh, D.; Turpin, W.; Croitoru, K.; Bonder, M.J.; Jackson, M.A.; Medina-Gomez, C.; Frost, F.; Homuth, G.; et al. Meta-analysis of human genome-microbiome association studies: The MiBioGen consortium initiative. Microbiome 2018, 6. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
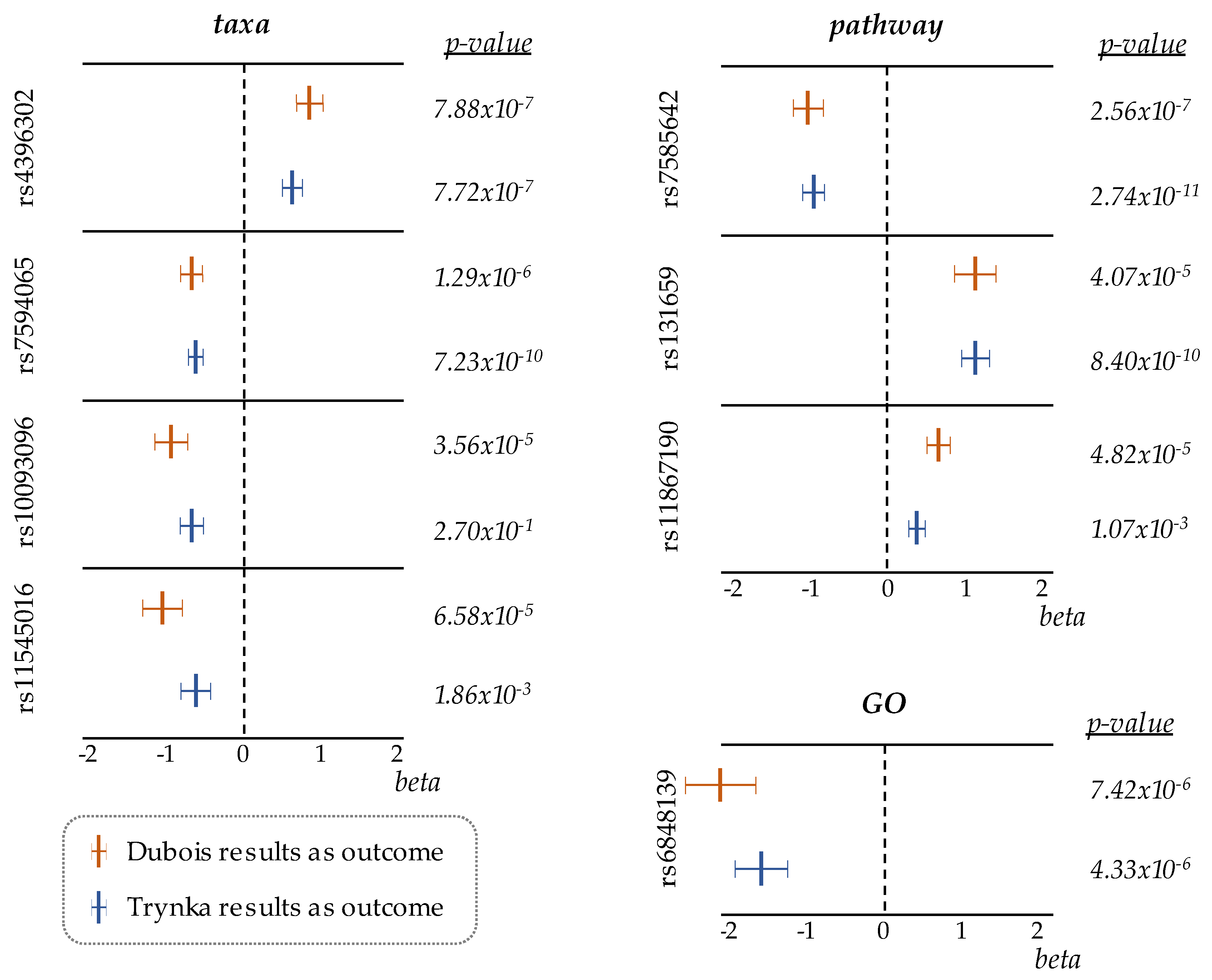
| SNP | Effect/Other Alleles | Chr. | Position | p-Value | Effect size ± SE | FDR |
|---|---|---|---|---|---|---|
| taxa | ||||||
| rs4396302 | A/G | 11 | 128420926 | 7.88 × 10−7 | 0.80 ± 0.16 | 0.001 |
| rs7594065 | T/C | 2 | 204814676 | 1.29 × 10−6 | −0.61 ± 0.13 | 0.001 |
| rs10093096 | C/T | 8 | 64907701 | 3.56 × 10−5 | −0.84 ± 0.20 | 0.027 |
| rs11545016 | T/C | 8 | 22438313 | 6.58 × 10−5 | −0.96 ± 0.24 | 0.037 |
| rs12913063 | T/C | 15 | 75424593 | 9.97 × 10−5 | 1.09 ± 0.28 | 0.044 |
| pathway | ||||||
| rs7585642 | A/C | 2 | 61217542 | 2.56 × 10−7 | −0.92 ± 0.18 | 0.001 |
| rs131659 | G/A | 22 | 21964761 | 4.07 × 10−5 | 1.04 ± 0.25 | 0.046 |
| rs11867190 | A/G | 17 | 5261220 | 4.82 × 10−5 | 0.59 ± 0.14 | 0.046 |
| GO | ||||||
| rs6848139 | C/A | 4 | 123395041 | 7.42 × 10−6 | −1.95 ± 0.43 | 0.021 |
| SNP | Associated Microbiota Trait |
|---|---|
| taxa | |
| rs4396302 | Firmicutes (p), Clostridia (c), Clostridiales (o), Peptostreptococcaceae (f), Peptostreptococcaceae (g), Peptostreptococcaceae unclassified (s) |
| rs7594065 | Firmicutes (p), Clostridia (c), Clostridiales (o), Clostridiales noname (f), Pseudoflavonifractor (g) |
| rs10093096 | Proteobacteria (p) |
| rs11545016 | Firmicutes (p), Clostridia (c), Clostridiales (o), Lachnospiraceae (f), Lachnospiraceae noname (g) |
| pathway | |
| rs7585642 | PWY-6060 (malonate degradation II, biotin-dependent) |
| rs131659 | ARG+POLYAMINE-SYN (super pathway of arginine and polyamine biosynthesis) |
| rs11867190 | PWY-3081 (L-lysine biosynthesis V) |
| GO | |
| rs6848139 | GO:0016831 (MF, carboxy-lyase activity) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Santisteban, I.; Cilleros-Portet, A.; Moyua-Ormazabal, E.; Kurilshikov, A.; Zhernakova, A.; Garcia-Etxebarria, K.; Fernandez-Jimenez, N.; Bilbao, J.R. A Two-Sample Mendelian Randomization Analysis Investigates Associations Between Gut Microbiota and Celiac Disease. Nutrients 2020, 12, 1420. https://doi.org/10.3390/nu12051420
García-Santisteban I, Cilleros-Portet A, Moyua-Ormazabal E, Kurilshikov A, Zhernakova A, Garcia-Etxebarria K, Fernandez-Jimenez N, Bilbao JR. A Two-Sample Mendelian Randomization Analysis Investigates Associations Between Gut Microbiota and Celiac Disease. Nutrients. 2020; 12(5):1420. https://doi.org/10.3390/nu12051420
Chicago/Turabian StyleGarcía-Santisteban, Iraia, Ariadna Cilleros-Portet, Elisabet Moyua-Ormazabal, Alexander Kurilshikov, Alexandra Zhernakova, Koldo Garcia-Etxebarria, Nora Fernandez-Jimenez, and Jose Ramon Bilbao. 2020. "A Two-Sample Mendelian Randomization Analysis Investigates Associations Between Gut Microbiota and Celiac Disease" Nutrients 12, no. 5: 1420. https://doi.org/10.3390/nu12051420
APA StyleGarcía-Santisteban, I., Cilleros-Portet, A., Moyua-Ormazabal, E., Kurilshikov, A., Zhernakova, A., Garcia-Etxebarria, K., Fernandez-Jimenez, N., & Bilbao, J. R. (2020). A Two-Sample Mendelian Randomization Analysis Investigates Associations Between Gut Microbiota and Celiac Disease. Nutrients, 12(5), 1420. https://doi.org/10.3390/nu12051420