
Administration of Linoleoylethanolamide Reduced Weight Gain, Dyslipidemia, and Inflammation Associated with High-Fat-Diet-Induced Obesity
 , , , , , , , , and
, , , , , , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Populational Study: Estimating Circulating Linoleoylethanolamide Concentrations in Normal and Overweight Subjects
2.2. Quantification of Acylethanolamides in Plasma and Liver
2.3. Animals and Diets
2.4. Glucose Tolerance Test
2.5. Drugs and Chronic Treatment
2.6. Sample Collection
2.7. Measurement of Metabolites, Hepatic Enzymes, Insulin, and Inflammatory Mediators in Plasma
2.8. Hepatic Lipid Extraction and Fat Content
2.9. Protein Extraction and Western Blot Analysis
2.10. Statistical Analysis
3. Results
3.1. Population Characteristics and Plasma Concentrations of Linoleoylethanolamide in Normal and Overweight People
3.2. Effects of Subchronic Administration of Linoleoylethanolamide in Weight Gain and Food Intake in Animals Receiving a HFD
3.3. Effects of Chronic Administration of Linoleoylethanolamide in Plasma Biochemistry
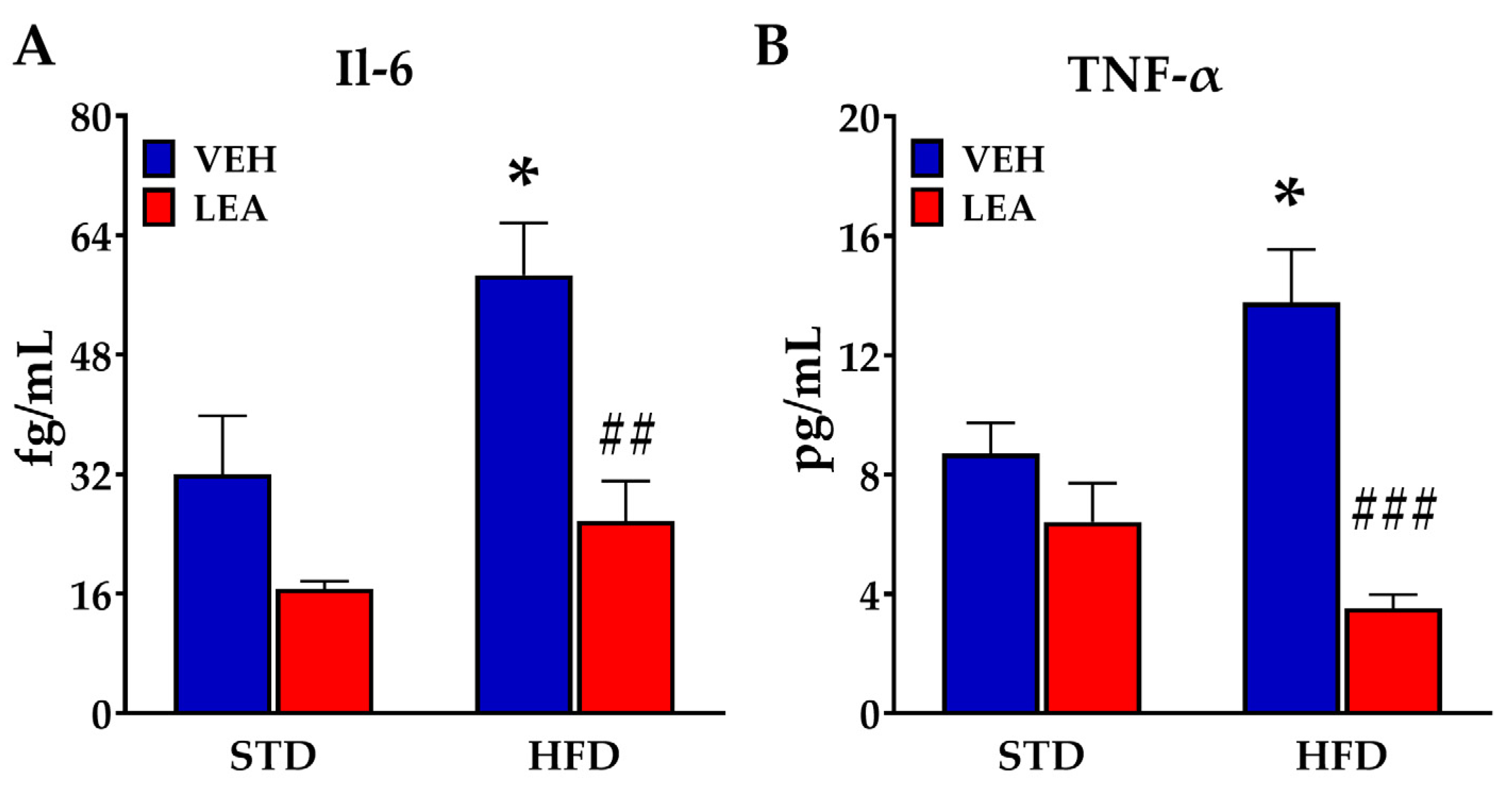
3.4. Effects of Sucbchronic Administration of Linoleoylethanolamide in Plasma Il-6 and Tnf-α
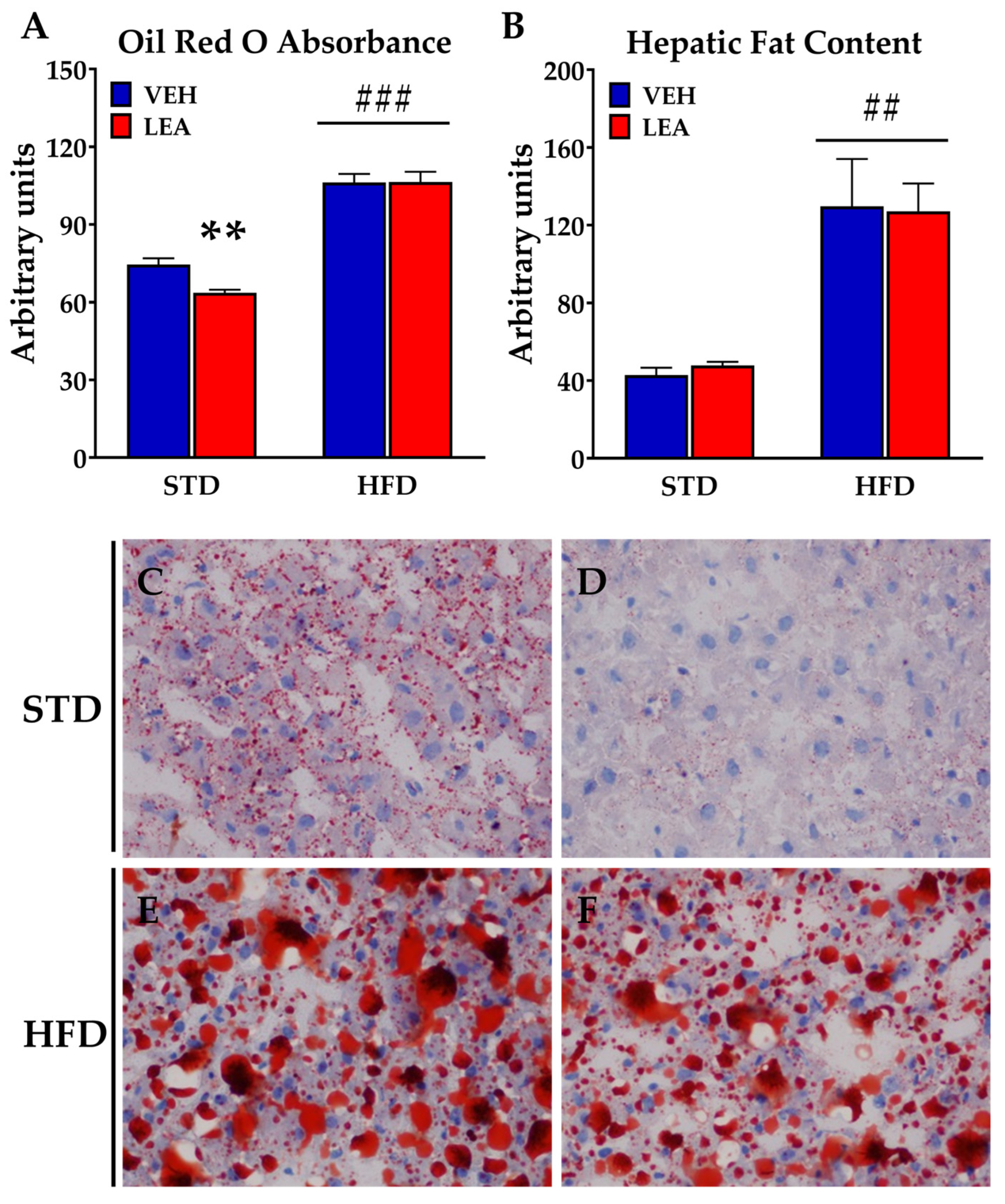
3.5. Effects of Chronic Administration of Linoleoylethanolamide on Liver Steatosis and the Expression of Lipid Metabolism-Related Enzymes in the Liver
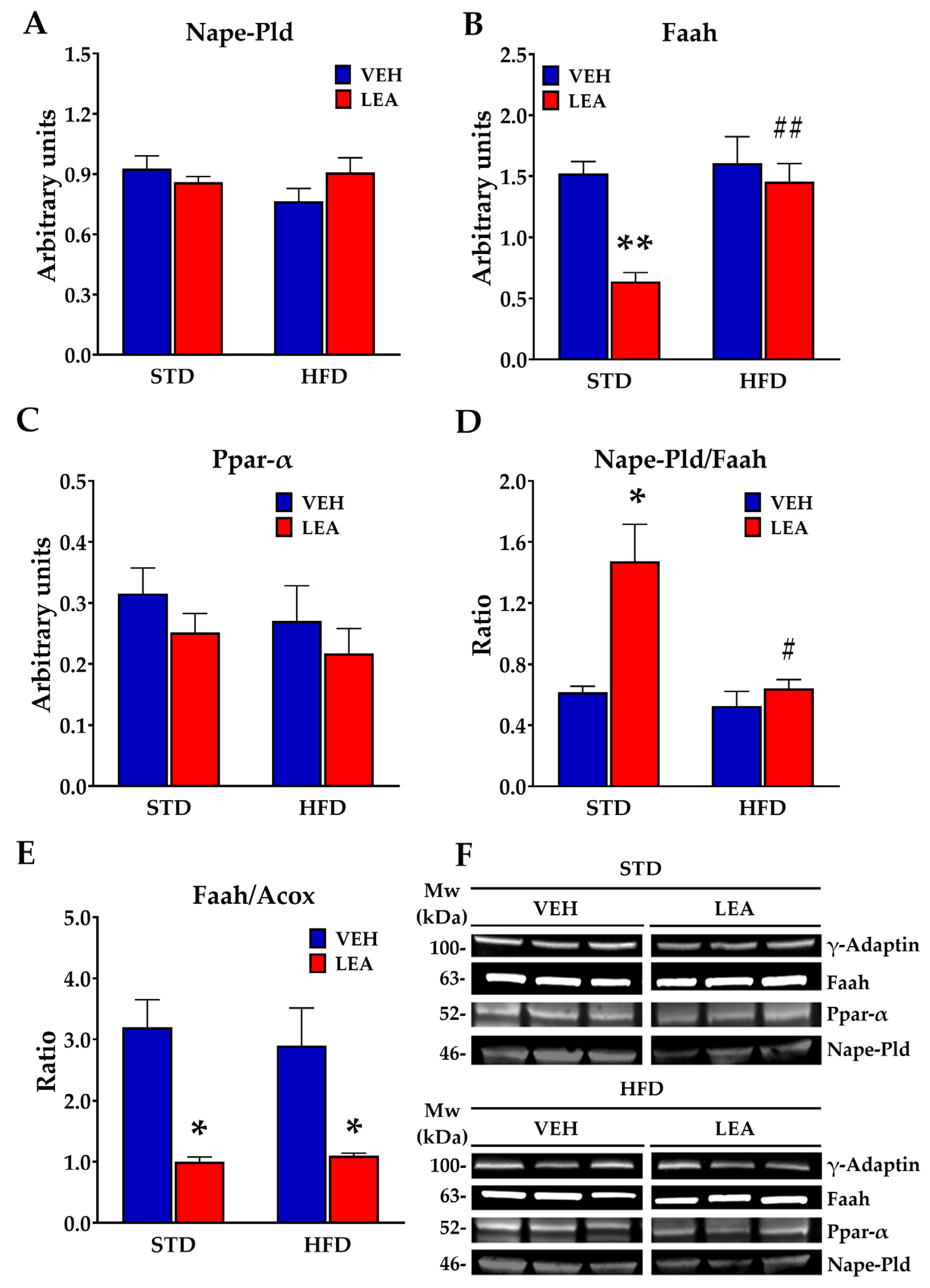
3.6. Effects of Chronic Administration of Linoleoylethanolamide on the Expression of Acylethanolamide-Related Enzymes in the Liver
3.7. Effects of Chronic Administration of Linoleoylethanolamide on Acylethanolamide Contents in the Liver
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Fonseca, F.R.; DEL Arco, I.; Bermudez-Silva, F.J.; Bilbao, A.; Cippitelli, A.; Navarro, M. The endocannabinoid system: Physiology and pharmacology. Alcohol. Alcohol. 2005, 40, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Mock, E.D.; Gagestein, B.; van der Stelt, M. Anandamide and other N-acylethanolamines: A class of signaling lipids with therapeutic opportunities. Prog. Lipid Res. 2023, 89, 101194. [Google Scholar] [CrossRef] [PubMed]
- Schmid, H.; Berdyshev, E. Cannabinoid receptor-inactive N -acylethanolamines and other fatty acid amides: Metabolism and function. Prostaglandins Leukot. Essent. Fat. Acids 2002, 66, 363–376. [Google Scholar] [CrossRef]
- Hansen, H.S. Role of anorectic N-acylethanolamines in intestinal physiology and satiety control with respect to dietary fat. Pharmacol. Res. 2014, 86, 18–25. [Google Scholar] [CrossRef] [PubMed]
- De Luca, L.; Ferracane, R.; Vitaglione, P. Food database of N-acyl-phosphatidylethanolamines, N-acylethanolamines and endocannabinoids and daily intake from a Western, a Mediterranean and a vegetarian diet. Food Chem. 2019, 300, 125218. [Google Scholar] [CrossRef]
- De Luca, L.; Ferracane, R.; Ramírez, N.C.; Vitaglione, P. N-Acylphosphatidylethanolamines and N-acylethanolamines increase in saliva upon food mastication: The influence of the individual nutritional status and fat type in food. Food Funct. 2020, 11, 3382–3392. [Google Scholar] [CrossRef]
- Liu, A.; Ma, T.; Xu, N.; Jin, H.; Zhao, F.; Kwok, L.-Y.; Zhang, H.; Zhang, S.; Sun, Z. Adjunctive Probiotics Alleviates Asthmatic Symptoms via Modulating the Gut Microbiome and Serum Metabolome. Microbiol. Spectr. 2021, 9, e0085921. [Google Scholar] [CrossRef]
- Rankin, L.; Fowler, C.J. The basal pharmacology of palmitoylethanolamide. Int. J. Mol. Sci. 2020, 21, 7942. [Google Scholar] [CrossRef]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef]
- Piomelli, D.; Scalvini, L.; Fotio, Y.; Lodola, A.; Spadoni, G.; Tarzia, G.; Mor, M. N-Acylethanolamine Acid Amidase (NAAA): Structure, Function, and Inhibition. J. Med. Chem. 2020, 63, 7475–7490. [Google Scholar] [CrossRef]
- Pertwee, R.G. Endocannabinoids and Their Pharmacological Actions. In Endocannabinoids; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 231, pp. 1–37. [Google Scholar] [CrossRef]
- Cota, D.; Marsicano, G.; Tschöp, M.; Grübler, Y.; Flachskamm, C.; Schubert, M.; Auer, D.; Yassouridis, A.; Thöne-Reineke, C.; Ortmann, S.; et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J. Clin. Investig. 2003, 112, 423–431. [Google Scholar] [CrossRef]
- De Fonseca, F.R.; Navarro, M.; Gómez, R.; Escuredo, L.; Nava, F.; Fu, J.; Murillo-Rodríguez, E.; Giuffrida, A.; LoVerme, J.; Gaetani, S.; et al. An anorexic lipid mediator regulated by feeding. Nature 2001, 414, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Gaetani, S.; Oveisi, F.; Verme, J.L.; Serrano, A.; de Fonseca, F.R.; Rosengarth, A.; Luecke, H.; Di Giacomo, B.; Tarzia, G.; et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature 2003, 425, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Overton, H.A.; Babbs, A.J.; Doel, S.M.; Fyfe, M.C.; Gardner, L.S.; Griffin, G.; Jackson, H.C.; Procter, M.J.; Rasamison, C.M.; Tang-Christensen, M.; et al. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006, 3, 167–175. [Google Scholar] [CrossRef]
- Bilbao, A.; Serrano, A.; Cippitelli, A.; Pavón, F.J.; Giuffrida, A.; Suárez, J.; García-Marchena, N.; Baixeras, E.; de Heras, R.G.; Orio, L.; et al. Role of the satiety factor oleoylethanolamide in alcoholism. Addict. Biol. 2016, 21, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Orio, L.; Alen, F.; Pavón, F.J.; Serrano, A.; García-Bueno, B. Oleoylethanolamide, Neuroinflammation, and Alcohol Abuse. Front. Mol. Neurosci. 2019, 11, 490. [Google Scholar] [CrossRef]
- Schwartz, G.J.; Fu, J.; Astarita, G.; Li, X.; Gaetani, S.; Campolongo, P.; Cuomo, V.; Piomelli, D. The Lipid Messenger OEA Links Dietary Fat Intake to Satiety. Cell Metab. 2008, 8, 281–288. [Google Scholar] [CrossRef]
- Serrano, A.; del Arco, I.; Pavón, F.J.; Macías, M.; Perez-Valero, V.; de Fonseca, F.R. The cannabinoid CB1 receptor antagonist SR141716A (Rimonabant) enhances the metabolic benefits of long-term treatment with oleoylethanolamide in Zucker rats. Neuropharmacology 2008, 54, 226–234. [Google Scholar] [CrossRef]
- Solorzano, C.; Zhu, C.; Battista, N.; Astarita, G.; Lodola, A.; Rivara, S.; Mor, M.; Russo, R.; Maccarrone, M.; Antonietti, F.; et al. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc. Natl. Acad. Sci. USA 2009, 106, 20966–20971. [Google Scholar] [CrossRef]
- Artmann, A.; Petersen, G.; Hellgren, L.I.; Boberg, J.; Skonberg, C.; Nellemann, C.; Hansen, S.H.; Hansen, H.S. Influence of dietary fatty acids on endocannabinoid and N-acylethanolamine levels in rat brain, liver and small intestine. Biochim. Biophys. Acta 2008, 1781, 200–212. [Google Scholar] [CrossRef]
- Tagliamonte, S.; Gill, C.I.R.; Pourshahidi, L.K.; Slevin, M.M.; Price, R.K.; Ferracane, R.; Lawther, R.; O’connor, G.; Vitaglione, P. Endocannabinoids, endocannabinoid-like molecules and their precursors in human small intestinal lumen and plasma: Does diet affect them? Eur. J. Nutr. 2021, 60, 2203–2215. [Google Scholar] [CrossRef]
- Li, Z.; Dong, F.; Sun, Y.; Sun, Z.; Song, X.; Dong, Y.; Huang, X.; Zhong, J.; Zhang, R.; Wang, M.; et al. Qualitative and Quantitative Analysis of Six Fatty Acid Amides in 11 Edible Vegetable Oils Using Liquid Chromatography–Mass Spectrometry. Front. Nutr. 2022, 9, 857858. [Google Scholar] [CrossRef]
- Ho, M.; Anderson, G.H.; Lin, L.; Bazinet, R.P.; Kubant, R. Ethanolamides of essential α-linolenic and linoleic fatty acids suppress short-term food intake in rats. Food Funct. 2020, 11, 3066–3072. [Google Scholar] [CrossRef]
- Xia, X.; Li, X.; Xie, F.; Yuan, G.; Cheng, D.; Peng, C. Non-targeted metabonomic analysis of plasma in patients with atherosclerosis by liquid chromatography-mass spectrometry. Ann. Transl. Med. 2022, 10, 133. [Google Scholar] [CrossRef]
- He, J.; Tang, D.; Liu, D.; Hong, X.; Ma, C.; Zheng, F.; Zeng, Z.; Chen, Y.; Du, J.; Kang, L.; et al. Serum proteome and metabolome uncover novel biomarkers for the assessment of disease activity and diagnosing of systemic lupus erythematosus. Clin. Immunol. 2023, 251, 109330. [Google Scholar] [CrossRef]
- Rubio-Martín, E.; Soriguer, F.; Gutiérrez-Repiso, C.; Garrido-Sánchez, L.; de Adana, M.S.R.; García-Fuentes, E.; Morcillo, S.; Esteva, I.; Chaves, F.J.; Rojo-Martinez, G. C-reactive protein and incidence of type 2 diabetes in the Pizarra study. Eur. J. Clin. Investig. 2013, 43, 159–167. [Google Scholar] [CrossRef]
- Soriguer, F.; Rojo-Martínez, G.; Almaraz, M.C.; Esteva, I.; de Adana, M.S.R.; Morcillo, S.; Valdés, S.; García-Fuentes, E.; García-Escobar, E.; Cardona, I.; et al. Incidence of type 2 diabetes in southern Spain (Pizarra Study). Eur. J. Clin. Investig. 2008, 38, 126–133. [Google Scholar] [CrossRef]
- Pastor, A.; Farré, M.; Fitó, M.; Fernandez-Aranda, F.; de la Torre, R. Analysis of ECs and related compounds in plasma: Artifactual isomerization and ex vivo enzymatic generation of 2-MGs. J. Lipid Res. 2014, 55, 966–977. [Google Scholar] [CrossRef]
- di Tomaso, E.; Beltramo, M.; Piomelli, D. Brain cannabinoids in chocolate. Nature 1996, 382, 677–678. [Google Scholar] [CrossRef]
- Parksepp, M.; Haring, L.; Kilk, K.; Koch, K.; Uppin, K.; Kangro, R.; Zilmer, M.; Vasar, E. The Expanded Endocannabinoid System Contributes to Metabolic and Body Mass Shifts in First-Episode Schizophrenia: A 5-Year Follow-Up Study. Biomedicines 2022, 10, 243. [Google Scholar] [CrossRef]
- Keereetaweep, J.; Blancaflor, E.B.; Hornung, E.; Feussner, I.; Chapman, K.D. Lipoxygenase-derived 9-hydro(pero)xides of linoleoylethanolamide interact with ABA signaling to arrest root development during Arabidopsis seedling establishment. Plant J. 2015, 82, 315–327. [Google Scholar] [CrossRef]
- Al-Khelaifi, F.; Diboun, I.; Donati, F.; Botrè, F.; Abraham, D.; Hingorani, A.; Albagha, O.; Georgakopoulos, C.; Suhre, K.; Yousri, N.A.; et al. Metabolic GWAS of elite athletes reveals novel genetically-influenced metabolites associated with athletic performance. Sci. Rep. 2019, 9, 19889, Erratum in Sci. Rep. 2020, 10, 10473. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, R.; Salvati, A.; Olinga, P.; Boersma, Y.L. Vanin 1: Its Physiological Function and Role in Diseases. Int. J. Mol. Sci. 2019, 20, 3891. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.S.; Diep, T.A. N-acylethanolamines, anandamide and food intake. Biochem. Pharmacol. 2009, 78, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Fernandez, M.E.; Giles, D.A.; Stankiewicz, T.E.; Sheridan, R.; Karns, R.; Cappelletti, M.; Lampe, K.; Mukherjee, R.; Sina, C.; Sallese, A.; et al. Peroxisomal β-oxidation regulates whole body metabolism, inflammatory vigor, and pathogenesis of nonalcoholic fatty liver disease. JCI Insight 2018, 3, e93626. [Google Scholar] [CrossRef]
- Ding, L.; Sun, W.; Balaz, M.; He, A.; Klug, M.; Wieland, S.; Caiazzo, R.; Raverdy, V.; Pattou, F.; Lefebvre, P.; et al. Peroxisomal β-oxidation acts as a sensor for intracellular fatty acids and regulates lipolysis. Nat. Metab. 2021, 3, 1648–1661. [Google Scholar] [CrossRef]
- Ježek, P.; Olejár, T.; Smolková, K.; Ježek, J.; Dlasková, A.; Plecitá-Hlavatá, L.; Zelenka, J.; Špaček, T.; Engstová, H.; Reguera, D.P.; et al. Antioxidant and Regulatory Role of Mitochondrial Uncoupling Protein UCP2 in Pancreatic β-cells. Physiol. Res. 2014, 63 (Suppl. S1), S73–S91, Erratum in Physiol. Res. 2015, 64, 277. [Google Scholar] [CrossRef]
- Di Bonaventura, M.V.M.; Coman, M.M.; Tomassoni, D.; Di Bonaventura, E.M.; Botticelli, L.; Gabrielli, M.G.; Rossolini, G.M.; Di Pilato, V.; Cecchini, C.; Amedei, A.; et al. Supplementation with Lactiplantibacillus plantarum IMC 510 Modifies Microbiota Composition and Prevents Body Weight Gain Induced by Cafeteria Diet in Rats. Int. J. Mol. Sci. 2021, 22, 11171. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | BMI (Kg/m2) < 25 (N = 13) | BMI (Kg/m2) > 25 (N = 34) | p Value |
|---|---|---|---|
| Age | 42.6 ± 3.0 | 44.5 ± 3.4 | n.s. |
| BMI | 23.7 ± 1.3 | 28.5 ± 2.1 ** | <0.0001 |
| Basal Glucose (mg/dL) | 126.0 ± 18.5 | 113.7 ± 57.3 | n.s. |
| % HBA1c | 5.2 ± 0.2 | 5.4 ± 0.4 | n.s. |
| Cholesterol (mg/dL) | 185.4 ± 21.8 | 208.9 ± 37.8 | n.s. |
| LDL Cholesterol (mg/dL) | 116.9 ± 20.1 | 130.8 ± 39.1 | n.s. |
| HDL Cholesterol (mg/dL) | 52.9 ± 7.9 | 45.1 ± 16.4 # | <0.05 |
| Triglycerides (mg/dL) | 77.7 ± 27.2 | 166.1 ± 143.1 # | <0.05 |
| Plasma Metabolites | Standard Diet (STD) | High-Fat Diet (HFD) | ||
|---|---|---|---|---|
| VEH | LEA | VEH | LEA | |
| Glucose (mg/dL) | 164.00 ± 8.45 | 157.33 ± 7.34 | 311.80 ± 42.3 *** | 168.57 ± 4.49 # |
| Insulin (ng/mL) | 3.94 ±0.55 | 3.28 ± 0.57 | 6.92 ± 1.51 * | 9.10 ± 1.48 * |
| Triglycerides (mg/dL) | 173.00 ± 7.68 | 146.57 ± 13.29 | 414.3 ± 93.7 ** | 150.29 ± 12.26 # |
| Cholesterol (mg/dL) | 68.38 ± 4.80 | 75.2 ± 8.88 | 211.00 ± 35.35 *** | 48.28 ± 3.76 # |
| Urea (mg/dL) | 16.86 ± 1.40 | 14.00 ± 1.29 | 72.92 ± 3.29 *** | 62.00 ± 13.66 ** |
| Uric acid (mg/dL) | 2.28 ± 0.34 | 1.63 ± 0.24 | 9.38 ± 1.95 *** | 2.27 ± 0.33 # |
| Creatinine (mg/dL) | 0.75 ± 0.09 | 0.69 ± 0.20 | 1.74 ± 0.12 ** | 0.71 ± 0.05 # |
| Bilirubin (mg/dL) | 0.18 ± 0.02 | 0.21 ± 0.03 | 0.99 ± 0.31 ** | 0.25 ± 0.13 |
| AST (UI) | 150.57 ± 19.91 | 138.00 ± 27.06 | 553.00 ± 41.37 ** | 288.33 ± 34.16 # |
| ALT (UI) | 53.14 ± 9.66 | 85.00 ± 3.56 | 206.33 ± 28.93 ** | 192.17 ± 27.64 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tovar, R.; de Ceglia, M.; Ubaldi, M.; Rodríguez-Pozo, M.; Soverchia, L.; Cifani, C.; Rojo, G.; Gavito, A.; Hernandez-Folgado, L.; Jagerovic, N.; et al. Administration of Linoleoylethanolamide Reduced Weight Gain, Dyslipidemia, and Inflammation Associated with High-Fat-Diet-Induced Obesity. Nutrients 2023, 15, 4448. https://doi.org/10.3390/nu15204448
Tovar R, de Ceglia M, Ubaldi M, Rodríguez-Pozo M, Soverchia L, Cifani C, Rojo G, Gavito A, Hernandez-Folgado L, Jagerovic N, et al. Administration of Linoleoylethanolamide Reduced Weight Gain, Dyslipidemia, and Inflammation Associated with High-Fat-Diet-Induced Obesity. Nutrients. 2023; 15(20):4448. https://doi.org/10.3390/nu15204448
Chicago/Turabian StyleTovar, Rubén, Marialuisa de Ceglia, Massimo Ubaldi, Miguel Rodríguez-Pozo, Laura Soverchia, Carlo Cifani, Gema Rojo, Ana Gavito, Laura Hernandez-Folgado, Nadine Jagerovic, and et al. 2023. "Administration of Linoleoylethanolamide Reduced Weight Gain, Dyslipidemia, and Inflammation Associated with High-Fat-Diet-Induced Obesity" Nutrients 15, no. 20: 4448. https://doi.org/10.3390/nu15204448