
CRABP1 in Non-Canonical Activities of Retinoic Acid in Health and Diseases


Department of Pharmacology, University of Minnesota, Minneapolis, MN 55455, USA
*
Author to whom correspondence should be addressed.
Nutrients 2022, 14(7), 1528; https://doi.org/10.3390/nu14071528
Submission received: 28 February 2022
/
Revised: 1 April 2022
/
Accepted: 3 April 2022
/
Published: 6 April 2022
(This article belongs to the Special Issue Recent Retinoid Research: Implications for Human Health)
Abstract
:In this review, we discuss the emerging role of Cellular Retinoic Acid Binding Protein 1 (CRABP1) as a mediator of non-canonical activities of retinoic acid (RA) and relevance to human diseases. We first discuss the role of CRABP1 in regulating MAPK activities and its implication in stem cell proliferation, cancers, adipocyte health, and neuro-immune regulation. We then discuss an additional role of CRABP1 in regulating CaMKII activities, and its implication in heart and motor neuron diseases. Through molecular and genetic studies of Crabp1 knockout (CKO) mouse and culture models, it is established that CRABP1 forms complexes with specific signaling molecules to function as RA-regulated signalsomes in a cell context-dependent manner. Gene expression data and CRABP1 gene single nucleotide polymorphisms (SNPs) of human cancer, neurodegeneration, and immune disease patients implicate the potential association of abnormality in CRABP1 with human diseases. Finally, therapeutic strategies for managing certain human diseases by targeting CRABP1 are discussed.
Keywords:
CRABP1; retinoic acid; neurodegeneration; inflammation; metabolism; cancer; human disease; non-canonical; MAPK; CAMKII1. Introduction: Canonical and Non-Canonical Activities of All-Trans Retinoic Acid (atRA)
Vitamin A (also known as retinol) is an essential nutrient required for almost all physiological processes [1]. The profound effects of vitamin A are elicited mainly through atRA, as well as its various isomers. Through decades of studies, it has been established that atRA, as the principal active metabolite of vitamin A, executes its activities through binding to its nuclear receptors, RA receptors (RARs), which usually pair with Retinoid X Receptors (RXRs), can bind the cis isomers of atRA. These RAR/RXR pairs, in various combinations, act to regulate the transcription of numerous target genes that harbor RA response elements (RAREs) in their regulatory regions [2,3]. RAR/RXR pairs often also act together with other transcription factors to confer further specificity in the expression of target genes, resulting in the tight regulation of specific cellular processes such as proliferation [4], differentiation [5], apoptosis [6], and other physiological functions. These ultimately ensure the homeostasis of most organ systems/physiological processes [2,7]. Dysregulated RA signaling often leads to disease conditions [8,9,10]. These RAR-mediated activities of RA, which occur in the nucleus to regulate the execution of genetic programs, generally span an extended period of time (days to years) and are referred to as canonical activities of RA [11]. An extensive body of work has determined that the cellular retinoid-binding proteins (CRABPs) I and II facilitate these canonical activities of RA. Rigorous biochemical studies have characterized classical CRABP1 functions in RA binding, sequestration, and metabolism via cytochrome (CYP) P-450 enzymes [12,13], (reviewed in-depth in [14,15,16,17]) while CRABP2 is responsible for the transport of RA to the nucleus [14,15,18,19].
In 2008, a study first reported a novel activity (effect) of atRA that occurred rapidly (within minutes) to alter the protein phosphorylation status of a transcription factor TR2 in maintaining stem cell proliferation and stemness potential [20]. Subsequent studies [21,22,23] further documented similar activities of atRA that shared several features: (1) RAR-independence, (2) occurring in the cytosol without altering gene expression, and (3) rapid (typically within minutes) action. These novel activities of atRA were collectedly referred to as “non-canonical” and were later found to be mediated by the Cellular Retinoic Acid Binding Protein 1 (CRABP1) [22]. These CRABP1-mediated non-canonical activities of atRA were ultimately validated in careful studies of Crabp1 knockout (CKO) mice and cultures, which also revealed the physiological/disease relevance of CRABP1 [24,25,26,27,28,29,30,31,32,33].
Extensive molecular and cell biological studies have identified specific cytosolic signaling pathways that can be targeted by CRABP1 in a cell context-dependent manner. It is believed that CRABP1 functions as a signal integrator by forming various specific RA-regulated signaling protein complexes (signalsomes) in different cells to modulate specific cellular processes/functions. Below we summarize two validated CRABP1-mediated, non-canonical RA signaling pathways, and discuss evidence/implications for the role of CRABP1-signalsome in human diseases.
2. CRABP1-Signalsomes
CRABP1 is the most highly conserved retinoid-binding protein among all the known binding proteins and nuclear receptors for retinoids. CRABP1 binds, specifically, atRA with a high affinity (<1 nM) [34,35,36,37]. Given its high affinity toward atRA and cytosolic distribution, CRABP1 has been proposed and shown to sequester the poorly soluble RA from the aqueous cytosolic environment [12,13,14,15,16,17,30]. This led to the belief that CRABP1 would function to control RA availability in the cell, which indeed was supported by several molecular studies, by altering the expression level of CRABP1, that documented subsequent changes in the expression of RA-responding genes [38,39]. As introduced earlier, CRABP1 could participate in RA metabolism by delivering RA to CYP P-450 metabolic enzymes and microsomes via protein-protein interactions and substrate channeling [15,40]. However, the physiological role of CRABP1 in mediating the newly observed, non-canonical activity has remained largely elusive. Only recently, studies of CKO mice and cultures in various physiological/pathological conditions (see the following section) began to shed light on multiple functional roles of CRABP1 in modulating specific cellular processes, which contributed to the “non-canonical” activity. The fact that CRABP1 is important for multiple signaling pathways is consistent with the extremely high conservation of its amino acid sequence across animal species. Figure 1 shows the reported amino acid sequence alignment of CRABP1 among five animal species including human [41], pig [42], rat [43], mouse [44], and bovine [45]. Importantly, there is only a single residue, at position 86, that is not conserved, with alanine in human and pig sequences and proline in mouse, rat, and bovine sequences (Figure 1).
The extreme conservation of CRABP1 during evolution would suggest important functional constraints. The evidence for this notion was obtained in careful studies of CKO mouse phenotypes (see later). Mechanistic details were provided in biochemical and cellular studies that first revealed specific context-dependent “binding partners” of CRABP1, which were rigorously defined according to at least two criteria: (a) specific and direct binding to CRABP1, which could be validated in vitro, and (b) forming specific cytosolic protein complexes that could be validated in vivo. Functional consequences of these CRABP1-containing protein complexes were each found to be capable of modulating certain specific cytosolic signaling pathways in a particular cell type. These CRABP1-containing protein complexes are therefore referred to as CRABP1-signalsomes. Currently, two types of CRABP1-signalsomes have been identified, which are discussed in the following sections.
2.1. CRABP1-MAPK (RAF-MEK-ERK) Signalsome in Stem Cells, Cancers
A specific Crabp1-signaling complex was first proposed after studying embryonal carcinoma (EC) and embryonic stem (ES) cells that were stimulated by a physiological concentration (10 nM) of atRA to modulate their proliferation/differentiation (reviewed in [46,47]). The initial study detected a very rapid (within minutes) response of these cells to atRA administration, which occurred in the cytosol and involved a mitogen-activated protein kinase (MAPK) pathway to modify target proteins for specific post-translational modifications [20,21,22,23]. This atRA-elicited signal was found to involve CRABP1, and could rapidly (within minutes) alter (dampen) the activity of the initiating kinase of the MAPK pathway, which is the rapidly Accelerated Fibrosarcoma (RAF) kinase and is a cell membrane-anchored kinase activated by the mitogenic signal Ras GTPase [48]. The MAPK kinase signaling cascade is comprised of Ras GTPase which activates RAF, then mitogen-activated protein kinase kinase (MEK), and then extracellular-signal-regulated kinase (ERK). Activation of this signaling pathway generally leads to cell proliferation and growth for stem/progenitor cells [48]. Through biochemical and molecular studies, it is now established that CRABP1 competes with Ras by directly interacting with RAF at its Ras-binding domain, thereby dampening MAPK signal propagation and ultimately modulating (reducing) cell proliferation of ES, EC, and neural stem cell (NSC) [22,25,29]. The proposed mechanistic model for CRABP1-MAPK signalsome is shown in Figure 2.
To this end, the physiological/pathological relevance of CRABP1 is most evident in cancers. For instance, the CRABP1 gene has been reported as a tumor suppressor or an oncogene in animals and humans [15,49,50,51,52,53,54,55,56,57,58,59,60,61]. In comparing CKO and wild-type ESCs, as well as in gain- and loss-of-functional studies of cancer cell models, it was found that CRABP1 was involved in modulating cell cycle control [22]. By competing with Ras for forming complexes with RAF/MEK, atRA-CRABP1 dampened mitogen-activated ERK activity and suppressed cell cycle progression by expanding the G1 phase [22,29]. This supports the notion that CRABP1 can be a tumor suppressor. Additional evidence supporting a functional role for CRABP1 in stem cell proliferation was obtained from studying CKO mice that were found to have expanded NSC pools (as a result of enhanced NSC proliferation in CRABP1-deleted hippocampus), which was consistent with the CKO mouse behavior indicating improved memory function [25]. Importantly, the hippocampus is among the tissues where CRABP1 is most highly expressed, especially in the NSC-rich region of the dentate gyrus. Thus, CRABP1 can participate in the homeostatic control of the NSC pool in the brain. Readers are referred to an in-depth review of this CRABP1-regulated signaling pathway by Nagpal and Wei [62].
2.2. Crabp1-MAPK Signalsome in Metabolism and Immunity
Lin et al. first observed that CKO mice exhibited increased high-fat diet (HFD)-induced obesity and insulin resistance (IR), suggesting a protective role for CRABP1 against the development of metabolic disorders. A molecular study of CKO mice elucidated an underlying mechanism for this metabolic phenotype that, in normal adipocytes, CRABP1 could negatively regulate ERK activity to inhibit adipogenesis and adipose hypertrophy [28]. Therefore, CKO mice are more prone to HFD-induced obesity and IR. To this end, it has been reported that pharmacological doses of RA could inhibit adipogenesis and protect against obesity, and this was attributed, primarily, to RAR-mediated activities [63,64,65,66,67]. These recent studies of CKO models revealed CRABP1 as an additional player in mediating physiological activities of atRA regarding metabolic homeostasis and the maintenance of healthy adipose tissue [28].
In examining the systemic inflammatory status/potential of CKO mice, it was found that HFD-fed CKO mice all had increased systemic inflammation, indicated by invading immune cells in adipose tissue [28], increased inflammatory driver Receptor Interacting Protein 140 (RIP140) (gene name Nrip1) [68] in the blood [31], elevation in inflammatory cytokines, and significantly enhanced macrophage M1 polarization (unpublished). Previous studies also indicated that CKO mice had overall increased inflammation in the heart, indicated by increased cardiac fibrosis [26], and an altered anxiety and stress response in their HPA axis [32]. To this end, CRABP1 was found to be involved in exosome secretion from CRABP1-expressing neurons. Specifically, the RIP140-containing exosome population was significantly expanded in the blood and cerebral spinal fluid (CSF) of CKO mice, due to, in part, increased exosome secretion from CKO neurons [31]. Importantly, these neuron-derived RIP140-containing exosomes could be engulfed by macrophages to increase their inflammatory M1 polarization, thereby increasing systemic inflammation. This study, by monitoring the intercellular transfer of the inflammatory driver, RIP140, demonstrates exosome secretion as a potent means to transfer neuronal inflammation into systemic inflammation; mechanistically, this study identifies CRABP1 as an important regulator of exosome secretion from specific CRABP1-expressing neurons, which also involves the MAPK-ERK signaling in these neurons [31].
2.3. CRABP1-CaMKII Signalsome in Cardiomyocytes and Motor Neurons (MNs)
A different CRABP1-signaling complex was identified from studying deteriorated heart function of CKO mice [26,27], and their premature weakening in motor function [33]. The expression study confirmed CRABP1 expression in cardiomyocytes [26] (relevant to the CKO heart phenotype) and motor neurons (relevant to the CKO motor function phenotype) [33]. This signaling complex is comprised of CRABP1 and calcium-calmodulin-dependent kinase 2 (CaMKII), an enzyme critical to calcium signaling/handling and highly enriched in both cardiomyocytes [69] and neurons [70,71]. It is known that CaMKII regulates contraction in cardiomyocytes [69] and long-term potentiation in neurons [70,71], respectively. Both types of cells are highly dependent upon calcium homeostasis for their functions where CaMKII is a key mediator of calcium signaling [72]. All the CaMKII isoforms have a conserved architecture comprised of the kinase, regulatory, and association/oligomerization domains, and share the same activation mechanism through the binding of calmodulin to the calmodulin-binding domain (CaMBD) within its regulatory domain. CaMKII activation occurs when intracellular (Ca2+) increases and binds calmodulin. Ca2+-calmodulin then binds and activates CaMKII, which is often marked by phosphorylation at threonine 286/7 (T286/7), depending on the CaMKII isoform [73,74]. In vitro data showed that CRABP1competes with calmodulin by directly interacting with CaMKII at the CaMBD [26,27]. Therefore, CRABP1 could dampen Ca2+/Calmodulin activated CaMKII activity. Since over-activation of CaMKII is a major trigger of the death/damage of cardiomyocytes [75] and neurons [76], by dampening CaMKII over-activation, CRABP1 can play a protective role in maintaining the health of both the heart and neurons. These are elaborated on in the following section. The proposed mechanistic model for CRABP1-CaMKII signalsome is shown in Figure 3.
2.3.1. CRABP1-CaMKII Signalsome in Cardiomyocytes
CKO mice naturally and gradually exhibited cardiac hypertrophy, reflected in their significantly depressed heart function in older animals [26]. Using the isoproterenol (ISO)-induced cardiomyopathy model for heart failure [77,78], studies showed that CKO mice were more sensitive/vulnerable to ISO treatment. Acute, high-dose ISO treatment activates beta-adrenergic receptors to induce acute cardiomyocyte contractions, triggering a pathological condition of heart overactivation. Chronic ISO treatment induces more severe cardiac hypertrophy, and, eventually, fibrosis and necrosis occur, mimicking heart failure in human patients [77,78]. Interestingly, in the acute ISO treated model, CKO mice were more sensitive and exhibited a significantly increased CaMKII activity marked by elevated T286 phospho-status and phosphorylation of the CaMKII cardiac substrate, PLN. These were supported by molecular studies described above, that CRABP1 dampened CaMKII activation by competing with calmodulin for its binding to CaMKII [26]. In a subsequent study [19], it was found that pretreatment with atRA before chronic ISO administration could attenuate ISO-induced heart damage and CaMKII activity in the wild type, but not the CKO mice [27]. These studies clearly demonstrated a protective role for CRABP1, as well as the potential application of CRABP1-ligand such as RA, in certain heart damage/diseased conditions.
2.3.2. Crabp1-CaMKII Signalsome in MNs
CRABP1 expression is tissue and cell-type specific. In the central nervous system, it is specifically and highly expressed in spinal cord MNs [33]. These neurons project to and innervate, primarily, muscles to form tightly regulated structures called neuromuscular junctions (NMJs) [79]. Neuronal activity from MNs is propagated through NMJs to elicit muscle contraction, and calcium signaling/handling (mediated by CaMKII) is critical to the function of both presynaptic (MN) [80,81,82] and post-synaptic (muscle) [83,84] compartments. MNs release neurotransmitters such as acetylcholine to induce muscle contraction, and express Agrin, a proteoglycan essential for NMJ development and maintenance [79,85]. Crabp1 is specifically expressed in the presynaptic compartment, comprised of MNs, but is not expressed in the post-synaptic muscle compartment [33]. This study identified CRABP1-CaMKII signaling in MNs, which contributed to the regulation of Agrin expression and its downstream target, the muscular LRP4-MuSK signaling that maintained AChR clusters and healthy NMJ [79,86]. By comparing to wild-type mice, CKO mice were found to exhibit age-dependent more profound motor deterioration, reflected in their significantly reduced grip strength compared to the age-controlled group. Detailed histological studies revealed more severely damaged NMJs in CKO mice as compared to wild-type mice, characterized by irregular NMJ morphology, fragmentation, and reduced number. Consistently, in the CKO spinal cord tissues, CaMKII activity was significantly increased as compared to WT spinal tissues. Pathological CaMKII activation (overactivation) occurs in multiple disease states of the nervous system, frequently referred to as excitotoxicity [76]. In MN1 culture (a spinal MN cell line), inhibiting CaMKII via KN-93 (mimicking CRABP1 dampening effect) increased their Agrin expression, consistent with the reduction in Agrin detected in CKO NMJ tissues. It is concluded that CRABP1, in MNs, can target CaMKII to dampen its over-activation, which provides a protective mechanism against over-activation of CaMKII that could lead to MN degeneration. Importantly, re-expressing CRABP1 in young (before disease onset) CKO mice could partially rescue their motor deficits and correct CaMKII activity and Agrin expression.
3. Crabp1 in Two Common Human Diseases: Cancer and Neurodegeneration
CRABP1 has been studied mostly in the context of nutrition, in particular vitamin A metabolism and homeostasis. The increasingly reported biological functions of CRABP1 as described above are all very different from the canonical RAR-mediated effects that typically alter genome programming and gene expression. The physiological relevance of these CRABP1-mediated effects has been illustrated in both CKO mice and tissue culture systems which model various human diseases. The multiple functions of CRABP1 would predict numerous disease conditions where CRABP1 can be involved. Indeed, CKO mice exhibited multiple phenotypes mimicking human diseases [24,25,26,27,28,29,30,31,32]. In tissue cultures, it is possible to examine the effects of its best-known ligand, atRA, in eliciting non-canonical activities through CRABP1, and to demonstrate holo- and apo-CRABP1’s functions in specific cell types. In a genetically manipulated mouse model such as CKO, it is possible to illustrate how CRABP1 can participate in physiological processes and prevent diseased conditions/progression. However, given the technical difficulty in manipulating vitamin A and RA status in mice, the contribution of endogenous RA to the prevention of diseases, via CRABP1, remains elusive. Nevertheless, the implication of CRABP1 in human diseases can be uncovered by mining the available human data sets and literature, which has yielded some interesting information supporting a potential role for CRABP1 in human diseases. Below, we discuss several human studies/data sets that have revealed altered expression or protein sequence of CRABP1 in human patients. First, the reported genetic association of CRABP1 in various human diseases is summarized in Table 1, followed by a discussion on specific implications in cancers, neurodegeneration, and other rare diseases. The relevant accession IDs of CRABP1 expression studies from the EMBL-EBI Expression Atlas Data Repository [87] are provided in Table 1.
3.1. CRABP1 in Cancers
Dysregulation of CRABP1 expression in cancers is a well-documented phenomenon (Table 1; [15,49,50,51,52,53,54,55,56,57,58,59,60,61]). Furthermore, cancer databases such as The Cancer Genome Atlas (TCGA) and cBioPortal [99,100] for Cancer Genomics have revealed numerous single nucleotide polymorphisms (SNPs) in patients across various cancers. These SNPs could result in various defects in CRABP1 such as synonymous mutation, splicing alternation, missense mutation, and augmented expression levels. Figure 4a lists SNPs present in patients from various cancer types that occurred in the −3 kb upstream regulatory region, which could affect CRABP1 expression levels; Figure 4b lists SNPs in the coding region that could alter the CRABP1 sequence. However, no experimental data have been provided to validate the “disease association” of these SNPs. Nevertheless, given the conservation of CRABP1 across mammals, any alterations in CRABP1 caused by these SNPs could potentially disturb CRABP1 functions and normal cellular processes especially proliferation which could impact tumor formation or progression.
3.2. CRABP1 in Neurodegeneration
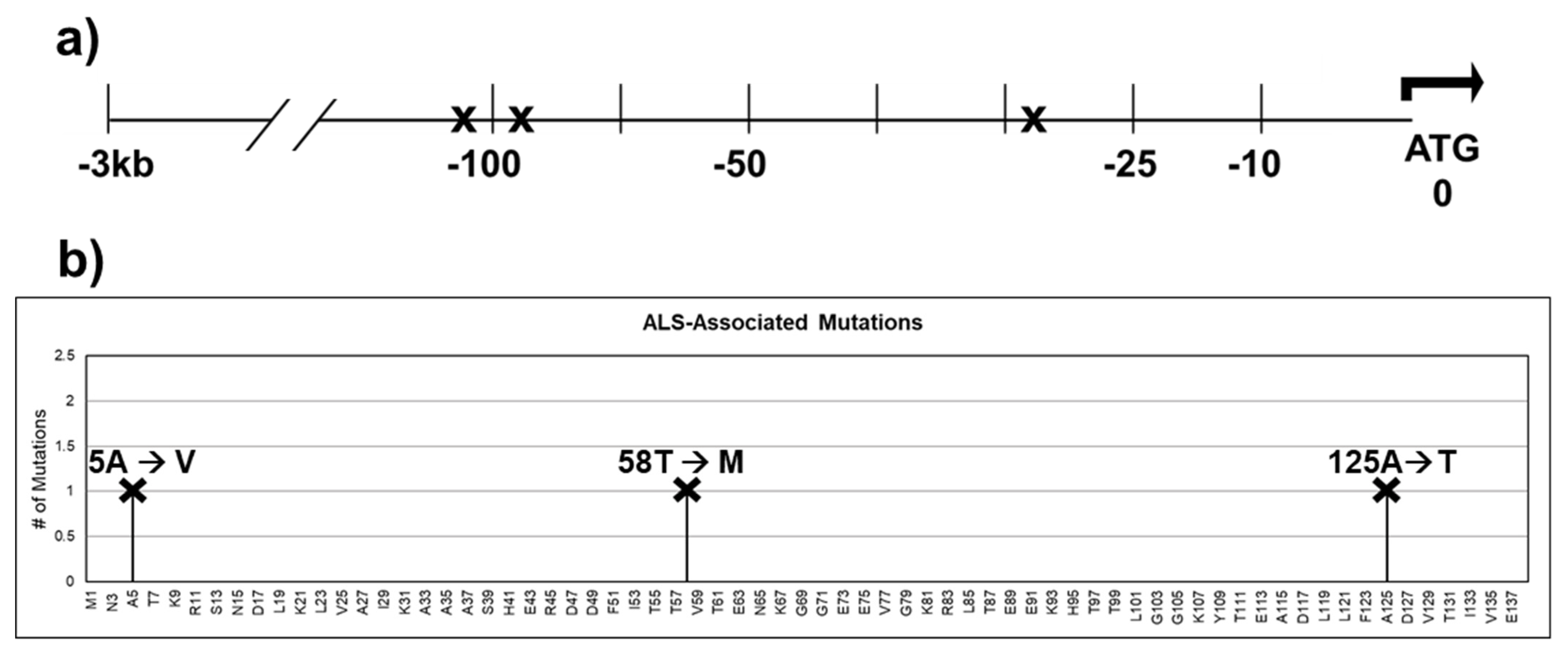
CRABP1 expression has been found to be reduced in the following neurodegenerative disease conditions: amyotrophic lateral sclerosis, spinal muscular dystrophy, and age-related macular degeneration. Data mining of the ALS Variant Server (http://als.umassmed.edu/, accessed on 31 January 2022) revealed several SNPs present in ALS patients that are located in the upstream regulatory region or in the coding region of CRABP1 (Figure 5a,b). These SNPs in ALS patients could potentially alter CRABP1 levels or functions, thereby contributing to disease initiation or progression. However, experiments are needed to verify the disease relevance of these SNPs. Interestingly, a study by Jiang et al. identified CRABP1 as the most significantly suppressed gene in ALS patients’ motor neurons (MNs) as compared to healthy subjects, suggesting that CRABP1 may play a role in ALS etiology. This is consistent with the severe motor degeneration phenotype of CKO mice in older age groups [33]. It would be interesting to experimentally examine the potential contribution of SNPs identified from the AVS database in various neurodegenerative diseases.
The importance of CRABP1 in neurons, particularly MNs, is further supported by the finding that the mouse Crabp1 gene is tightly regulated by sonic hedgehog (Shh) [30], a potent inducer of motor neuron differentiation [101]. It appears that Shh activates glioma-associated oncogene homolog 1 (Gli1) that binds the Gli target sequence in Crabp1′s regulatory region, thereby up-regulating Crabp1 expression [30]. Therefore, for MN differentiation and function, proper expression of CRABP1 is important.
3.3. CRABP1 in Rare Human Diseases
Altered CRABP1 level or function has also been observed in other diseases. In Moyamoya Disease (MMD), a vascular disease characterized by progressive occlusion of cerebral arteries [102], CRABP1 protein level was found to be increased in the CSF of MMD patients [96]. Kim et al. speculated that the increase in CRABP1 during MMD progression might disrupt the regulatory activity of retinoids on growth factor signaling responsible for arterial occlusion [96]. A study by Hur et al. also speculated an increase in CRABP1 as a potential biomarker of diabetic neuropathy [97]. CRABP1 has also been implicated in HIV therapy associated with lipodystrophy and metabolic disorder. Carr et al. proposed that the toxic effects on adipose and metabolism associated with the use of HIV-1 protease inhibitors were, in part, due to these inhibitors’ direct binding and inhibiting CRABP1 function [98]. However, no experimental data have been presented to substantiate or support a role for CRABP1 in these rare human diseases.
4. Conclusions and Future Directions
The CRABPs have been established as key players in RA binding, sequestration, metabolism, and nuclear transport to RARs. In addition to these classical functions, novel roles in the CRABPs have also been observed, such as CRABPII in RNA transcript stabilization [103,104] and as a tumor suppressor in breast cancer [105,106]. Here, we have reviewed the novel functions of the CRABP1 as signalsomes, particularly in the physiological contexts of (1) MAPK regulation in growth, cancer, metabolism, and immunity and (2) CaMKII regulation in cardiomyocyte and motor neuronal function.
Clinically, RA and its analogs have long been proposed for therapeutic applications in managing different diseases [107,108]. Through decades of studies, most of these efforts have not proven to be fruitful because of the wide spectrum of retinoid toxicities. This has presented a particularly serious concern in using retinoids to manage chronic diseases such as metabolic/inflammatory/neurological diseases [109,110,111,112,113]. The most efficacious application of retinoids has been in topical application, such as for treating acne vulgaris [114] and in aggressively treating severe or end-stage cancer patients, particularly an aggressive form of leukemia (acute myeloid leukemia) [115]. Most other attempts have proven to be not successful due to toxic side-effects which are caused by the RAR/RXR-mediated activities. Given that CRABP1 is specifically expressed in limited types of cells and only in certain stages of cell differentiation, and that CRABP1 participates in very specific signaling pathways, it might be more feasible by targeting CRABP1 using selective ligands that do not act on RARs or RXRs. This strategy exploits the collected evidence, as reviewed here, that CRABP1 mediates non-canonical RA signaling pathways that are cell type- and context-specific.
Currently, two novel CRABP1-selective compounds, C3 and C4, have been documented, which have been shown to modulate, specifically, the MAPK signaling pathway in CRABP1-expressing cells [24,31]. The efficacy of C3 and C4 has been demonstrated to induce apoptosis (in cancer cells) [24] and regulate exosome secretion (in neurons) [31]. These in vitro results would encourage further exploitation of this potential therapeutic strategy, such as in managing cancers and inflammation. Other groups have recently explored the use of synthetic ligands to target CRABP1. Tomlinson et al. determined the crystal structures of CRABP1 bound to fatty acids and a synthetic retinoid, DC645 [116]. DC645 appeared to bind CRABP1 in a manner similar to that of RA, and the binding resulted in minimal structural changes in CRABP1. Interestingly upon ligand binding, side-chains on the beta-sheet surface underwent conformational re-arrangements. Therefore, structural information obtained from these biophysical studies supports our fundamental hypothesis that CRABP1 signalsome acts, primarily, through its surface interactions that involve the beta-sheet face of CRABP1. Zheng et al. determined that Maprotiline can directly bind and inhibit CRABP1, resulting in dampened ERK-mediated SREBP2 activity and ultimately reducing tumor growth in a hepatocarcinoma xenograft model [117].
Additionally, the possibility of targeting CRABP1, such as by gene or cell therapy, is underscored by clinical data of human studies which have clearly implicated that a reduction in CRABP1 level was correlated with disease severity or its progression. To correct this deficiency, gene therapy (to deliver CRABP1-expressing vector) may be carried out to target implicated tissues. Further, a cell therapy-based strategy may also be feasible. For instance, CRABP1-expressing adipocytes may be locally delivered to adipose tissues to help correct the abnormally expanded obesity.
For future studies, the most important task would be to identify and develop CRABP1-selective and signaling pathway-specific ligands that do not elicit RAR-mediated toxicity. In addition to synthetic compounds, it would be of great interest to identify and study compounds derived from naturally occurring sources, such as plants and meats. These naturally occurring ligands, if present, would be very useful to the understanding and application of nutrients that may enhance the potential physiological and protective functions of CRABP1 signalsomes. It would also be of great interest to identify other components and networks that may comprise new CRABP1 signalsomes which remain to be uncovered. Finally, a more systemic investigation into human diseases where CRABP1 could play a role is needed. Given that CKO mice have CRABP1 deleted from birth, their disease spectrum may not reflect the entire spectrum of human diseases involving CRABP1. Human genetic association studies can provide important clues into this important research direction, and may uncover more physiologically important CRABP1-signalsomes that can also deliver non-canonical activities of RA.
Author Contributions
J.N., L.-N.W.; writing—original draft preparation, Y.-L.L.; writing—review and editing, L.-N.W.; supervision, project administration, and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported by NIH grants DK54733, DK60521, and the Dean’s Commitment and the Distinguished McKnight Professorship of the University of Minnesota to L.N.W., J.N. is supported by NIH fellowship F31DK123999.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank the ALS Variant Server (als.umassmed.edu) which is supported by funds from NIH/NINDS (1R01NS065847), AriSLA (EXOMEFALS, NOVALS), the ALS Association, and the Motor Neurone Disease Association. The results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. accessed on 7 February 2022.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tanumihardjo, S.A.; Russell, R.M.; Stephensen, C.B.; Gannon, B.M.; Craft, N.E.; Haskell, M.J.; Lietz, G.; Schulze, K.; Raiten, D.J. Biomarkers of nutrition for development (BOND)-vitamin A review. J. Nutr. 2016, 146, 1816S–1848S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, T.J.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef] [Green Version]
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, G. Retinoic acid as cause of cell proliferation or cell growth inhibition depending on activation of one of two different nuclear receptors. Nutr. Rev. 2008, 66, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Gudas, L.J.; Wagner, J.A. Retinoids regulate stem cell differentiation. J. Cell. Physiol. 2011, 226, 322–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, N. Between death and survival: Retinoic acid in regulation of apoptosis. Annu. Rev. Nutr. 2010, 30, 201–217. [Google Scholar] [CrossRef]
- Niederreither, K.; Dollé, P. Retinoic acid in development: Towards an integrated view. Nat. Rev. Genet. 2008, 9, 541–553. [Google Scholar] [CrossRef]
- Rothman, K.J.; Moore, L.L.; Singer, M.R.; Nguyen, U.-S.D.T.; Mannino, S.; Milunsky, A. Teratogenicity of High Vitamin A Intake. N. Engl. J. Med. 1995, 333, 1369–1373. [Google Scholar] [CrossRef]
- Shenefelt, R.E. Morphogenesis of malformations in hamsters caused by retinoic acid: Relation to dose and stage at treatment. Teratology 5:103-18. 1972. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 847–862. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.G.; Roth, C.B.; Warkany, J. An analysis of the syndrome of malformations induced by maternal vitamin a deficiency. Effects of restoration of vitamin a at various times during gestation. Am. J. Anat. 1953, 92, 189–217. [Google Scholar] [CrossRef]
- Duong, V.; Rochette-Egly, C. The molecular physiology of nuclear retinoic acid receptors. From health to disease. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 1023–1031. [Google Scholar] [CrossRef] [Green Version]
- Topletz, A.R.; Thatcher, J.E.; Zelter, A.; Lutz, J.D.; Tay, S.; Nelson, W.L.; Isoherranen, N. Comparison of the function and expression of CYP26A1 and CYP26B1, the two retinoic acid hydroxylases. Biochem. Pharmacol. 2012, 83, 149–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topletz, A.R.; Tripathy, S.; Foti, R.S.; Shimshoni, J.A.; Nelson, W.L.; Isoherranen, N. Induction of CYP26A1 by metabolites of retinoic acid: Evidence that CYP26A1 is an important enzyme in the elimination of active retinoids. Mol. Pharmacol. 2015, 87, 430–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, J.L. Functions of intracellular retinoid binding-proteins. Subcell. Biochem. 2016, 81, 21–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, J.L. Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmacol Ther 2017, 173, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevison, F.; Jing, J.; Tripathy, S.; Isoherranen, N. Role of Retinoic Acid-Metabolizing Cytochrome P450s, CYP26, in Inflammation and Cancer. Adv. Pharmacol. 2015, 74, 373–412. [Google Scholar]
- Thatcher, J.E.; Isoherranen, N. The role of CYP26 enzymes in retinoic acid clearance. Expert Opin. Drug Metab. Toxicol. 2009, 5, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Dong, D.; Ruuska, S.E.; Levinthal, D.J.; Noy, N. Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J. Biol. Chem. 1999, 274, 23695–23698. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, A.; Petrescu, A.D.; Xiong, Y.; Noys, N. Nuclear translocation of cellular retinoic acid-binding protein II is regulated by retinoic acid-controlled SUMOylation. J. Biol. Chem. 2011, 286, 42749–42757. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Ho, P.-C.; Huq, M.M.; Ha, S.G.; Park, S.W.; Khan, A.A.; Tsai, N.-P.; Wei, L.-N. Retinoic acid-stimulated sequential phosphorylation, PML recruitment, and SUMOylation of nuclear receptor TR2 to suppress Oct4 expression. Proc. Natl. Acad. Sci. USA 2008, 105, 11424–11429. [Google Scholar] [CrossRef] [Green Version]
- Chuang, Y.-S.S.; Huang, W.-H.H.; Park, S.W.; Persaud, S.D.; Hung, C.-H.H.; Ho, P.-C.C.; Wei, L.-N.N. Promyelocytic leukemia protein in retinoic acid-induced chromatin remodeling of Oct4 gene promoter. Stem Cells 2011, 29, 660–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persaud, S.D.; Lin, Y.-W.; Wu, C.-Y.; Kagechika, H.; Wei, L.-N. Cellular retinoic acid binding protein I mediates rapid non-canonical activation of ERK1/2 by all-trans retinoic acid. Cell. Signal. 2013, 25, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.Y.; Persaud, S.D.; Wei, L.N. Retinoic Acid Induces Ubiquitination-Resistant RIP140/LSD1 Complex to Fine-Tune Pax6 Gene in Neuronal Differentiation. Stem Cells 2016, 34, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persaud, S.D.; Park, S.W.; Ishigami-Yuasa, M.; Koyano-Nakagawa, N.; Kagechika, H.; Wei, L.N. All trans-retinoic acid analogs promote cancer cell apoptosis through non-genomic Crabp1 mediating ERK1/2 phosphorylation. Sci. Rep. 2016, 6, 22396. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.L.; Persaud, S.D.; Nhieu, J.; Wei, L.N. Cellular Retinoic Acid-Binding Protein 1 Modulates Stem Cell Proliferation to Affect Learning and Memory in Male Mice. Endocrinology 2017, 158, 3004–3014. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W.; Persaud, S.D.; Ogokeh, S.; Meyers, T.A.; Townsend, D.W.; Wei, L.N. CRABP1 protects the heart from isoproterenol-induced acute and chronic remodeling. J. Endocrinol. 2018, 236, 151–165. [Google Scholar] [CrossRef]
- Park, S.W.; Nhieu, J.; Lin, Y.W.; Wei, L.N. All-trans retinoic acid attenuates isoproterenol-induced cardiac dysfunction through Crabp1 to dampen CaMKII activation. Eur. J. Pharmacol. 2019, 858, 172485. [Google Scholar] [CrossRef]
- Lin, Y.W.; Park, S.W.; Lin, Y.L.; Burton, F.H.; Wei, L.N. Cellular retinoic acid binding protein 1 protects mice from high-fat diet-induced obesity by decreasing adipocyte hypertrophy. Int. J. Obes. 2020, 44, 466–474. [Google Scholar] [CrossRef]
- Wook Park, S.; Nhieu, J.; Persaud, S.D.; Miller, M.C.; Xia, Y.; Lin, Y.W.; Lin, Y.L.; Kagechika, H.; Mayo, K.H.; Wei, L.N. A new regulatory mechanism for Raf kinase activation, retinoic acid-bound Crabp1. Sci. Rep. 2019, 9, 10929. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.W.Y.L.W.; Nhieu, J.; Zhang, X.; Wei, L.N. Sonic hedgehog-gli1 signaling and cellular retinoic acid binding protein 1 gene regulation in motor neuron differentiation and diseases. Int. J. Mol. Sci. 2020, 21, 4125. [Google Scholar] [CrossRef]
- Lin, Y.W.; Nhieu, J.; Wei, C.W.; Lin, Y.L.; Kagechika, H.; Wei, L.N. Regulation of exosome secretion by cellular retinoic acid binding protein 1 contributes to systemic anti-inflammation. Cell Commun. Signal. 2021, 19, 69. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Wei, C.W.; Lerdall, T.A.; Nhieu, J.; Wei, L.N. Crabp1 modulates hpa axis homeostasis and anxiety-like behaviors by altering fkbp5 expression. Int. J. Mol. Sci. 2021, 22, 12240. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-L.Y.-W.; Nhieu, J.; Liu, P.-Y.; Le, G.; Lee, D.J.; Wei, C.-W.; Lin, Y.-L.Y.-W.; Oh, S.-H.; Lowe, D.; Wei, L.-N. CRABP1-CaMKII-Agrn regulates the maintenance of neuromuscular junction in spinal motor neuron. Cell Death Differ. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ong, D.E.; Chytil, F. Cellular retinoic acid-binding protein from rat testis. Purification and characterization. J. Biol. Chem. 1978, 253, 4551–4554. [Google Scholar] [CrossRef]
- Fiorella, P.D.; Giguère, V.; Napoli, J.L. Expression of cellular retinoic acid-binding protein (type II) in Escherichia coli: Characterization and comparison to cellular retinoic acid-binding protein (type I). J. Biol. Chem. 1993, 268, 21545–21552. [Google Scholar] [CrossRef]
- Norris, A.W.; Cheng, L.; Giguère, V.; Rosenberger, M.; Li, E. Measurement of subnanomolar retinoic acid binding affinities for cellular retinoic acid binding proteins by fluorometric titration. Biochim. Biophys. Acta 1994, 1209, 10–18. [Google Scholar] [CrossRef]
- Wang, L.; Li, Y.; Yan, H. Structure-function relationships of cellular retinoic acid-binding proteins: Quantitative analysis of the ligand binding properties of the wild-type proteins and site-directed mutants. J. Biol. Chem. 1997, 272, 1541–1547. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.N.; Chang, L.; Lee, C.H. Studies of over-expressing cellular retinoic acid binding protein-I in cultured cells and transgenic mice. Transgenics 1997, 2, 201–209. [Google Scholar]
- Wei, L.N.; Chang, L.; Hu, X. Studies of the type I cellular retinoic acid-binding protein mutants and their biological activities. Mol. Cell. Biochem. 1999, 200, 69–76. [Google Scholar] [CrossRef]
- Nelson, C.H.; Peng, C.C.; Lutz, J.D.; Yeung, C.K.; Zelter, A.; Isoherranen, N. Direct protein–protein interactions and substrate channeling between cellular retinoic acid binding proteins and CYP26B1. FEBS Lett. 2016, 590, 2527–2535. [Google Scholar] [CrossRef] [Green Version]
- Eller, M.S.; Oleksiak, M.F.; McQuaid, T.J.; McAfee, S.G.; Gilchrest, B.A. The molecular cloning and expression of two CRABP cDNAs from human skin. Exp. Cell Res. 1992, 198, 328–336. [Google Scholar] [CrossRef]
- Tang, Z.; Li, Y.; Wan, P.; Li, X.; Zhao, S.; Liu, B.; Fan, B.; Zhu, M.; Yu, M.; Li, K. LongSAGE analysis of skeletal muscle at three prenatal stages in Tongcheng and Landrace pigs. Genome Biol. 2007, 8, R115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, J.M.; Curley, R.W. Affinity purification of retinoic acid-binding proteins using immobilized 4-(2-Hydroxyethoxy)retinoic acid. Protein Expr. Purif. 1990, 1, 63–69. [Google Scholar] [CrossRef]
- Stoner, C.M.; Gudas, L.J. Mouse Cellular Retinoic Acid Binding Protein: Cloning, Complementary DNA Sequence, and Messenger RNA Expression during the Retinoic Acid-induced Differentiation of F9 Wild Type and RA-3-10 Mutant Teratocarcinoma Cells. Cancer Res. 1989, 49, 1497–1504. [Google Scholar] [PubMed]
- Nilsson, M.H.L.; Spurr, N.K.; Saksena, P.; Busch, C.; Nordlinder, H.; Peterson, P.A.; Rask, L.; Sundelin, J. Isolation and characterization of a cDNA clone corresponding to bovine cellular retinoic-acid-binding protein and chromosomal localization of the corresponding human gene. Eur. J. Biochem. 1988, 173, 45–51. [Google Scholar] [CrossRef]
- Wei, L.-N. Non-canonical activity of retinoic acid in epigenetic control of embryonic stem cell. Transcription 2013, 4, 158–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.N. Cellular retinoic acid binding proteins: Genomic and non-genomic functions and their regulation. Subcell. Biochem. 2016, 81, 163–178. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T.; LIU, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Guidez, F.; Parks, S.; Wong, H.; Jovanovic, J.V.; Mays, A.; Gilkes, A.F.; Mills, K.I.; Guillemin, M.-C.C.; Hobbs, R.M.; Pandolfi, P.P.; et al. RARalpha-PLZF overcomes PLZF-mediated repression of CRABPI, contributing to retinoid resistance in t(11;17) acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2007, 104, 18694–18699. [Google Scholar] [CrossRef] [Green Version]
- Pfoertner, S.; Goelden, U.; Hansen, W.; Toepfer, T.; Geffers, R.; Ukena, S.N.; von Knobloch, R.; Hofmann, R.; Buer, J.; Schrader, A.J. Cellular retinoic acid binding protein I: Expression and functional influence in renal cell carcinoma. Tumour Biol. 2005, 26, 313–323. [Google Scholar] [CrossRef]
- Tanaka, K.; Imoto, I.; Inoue, J.; Kozaki, K.; Tsuda, H.; Shimada, Y.; Aiko, S.; Yoshizumi, Y.; Iwai, T.; Kawano, T.; et al. Frequent methylation-associated silencing of a candidate tumor-suppressor, CRABP1, in esophageal squamous-cell carcinoma. Oncogene 2007, 26, 6456–6468. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Ueda, Y.; Matsuzaki, S.; Miyatake, T.; Yoshino, K.; Fujita, M.; Nomura, T.; Enomoto, T.; Kimura, T. CRABP1-reduced expression is associated with poorer prognosis in serous and clear cell ovarian adenocarcinoma. J. Cancer Res. Clin. Oncol. 2011, 137, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Lothe, R.A.; Ahlquist, T.; Silins, I.; Tropé, C.G.; Micci, F.; Nesland, J.M.; Suo, Z.; Lind, G.E. DNA methylation profiling of ovarian carcinomas and their in vitro models identifies HOXA9, HOXB5, SCGB3A1, and CRABP1 as novel targets. Mol. Cancer 2007, 6, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawthorn, L.; Stein, L.; Varma, R.; Wiseman, S.; Loree, T.; Tan, D.F. TIMP1 and SERPIN-A overexpression and TFF3 and CRABP1 underexpression as biomarkers for papillary thyroid carcinoma. Head Neck 2004, 26, 1069–1083. [Google Scholar] [CrossRef]
- Celestino, R.; Nome, T.; Pestana, A.; Hoff, A.M.; Gonçalves, A.P.; Pereira, L.; Cavadas, B.; Eloy, C.; Bjøro, T.; Sobrinho-Simões, M.; et al. CRABP1, C1QL1 and LCN2 are biomarkers of differentiated thyroid carcinoma, and predict extrathyroidal extension. BMC Cancer 2018, 18, 68. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; de la Chapelle, A.; Pellegata, N.S. Hypermethylation, but not LOH, is associated with the low expression of MT1G and CRABP1 in papillary thyroid carcinoma. Int. J. Cancer 2003, 104, 735–744. [Google Scholar] [CrossRef]
- Lind, G.E.; Kleivi, K.; Meling, G.I.; Teixeira, M.R.; Thiis-Evensen, E.; Rognum, T.O.; Lothe, R.A. ADAMTS1, CRABP1, and NR3C1 identified as epigenetically deregulated genes in colorectal tumorigenesis. Cell. Oncol. 2006, 28, 259–272. [Google Scholar] [CrossRef]
- Won, J.Y.; Nam, E.C.; Yoo, S.J.; Kwon, H.J.; Um, S.J.; Han, H.S.; Kim, S.H.; Byun, Y.; Kim, S.Y. The effect of cellular retinoic acid binding protein-I expression on the CYP26-mediated catabolism of all-trans retinoic acid and cell proliferation in head and neck squamous cell carcinoma. Metabolism 2004, 53, 1007–1012. [Google Scholar] [CrossRef]
- Kainov, Y.; Favorskaya, I.; Delektorskaya, V.; Chemeris, G.; Komelkov, A.; Zhuravskaya, A.; Trukhanova, L.; Zueva, E.; Tavitian, B.; Dyakova, N.; et al. CRABP1 provides high malignancy of transformed mesenchymal cells and contributes to the pathogenesis of mesenchymal and neuroendocrine tumors. Cell Cycle 2014, 13, 1530–1539. [Google Scholar] [CrossRef] [Green Version]
- Choi, N.; Park, J.; Lee, J.S.; Yoe, J.; Park, G.Y.; Kim, E.; Jeon, H.; Cho, Y.M.; Roh, T.Y.; Lee, Y. miR-93/miR-106b/miR-375-CIC-CRABP1: A novel regulatory axis in prostate cancer progression. Oncotarget 2015, 6, 23533–23547. [Google Scholar] [CrossRef]
- Liu, R.Z.; Garcia, E.; Glubrecht, D.D.; Poon, H.Y.; Mackey, J.R.; Godbout, R. CRABP1 is associated with a poor prognosis in breast cancer: Adding to the complexity of breast cancer cell response to retinoic acid. Mol. Cancer 2015, 14, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpal, I.; Wei, L.N. All-trans retinoic acid as a versatile cytosolic signal modulator mediated by CRABP1. Int. J. Mol. Sci. 2019, 20, 3610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, D.C.; DeSantis, D.; Soltanian, H.; Croniger, C.M.; Noy, N. Retinoic acid upregulates preadipocyte genes to block adipogenesis and suppress diet-induced obesity. Diabetes 2012, 61, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, D.C.; Noy, N. All-trans-Retinoic Acid Represses Obesity and Insulin Resistance by Activating both Peroxisome Proliferation-Activated Receptor β/δ and Retinoic Acid Receptor. Mol. Cell. Biol. 2009, 29, 3286–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyakumar, S.M.; Vajreswari, A.; Sesikeran, B.; Giridharan, N.V. Vitamin A supplementation induces adipose tissue loss through apoptosis in lean but not in obese rats of the WNIN/Ob strain. J. Mol. Endocrinol. 2005, 35, 391–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamei, Y.; Kawada, T.; Mizukami, J.; Sugimoto, E. The prevention of adipose differentiation of 3T3-L1 cells caused by retinoic acid is elicited through retinoic acid receptor alpha. Life Sci. 1994, 55, PL307–PL312. [Google Scholar] [CrossRef]
- Murray, T.; Russell, T.R. Inhibition of adipose conversion in 3T3-L2 cells by retinoic acid. J. Supramol. Cell. Biochem. 1980, 14, 255–266. [Google Scholar] [CrossRef]
- Lee, B.; Ho, P.-C.; Wei, L.-N. Nuclear Receptor-Interacting Protein 1 (NRIP1). In Encyclopedia of Signaling Molecules; Springer: Cham, Switzerland, 2018; pp. 3606–3616. [Google Scholar]
- Erickson, J.R. Mechanisms of CaMKII activation in the heart. Front. Pharmacol. 2014, 5 APR, 59. [Google Scholar] [CrossRef] [Green Version]
- Lisman, J.; Schulman, H.; Cline, H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 2002, 3, 175–190. [Google Scholar] [CrossRef]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, M.; Karandur, D.; Kuriyan, J. Structural insights into the regulation of Ca2+/calmodulin-dependent protein kinase II (Camkii). Cold Spring Harb. Perspect. Biol. 2020, 12, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.M.; Chao, L.H.; Schulman, H.; Kuriyan, J. Structural studies on the regulation of Ca2+/calmodulin dependent protein kinase II. Curr. Opin. Struct. Biol. 2013, 23, 292–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P. CaMKII: The molecular villain that aggravates cardiovascular disease. Exp. Ther. Med. 2017, 13, 815–820. [Google Scholar] [CrossRef] [Green Version]
- Ashpole, N.M.; Hudmon, A. Excitotoxic neuroprotection and vulnerability with CaMKII inhibition. Mol. Cell. Neurosci. 2011, 46, 720–730. [Google Scholar] [CrossRef]
- Garg, M.; Khanna, D. Exploration of pharmacological interventions to prevent isoproterenol-induced myocardial infarction in experimental models. Ther. Adv. Cardiovasc. Dis. 2014, 8, 155–169. [Google Scholar] [CrossRef]
- Nichtova, Z.; Novotova, M.; Kralova, E.; Stankovicova, T. Morphological and functional characteristics of models of experimental myocardial injury induced by isoproterenol. Gen. Physiol. Biophys. 2012, 31, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Xiong, W.C.; Mei, L. Neuromuscular Junction Formation, Aging, and Disorders. Annu. Rev. Physiol. 2018, 80, 159–188. [Google Scholar] [CrossRef]
- Martinez-Pena, Y. Valenzuela, I.; Mouslim, C.; Akaaboune, M. Calcium/calmodulin kinase II-dependent acetylcholine receptor cycling at the mammalian neuromuscular junction in vivo. J. Neurosci. 2010, 30, 12455–12465. [Google Scholar] [CrossRef] [Green Version]
- Koh, Y.H.; Popova, E.; Thomas, U.; Griffith, L.C.; Budnik, V. Regulation of DLG localization at synapses by CaMKII-dependent phosphorylation. Cell 1999, 98, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, J.M.; Hodge, J.J.L. CASK regulates CaMKII autophosphorylation in neuronal growth, calcium signaling, and learning. Front. Mol. Neurosci. 2013, 6, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, E.R. Role of Ca 2+/calmodulin-dependent kinases in skeletal muscle plasticity. J. Appl. Physiol. 2005, 99, 414–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, A.J.; Kiens, B.; Richter, E.A. Ca2+-calmodulin-dependent protein kinase expression and signalling in skeletal muscle during exercise. J. Physiol. 2006, 574, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Taetzsch, T.; Valdez, G. NMJ maintenance and repair in aging. Curr. Opin. Physiol. 2018, 4, 57–64. [Google Scholar] [CrossRef]
- Glass, D.J.; Bowen, D.C.; Stitt, T.N.; Radziejewski, C.; Bruno, J.A.; Ryan, T.E.; Gies, D.R.; Shah, S.; Mattsson, K.; Burden, S.J.; et al. Agrin acts via a MuSK receptor complex. Cell 1996, 85, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Papatheodorou, I.; Moreno, P.; Manning, J.; Fuentes, A.M.P.; George, N.; Fexova, S.; Fonseca, N.A.; Füllgrabe, A.; Green, M.; Huang, N.; et al. Expression Atlas update: From tissues to single cells. Nucleic Acids Res. 2020, 48, D77–D83. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.M.; Yamamoto, M.; Kobayashi, Y.; Yoshihara, T.; Liang, Y.; Terao, S.; Takeuchi, H.; Ishigaki, S.; Katsuno, M.; Adachi, H.; et al. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 2005, 57, 236–251. [Google Scholar] [CrossRef]
- Rizzo, F.; Nizzardo, M.; Vashisht, S.; Molteni, E.; Melzi, V.; Taiana, M.; Salani, S.; Santonicola, P.; Di Schiavi, E.; Bucchia, M.; et al. Key role of SMN/SYNCRIP and RNA-Motif 7 in spinal muscular atrophy: RNA-Seq and motif analysis of human motor neurons. Brain 2019, 142, 276–294. [Google Scholar] [CrossRef]
- Nordgaard, C.L.; Berg, K.M.; Kapphahn, R.J.; Reilly, C.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 815–822. [Google Scholar] [CrossRef]
- Satoh, J.I.; Kino, Y. Expression profiles of RNA-Seq-based grey matter-specific genes versus white matter-specific genes in grey matter lesions of multiple sclerosis. Clin. Exp. Neuroimmunol. 2015, 6, 289–298. [Google Scholar] [CrossRef]
- Scholtissek, B.; Zahn, S.; Maier, J.; Klaeschen, S.; Braegelmann, C.; Hoelzel, M.; Bieber, T.; Barchet, W.; Wenzel, J. Immunostimulatory Endogenous Nucleic Acids Drive the Lesional Inflammation in Cutaneous Lupus Erythematosus. J. Investig. Dermatol. 2017, 137, 1484–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbari, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gonzalez, J.; Cueto, I.; Franks, A.G.; Krueger, J.G. Dominant Th1 and minimal Th17 skewing in discoid lupus revealed by transcriptomic comparison with psoriasis. J. Investig. Dermatol. 2014, 134, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regazzetti, C.; Joly, F.; Marty, C.; Rivier, M.; Mehul, B.; Reiniche, P.; Mounier, C.; Rival, Y.; Piwnica, D.; Cavalié, M.; et al. Transcriptional analysis of vitiligo skin reveals the alteration of WNT pathway: A promising target for repigmenting vitiligo patients. J. Investig. Dermatol. 2015, 135, 3105–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, I.; Silva, P.; da Mata, S.; Magro, F.; Carneiro, F.; Peixoto, A.; Silva, M.; Sousa, H.T.; Roseira, J.; Parra, J.; et al. Methylation patterns in dysplasia in inflammatory bowel disease patients. Scand. J. Gastroenterol. 2020, 55, 646–655. [Google Scholar] [CrossRef]
- Kim, S.K.; Yoo, J.I.; Cho, B.K.; Hong, S.J.; Kim, Y.K.; Moon, J.A.; Kim, J.H.; Chung, Y.N.; Wang, K.C. Elevation of CRABP-I in the Cerebrospinal Fluid of Patients with Moyamoya Disease. Stroke 2003, 34, 2835–2841. [Google Scholar] [CrossRef] [Green Version]
- Hur, J.; Sullivan, K.A.; Pande, M.; Hong, Y.; Sima, A.A.F.; Jagadish, H.V.; Kretzler, M.; Feldman, E.L. The identification of gene expression profiles associated with progression of human diabetic neuropathy. Brain 2011, 134, 3222–3235. [Google Scholar] [CrossRef]
- Carr, A.; Samaras, K.; Chisholm, D.J.; Cooper, D.A. Pathogenesis of HIV-1-protease inhibitor-associated peripheral lipodystrophy, hyperlipidaemia, and insulin resistance. Lancet 1998, 351, 1881–1883. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, 269. [Google Scholar] [CrossRef] [Green Version]
- Soundararajan, P.; Lindsey, B.W.; Leopold, C.; Rafuse, V.F. Easy and Rapid Differentiation of Embryonic Stem Cells into Functional Motoneurons Using Sonic Hedgehog-Producing Cells. Stem Cells 2007, 25, 1697–1706. [Google Scholar] [CrossRef]
- Scott, R.M.; Smith, E.R. Moyamoya Disease and Moyamoya Syndrome. N. Engl. J. Med. 2009, 360, 1226–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vreeland, A.C.; Yu, S.; Levi, L.; de Barros Rossetto, D.; Noy, N. Transcript Stabilization by the RNA-Binding Protein HuR Is Regulated by Cellular Retinoic Acid-Binding Protein 2. Mol. Cell. Biol. 2014, 34, 2135–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Vreeland, A.C.; Noy, N. RNA-binding protein HuR regulates nuclear import of protein. J. Cell Sci. 2016, 129, 4025–4033. [Google Scholar] [CrossRef] [Green Version]
- Budhu, A.S.; Noy, N. Direct Channeling of Retinoic Acid between Cellular Retinoic Acid-Binding Protein II and Retinoic Acid Receptor Sensitizes Mammary Carcinoma Cells to Retinoic Acid-Induced Growth Arrest. Mol. Cell. Biol. 2002, 22, 2632–2641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schug, T.T.; Berry, D.C.; Toshkov, I.A.; Cheng, L.; Nikitin, A.Y.; Noy, N. Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPARβ/δ to RAR. Proc. Natl. Acad. Sci. USA 2008, 105, 7546–7551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orfanos, C.E.; Ehlert, R.; Gollnick, H. The Retinoids: A Review of their Clinical Pharmacology and Therapeutic Use. Drugs 1987, 34, 459–503. [Google Scholar] [CrossRef]
- Ferreira, R.; Napoli, J.; Enver, T.; Bernardino, L.; Ferreira, L. Advances and challenges in retinoid delivery systems in regenerative and therapeutic medicine. Nat. Commun. 2020, 11, 4265. [Google Scholar] [CrossRef]
- Bremner, J.D.; Shearer, K.D.; McCaffery, P.J. Retinoic acid and affective disorders: The evidence for an association. J. Clin. Psychiatry 2012, 73, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Wood, E. The Retinoids, Volumes I and II. Biochem. Educ. 1985, 13, 147. [Google Scholar] [CrossRef]
- Hathcock, J.N.; Hattan, D.G.; Jenkins, M.Y.; McDonald, J.T.; Sundaresan, P.R.; Wilkening, V.L. Evaluation of vitamin A toxicity. Am. J. Clin. Nutr. 1990, 52, 183–202. [Google Scholar] [CrossRef]
- Bendich, A.; Langseth, L. Safety of vitamin A. Am. J. Clin. Nutr. 1989, 49, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Yob, E.H.; Pochi, P.E. Side Effects and Long-term Toxicity of Synthetic Retinoids. Arch. Dermatol. 1987, 123, 1375–1378. [Google Scholar] [CrossRef] [PubMed]
- Thielitz, A.; Gollnick, H. Topical retinoids in acne vulgaris: Update on efficacy and safety. Am. J. Clin. Dermatol. 2008, 9, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.-B.B.; Zhang, J.; Wang, Z.-Y.Y.; Chen, S.-J.J.; Chen, Z. Treatment of acute promyelocytic leukaemia with all-trans retinoic acid and arsenic trioxide: A paradigm of synergistic molecular targeting therapy. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 959–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, C.W.E.; Cornish, K.A.S.; Whiting, A.; Pohl, E. Structure-functional relationship of cellular retinoic acid-binding proteins i and II interacting with natural and synthetic ligands. Acta Crystallogr. Sect. D Struct. Biol. 2021, 77, 164–175. [Google Scholar] [CrossRef]
- Zheng, C.; Zhu, Y.; Liu, Q.; Luo, T.; Xu, W. Maprotiline Suppresses Cholesterol Biosynthesis and Hepatocellular Carcinoma Progression Through Direct Targeting of CRABP1. Front. Pharmacol. 2021, 12, 1243. [Google Scholar] [CrossRef]
Figure 1.
CRABP1 sequence alignment across mammals. Reported CRABP1 protein sequences from the Uniprot database of human (ID: P29762) [41], pig (ID: B3F0B7) [42], rat (ID: P62966) [43], mouse (ID: P62965) [44], and bovine (ID: P62964) [45] were aligned using the ClustalWS alignment algorithm in Jalview. Only the residue at position 86 (indicated by bold text and arrow ↓) is not conserved and exists either as an alanine (A) in human and pig sequences or as a proline (P) in bovine, rat, and mouse sequences.
Figure 1.
CRABP1 sequence alignment across mammals. Reported CRABP1 protein sequences from the Uniprot database of human (ID: P29762) [41], pig (ID: B3F0B7) [42], rat (ID: P62966) [43], mouse (ID: P62965) [44], and bovine (ID: P62964) [45] were aligned using the ClustalWS alignment algorithm in Jalview. Only the residue at position 86 (indicated by bold text and arrow ↓) is not conserved and exists either as an alanine (A) in human and pig sequences or as a proline (P) in bovine, rat, and mouse sequences.

Figure 2.
CRABP1-MAPK signalsome. The action of CRABP1-signalsome in growth-factor stimulated MAPK activity is mediated by its direct competition with Ras, resulting in dampened MAPK activation. CRABP1: Cellular Retinoic Acid Binding Protein 1, RA: retinoic acid, RAF: rapidly Accelerated Fibrosarcoma, MEK: mitogen-activated protein kinase kinase, ERK: extracellular-signal-regulated kinase.
Figure 2.
CRABP1-MAPK signalsome. The action of CRABP1-signalsome in growth-factor stimulated MAPK activity is mediated by its direct competition with Ras, resulting in dampened MAPK activation. CRABP1: Cellular Retinoic Acid Binding Protein 1, RA: retinoic acid, RAF: rapidly Accelerated Fibrosarcoma, MEK: mitogen-activated protein kinase kinase, ERK: extracellular-signal-regulated kinase.
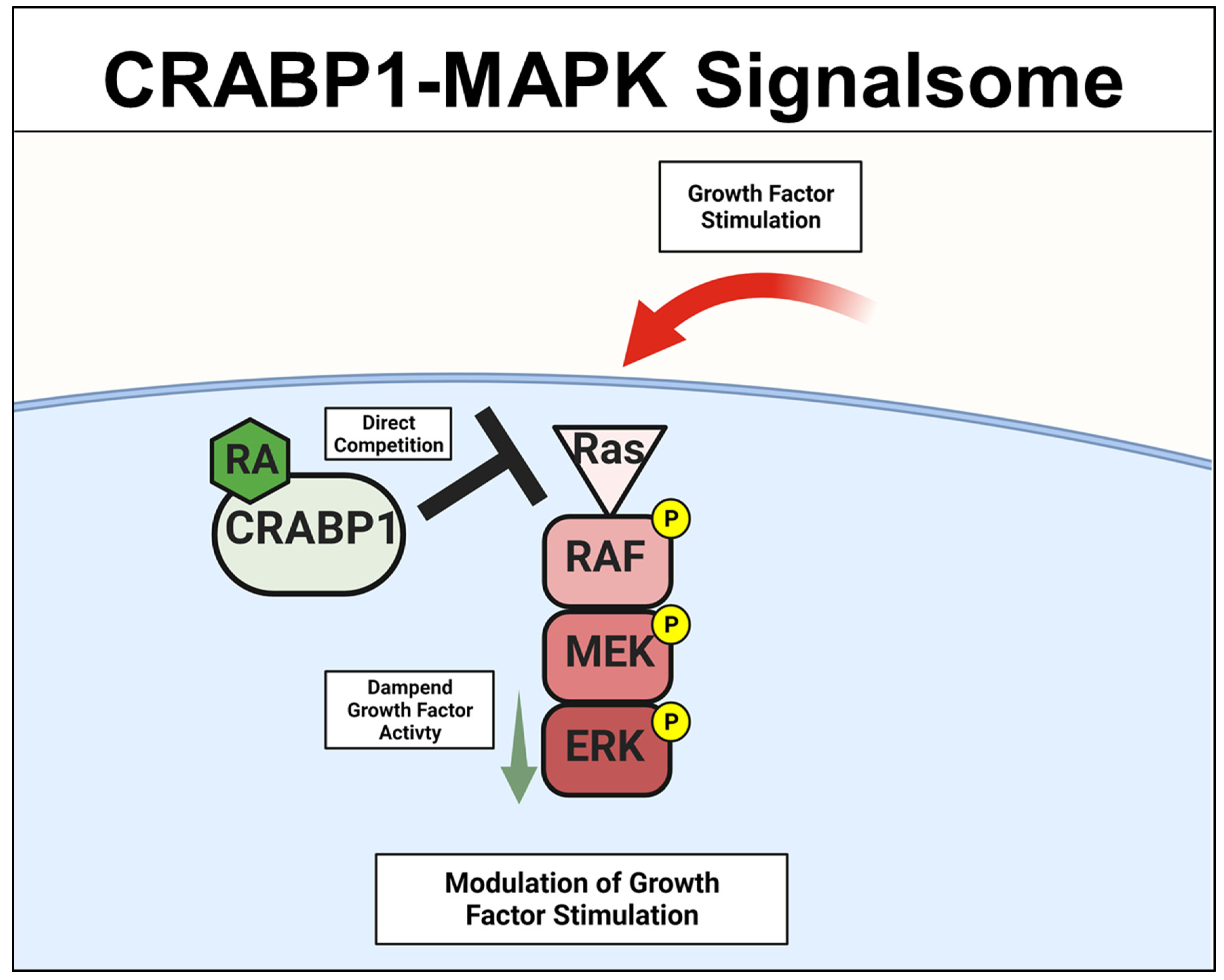
Figure 3.
CRABP1-CaMKII signalsome. Upon cardiac or neuronal stimulation and subsequent intracellular Ca2+ increase to activate CaMKII, CRABP1 directly competes with calmodulin (CaM) to dampen CaMKII enzyme activity to ultimately modulate cardiac and/or neuronal stimulation. CRABP1: Cellular Retinoic Acid Binding Protein 1, RA: retinoic acid, CaMKII: calcium-calmodulin-associated dependent kinase 2.
Figure 3.
CRABP1-CaMKII signalsome. Upon cardiac or neuronal stimulation and subsequent intracellular Ca2+ increase to activate CaMKII, CRABP1 directly competes with calmodulin (CaM) to dampen CaMKII enzyme activity to ultimately modulate cardiac and/or neuronal stimulation. CRABP1: Cellular Retinoic Acid Binding Protein 1, RA: retinoic acid, CaMKII: calcium-calmodulin-associated dependent kinase 2.

Figure 4.
SNPs identified in cancer patients within the 3 kb upstream regulatory region and the coding sequence of CRABP1. (a) Gene diagram of the 3 kb upstream region of CRABP1 with SNPs denoted by “●”. (b) Lollipop plot indicating the location of amino acid mutations as a consequence of cancer-associated SNPs.
Figure 4.
SNPs identified in cancer patients within the 3 kb upstream regulatory region and the coding sequence of CRABP1. (a) Gene diagram of the 3 kb upstream region of CRABP1 with SNPs denoted by “●”. (b) Lollipop plot indicating the location of amino acid mutations as a consequence of cancer-associated SNPs.
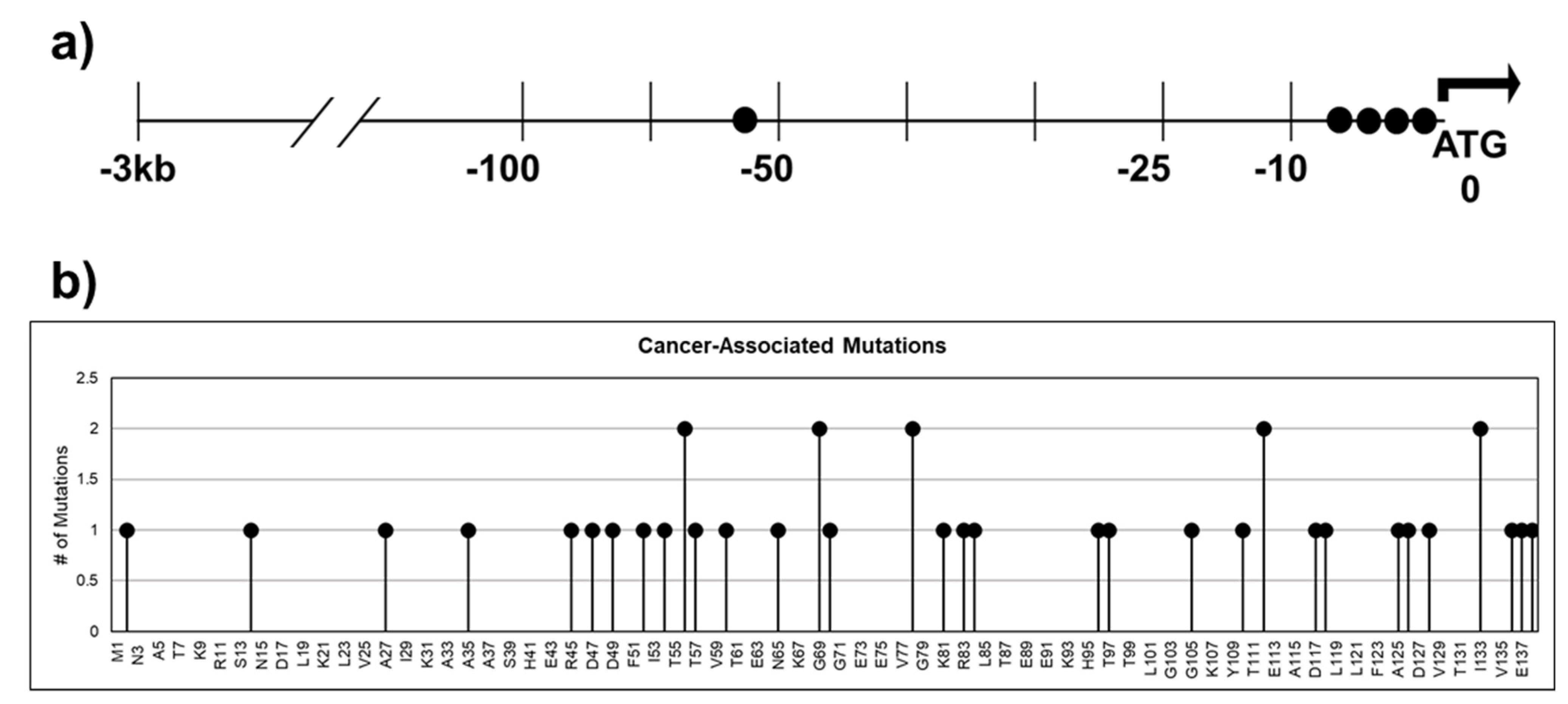
Figure 5.
SNPs identified within the 3 kb upstream regulatory region and the coding sequence of CRABP1 in ALS patients. (a) Gene diagram of the 3 kb upstream region of CRABP1 with SNPs denoted by “X”. (b) Lollipop plot indicating the location of amino acid mutations as a consequence of ALS-associated SNPs. Altered residues are marked above each lollipop.
Figure 5.
SNPs identified within the 3 kb upstream regulatory region and the coding sequence of CRABP1 in ALS patients. (a) Gene diagram of the 3 kb upstream region of CRABP1 with SNPs denoted by “X”. (b) Lollipop plot indicating the location of amino acid mutations as a consequence of ALS-associated SNPs. Altered residues are marked above each lollipop.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Changes in CRABP1 detected in human patients.
| Cancer Type | CRABP1 Status | Reference |
|---|---|---|
| Breast Cancer | Over-Expression | [61] |
| Prostate Cancer | Over-Expression | [60] |
| Mesenchymal & Neuroendocrine Tumors | Over-Expression | [59] |
| Head and Neck Squamous Cell Carcinoma (HNSCC) | Over-Expression | [58] |
| Colorectal Cancer | Silenced (Promoter Hypermethylation) | [57] |
| Thyroid Cancer | Silenced (Promoter Hypermethylation) | [56] |
| Reduced Expression | [54] | |
| Ovarian Cancer | Silenced (Promoter Hypermethylation) | [53] |
| Reduced Expression | [52] | |
| Esophageal Squamous-Cell Carcinoma (ESCC) | Silenced (Promoter Hypermethylation) | [51] |
| Renal Cell Carcinoma | Reduced Expression | [50] |
| Acute myeloid leukemia (AML) | Silenced (Promoter Hypermethylation) | [49] |
| Neurodegenerative Diseases | CRABP1 Status | Reference |
| Amyotrophic Lateral Sclerosis (ALS) | Reduced Expression | [88] |
| Spinal Muscular Atrophy (SMA) | Reduced Expression | [89] |
| Late-Stage Age-Related Macular Degeneration (AMD) | Reduced Expression | [90] |
| Immune Disorders | CRABP1 Status | Reference |
| Multiple Sclerosis | Reduced Expression | [91] |
| Cutaneous Lupus Erythematosus (CLE) | Reduced Expression | [92] # E-MTAB-5542 |
| Psoriasis | Reduced Expression | [93] # E-GEOD-52471 |
| Vitiligo | Reduced Expression | [94] # E-GEOD-65127 |
| Inflammatory Bowel Disease (IBD) | Silenced (Promoter Hypermethylation) | [95] |
| Other Diseases | CRABP1 Status | Reference |
| Moyamoya Disease (MMD) | Increased Protein Level | [96] |
| Diabetic Neuropathy | Increased Expression | [97] |
| HIV Therapy-Associated Lipodystrophy and Metabolic Syndrome | Inhibited Function | [98] |
# EMBL-EBI Expression Atlas Data Repository Accession ID.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nhieu, J.; Lin, Y.-L.; Wei, L.-N. CRABP1 in Non-Canonical Activities of Retinoic Acid in Health and Diseases. Nutrients 2022, 14, 1528. https://doi.org/10.3390/nu14071528
AMA Style
Nhieu J, Lin Y-L, Wei L-N. CRABP1 in Non-Canonical Activities of Retinoic Acid in Health and Diseases. Nutrients. 2022; 14(7):1528. https://doi.org/10.3390/nu14071528
Chicago/Turabian StyleNhieu, Jennifer, Yu-Lung Lin, and Li-Na Wei. 2022. "CRABP1 in Non-Canonical Activities of Retinoic Acid in Health and Diseases" Nutrients 14, no. 7: 1528. https://doi.org/10.3390/nu14071528
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.