
Selected Flavonols Targeting Cell Death Pathways in Cancer Therapy: The Latest Achievements in Research on Apoptosis, Autophagy, Necroptosis, Pyroptosis, Ferroptosis, and Cuproptosis


Abstract
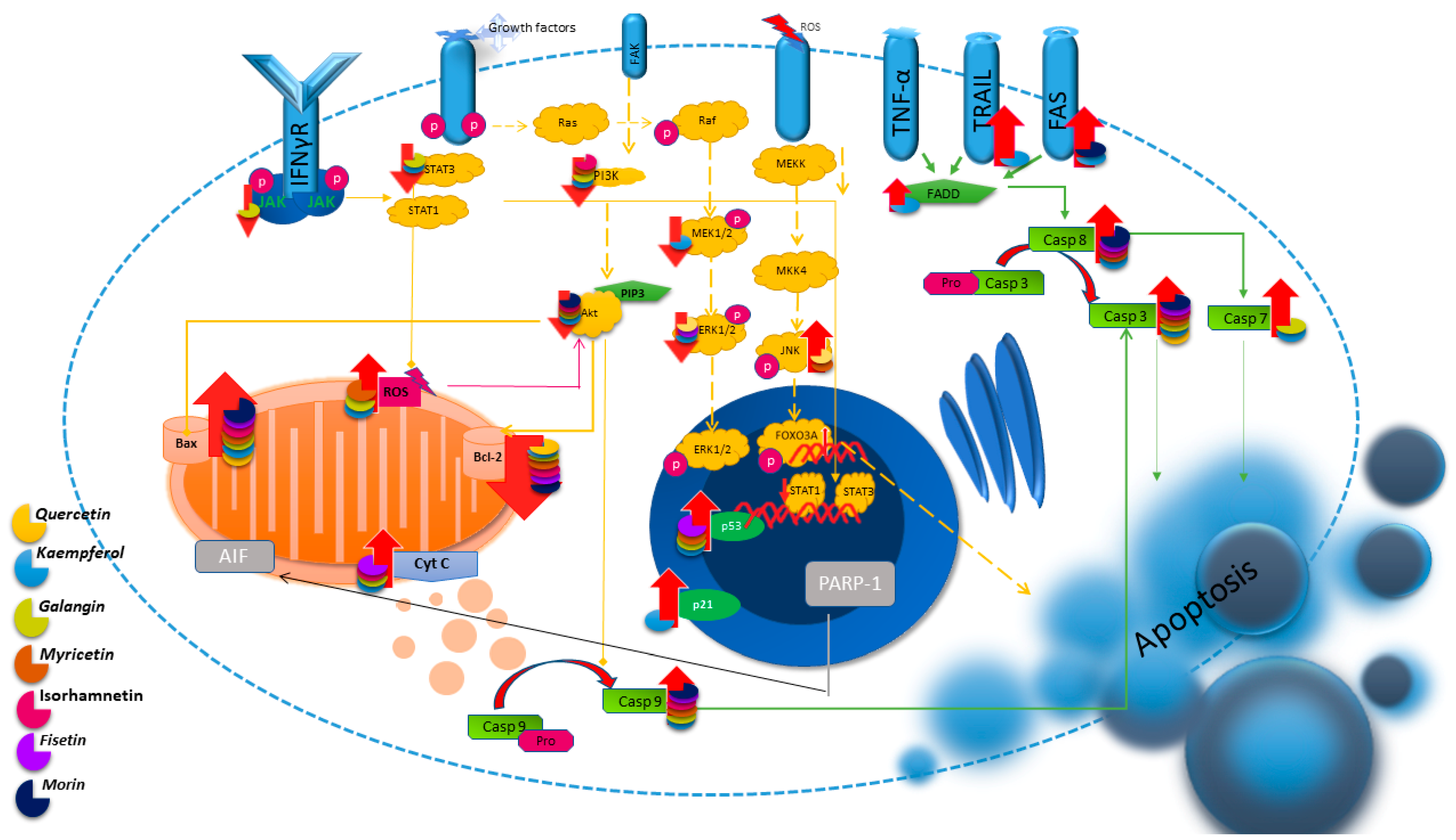
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flavonoid | Chemical Formula | Structure | Cell Death |
|---|---|---|---|
| Quercetin | C15H10O7 | 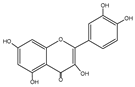 |
|
| Kaempferol | C15H10O6 |  |
|
| Galangin | C15H10O5 |  |
|
| Myricetin | C15H10O8 |  |
|
| Isorhamnetin | C16H12O7 | 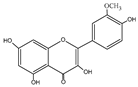 |
|
| Fisetin | C15H10O6 | 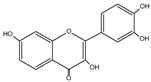 |
|
| Morin | C15H10O7 | 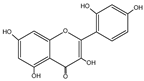 |
|
2. Apoptosis
2.1. Quercetin
2.2. Kaempferol
2.3. Galangin
2.4. Myricetin
2.5. Isorhamnetin
2.6. Fisetin
2.7. Morin
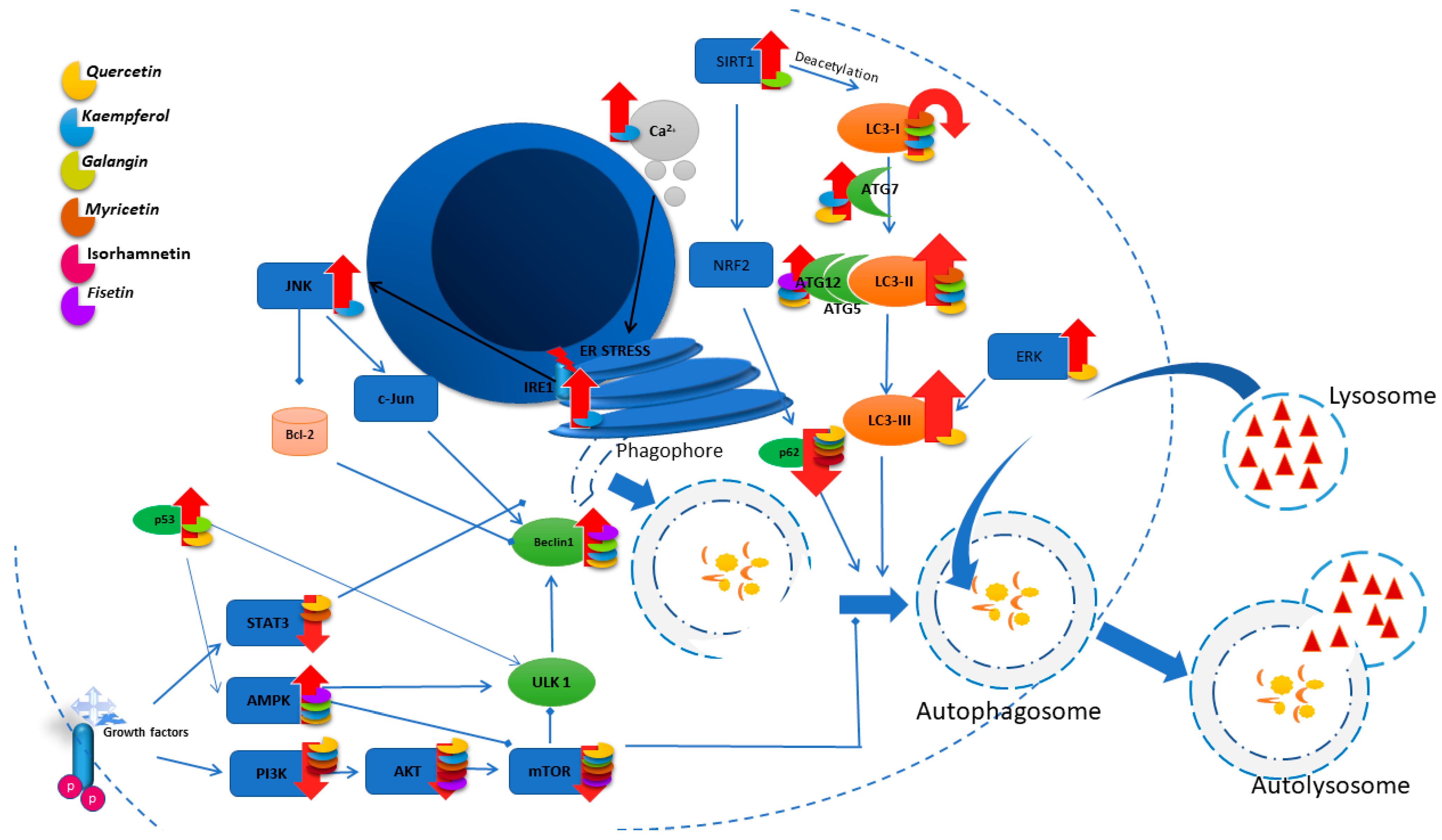
3. Autophagy
- (1)
- ADCD (autophagy-dependent cell death), which is independent of other forms of programmed death.
- (2)
- AMCD (autophagy-mediated cell death), where autophagic molecules interact directly with cell death molecules or where the autophagy is related to other cell death mechanisms such as apoptosis, necrosis, or ferroptosis through its dynamic processes.
3.1. Quercetin
3.2. Kaempferol
3.3. Galangin
3.4. Myricetin
3.5. Isorhamnetin
3.6. Fisetin
3.7. Morin
4. Pyroptosis
4.1. Kaempferol
4.2. Galangin
4.3. Myricetin
4.4. Fisetin
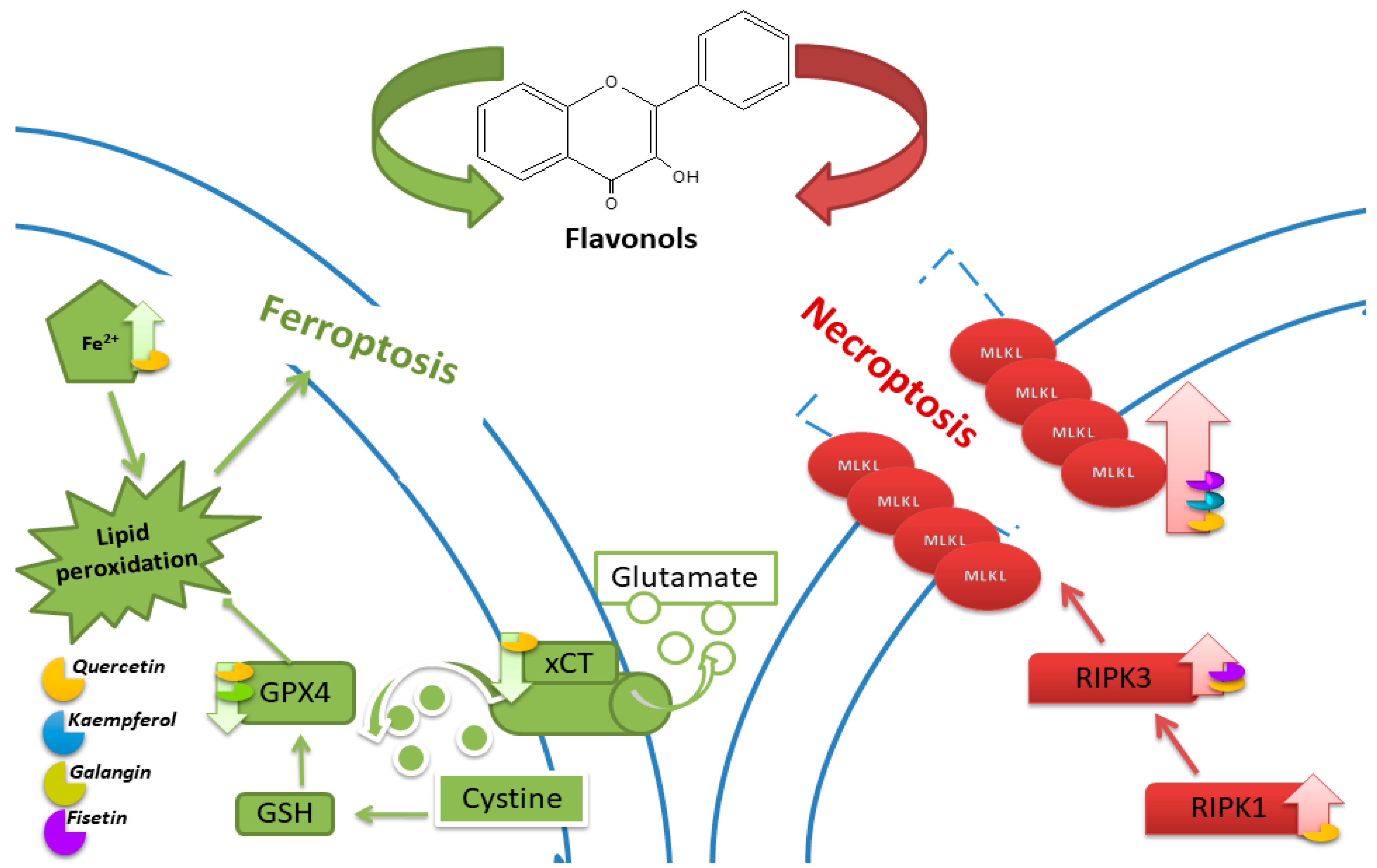
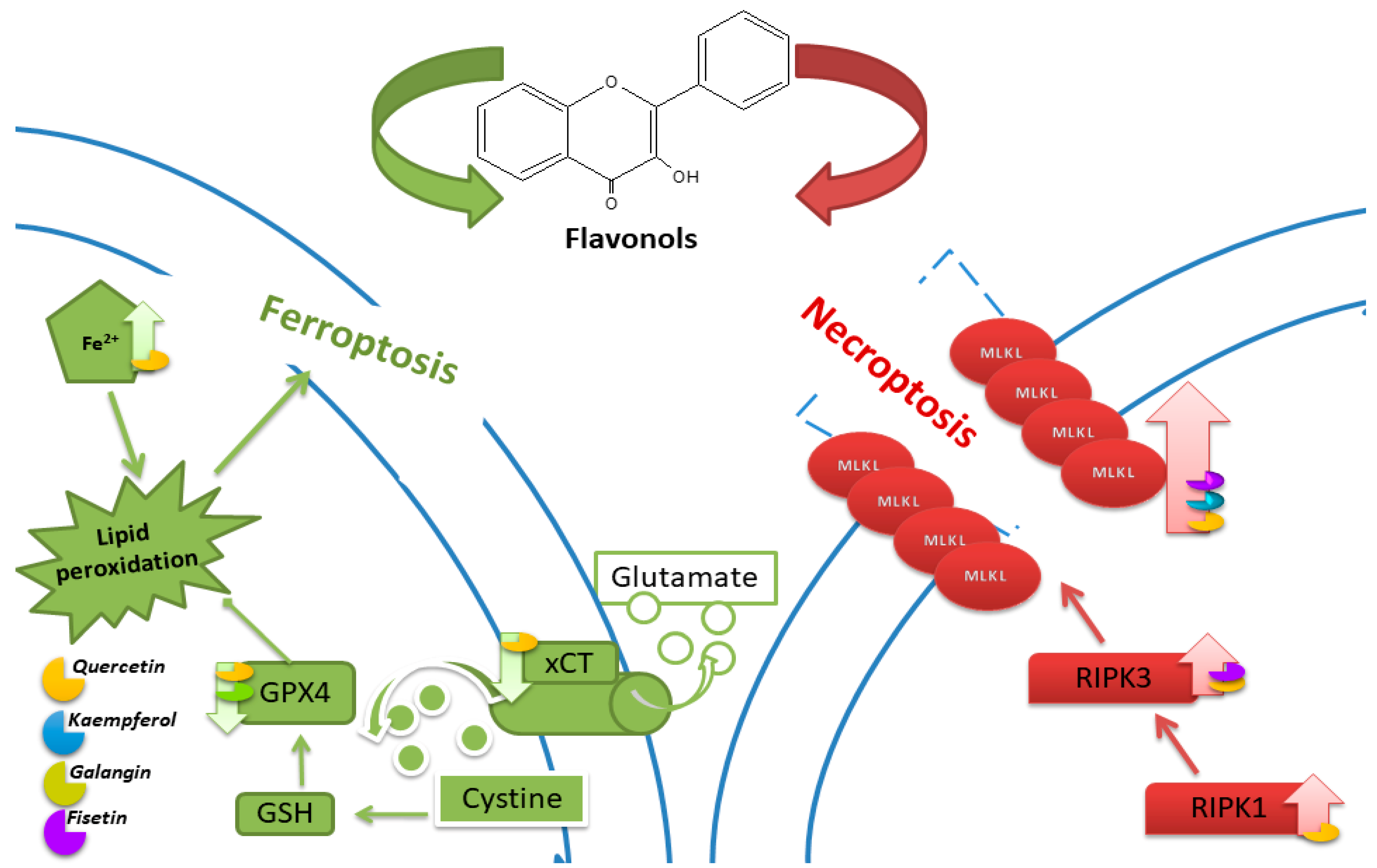
5. Ferroptosis
5.1. Quercetin
5.2. Kaempferol
5.3. Galangin
6. Necroptosis
6.1. Quercetin
6.2. Kaempferol
6.3. Fisetin
7. Cuproptosis
8. Protective Effects of Flavonoids against Cancer: In Vivo Evidence
8.1. Quercetin
8.2. Kaempferol
8.3. Galangin
8.4. Myricetin
8.5. Isorhamnetin
8.6. Fisetin
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Global Cancer Observatory. Available online: https://gco.iarc.fr/En (accessed on 19 March 2024).
- Kopustinskiene, D.M.; Jakstas, V.; Savickas, A.; Bernatoniene, J. Flavonoids as Anticancer Agents. Nutrients 2020, 12, 457. [Google Scholar] [CrossRef] [PubMed]
- Kubina, R.; Iriti, M.; Kabała-Dzik, A. Anticancer Potential of Selected Flavonols: Fisetin, Kaempferol, and Quercetin on Head and Neck Cancers. Nutrients 2021, 13, 845. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Tang, R.; Xiao, M.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Targeting Cell Death Pathways for Cancer Therapy: Recent Developments in Necroptosis, Pyroptosis, Ferroptosis, and Cuproptosis Research. J. Hematol. Oncol. 2022, 15, 174. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated Cell Death (RCD) in Cancer: Key Pathways and Targeted Therapies. Sig. Transduct. Target. Ther. 2022, 7, 286. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Forni, C.; Rossi, M.; Borromeo, I.; Feriotto, G.; Platamone, G.; Tabolacci, C.; Mischiati, C.; Beninati, S. Flavonoids: A Myth or a Reality for Cancer Therapy? Molecules 2021, 26, 3583. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, S.R.; Ebrahimzadeh, M.A. Quercetin Derivatives: Drug Design, Development, and Biological Activities, a Review. Eur. J. Med. Chem. 2022, 229, 114068. [Google Scholar] [CrossRef] [PubMed]
- Qaed, E.; Al-Hamyari, B.; Al-Maamari, A.; Qaid, A.; Alademy, H.; Almoiliqy, M.; Munyemana, J.C.; Al-Nusaif, M.; Alafifi, J.; Alyafeai, E.; et al. Fisetin’s Promising Antitumor Effects: Uncovering Mechanisms and Targeting for Future Therapies. Glob. Med. Genet. 2023, 10, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, G.M. Molecular Structure and Anti-Proliferative Effect of Galangin in HCT-116 Cells: In Vitro Study. Food Sci. Biotechnol. 2016, 25, 247–252. [Google Scholar] [CrossRef]
- Alkandahri, M.Y.; Pamungkas, B.T.; Oktoba, Z.; Shafirany, M.Z.; Sulastri, L.; Arfania, M.; Anggraeny, E.N.; Pratiwi, A.; Astuti, F.D.; Indriyani; et al. Hepatoprotective Effect of Kaempferol: A Review of the Dietary Sources, Bioavailability, Mechanisms of Action, and Safety. Adv. Pharmacol. Pharm. Sci. 2023, 2023, 1387665. [Google Scholar] [CrossRef]
- Semwal, D.; Semwal, R.; Combrinck, S.; Viljoen, A. Myricetin: A Dietary Molecule with Diverse Biological Activities. Nutrients 2016, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, L.; Yang, B.; Du, J.; Chen, L.; Li, Y.; Guo, F. Structures, Sources, Identification/Quantification Methods, Health Benefits, Bioaccessibility, and Products of Isorhamnetin Glycosides as Phytonutrients. Nutrients 2023, 15, 1947. [Google Scholar] [CrossRef] [PubMed]
- Chagas, M.D.S.S.; Behrens, M.D.; Moragas-Tellis, C.J.; Penedo, G.X.M.; Silva, A.R.; Gonçalves-de-Albuquerque, C.F. Flavonols and Flavones as Potential Anti-Inflammatory, Antioxidant, and Antibacterial Compounds. Oxidative Med. Cell. Longev. 2022, 2022, 9966750. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lai, Y.; Hua, Z.-C. Apoptosis and Apoptotic Body: Disease Message and Therapeutic Target Potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S. Apoptosis in Cancer: From Pathogenesis to Treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Morana, O.; Wood, W.; Gregory, C.D. The Apoptosis Paradox in Cancer. Int. J. Mol. Sci. 2022, 23, 1328. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Kim, B. Anti-Cancer Natural Products and Their Bioactive Compounds Inducing ER Stress-Mediated Apoptosis: A Review. Nutrients 2018, 10, 1021. [Google Scholar] [CrossRef] [PubMed]
- Kulbay, M.; Paimboeuf, A.; Ozdemir, D.; Bernier, J. Review of Cancer Cell Resistance Mechanisms to Apoptosis and Actual Targeted Therapies. J. Cell. Biochem. 2022, 123, 1736–1761. [Google Scholar] [CrossRef]
- Khorsandi, L.; Orazizadeh, M.; Niazvand, F.; Abbaspour, M.R.; Mansouri, E.; Khodadadi, A. Quercetin Induces Apoptosis and Necroptosis in MCF-7 Breast Cancer Cells. Bratisl. Lek. Listy 2017, 118, 123–128. [Google Scholar] [CrossRef]
- Niu, G.; Yin, S.; Xie, S.; Li, Y.; Nie, D.; Ma, L.; Wang, X.; Wu, Y. Quercetin Induces Apoptosis by Activating Caspase-3 and Regulating Bcl-2 and Cyclooxygenase-2 Pathways in Human HL-60 Cells. Acta Biochim. Biophys Sin. 2011, 43, 30–37. [Google Scholar] [CrossRef]
- Kim, M.C.; Lee, H.J.; Lim, B.; Ha, K.-T.; Kim, S.Y.; So, I.; Kim, B.J. Quercetin Induces Apoptosis by Inhibiting MAPKs and TRPM7 Channels in AGS Cells. Int. J. Mol. Med. 2014, 33, 1657–1663. [Google Scholar] [CrossRef]
- Kashafi, E.; Moradzadeh, M.; Mohamadkhani, A.; Erfanian, S. Kaempferol Increases Apoptosis in Human Cervical Cancer HeLa Cells via PI3K/AKT and Telomerase Pathways. Biomed. Pharmacother. 2017, 89, 573–577. [Google Scholar] [CrossRef]
- Kubina, R.; Krzykawski, K.; Dziedzic, A.; Kabała-Dzik, A. Kaempferol and Fisetin-Related Signaling Pathways Induce Apoptosis in Head and Neck Cancer Cells. Cells 2023, 12, 1568. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Si, L.; Jia, Y.; Jian, W.; Yu, Q.; Wang, M.; Lin, R. Kaempferol Exerts Anti-Proliferative Effects on Human Ovarian Cancer Cells by Inducing Apoptosis, G0/G1 Cell Cycle Arrest and Modulation of MEK/ERK and STAT3 Pathways. J. Buon 2019, 24, 975–981. [Google Scholar] [PubMed]
- Gao, Y.; Yin, J.; Rankin, G.; Chen, Y. Kaempferol Induces G2/M Cell Cycle Arrest via Checkpoint Kinase 2 and Promotes Apoptosis via Death Receptors in Human Ovarian Carcinoma A2780/CP70 Cells. Molecules 2018, 23, 1095. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, L.; Gao, M.; Wang, S.; Meng, L.; Guo, L. Molecular Docking and In Vitro Experiments Verified That Kaempferol Induced Apoptosis and Inhibited Human HepG2 Cell Proliferation by Targeting BAX, CDK1, and JUN. Mol. Cell Biochem. 2023, 478, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, L.; Qu, C.; Chen, L.; Geng, Y.; Cheng, C.; Yu, S.; Wang, D.; Yang, L.; Meng, Z.; et al. Kaempferol Induces ROS-Dependent Apoptosis in Pancreatic Cancer Cells via TGM2-Mediated Akt/mTOR Signaling. BMC Cancer 2021, 21, 396. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Meng, X.; Zheng, H.; Zeng, Q.; Chen, T.; Wang, W.; Zhang, X.; Su, J. Kaempferol Attenuates ROS-Induced Hemolysis and the Molecular Mechanism of Its Induction of Apoptosis on Bladder Cancer. Molecules 2018, 23, 2592. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Cho, H.; Yu, R.; Lee, K.; Chun, H.; Park, J. Mechanisms Underlying Apoptosis-Inducing Effects of Kaempferol in HT-29 Human Colon Cancer Cells. Int. J. Mol. Sci. 2014, 15, 2722–2737. [Google Scholar] [CrossRef]
- Zhu, L.; Xue, L. Kaempferol Suppresses Proliferation and Induces Cell Cycle Arrest, Apoptosis, and DNA Damage in Breast Cancer Cells. Oncol. Res. 2019, 27, 629–634. [Google Scholar] [CrossRef]
- Choi, J.-B.; Kim, J.-H.; Lee, H.; Pak, J.-N.; Shim, B.S.; Kim, S.-H. Reactive Oxygen Species and P53 Mediated Activation of P38 and Caspases Is Critically Involved in Kaempferol Induced Apoptosis in Colorectal Cancer Cells. J. Agric. Food Chem. 2018, 66, 9960–9967. [Google Scholar] [CrossRef] [PubMed]
- Fouzder, C.; Mukhuty, A.; Kundu, R. Kaempferol Inhibits Nrf2 Signalling Pathway via Downregulation of Nrf2 mRNA and Induces Apoptosis in NSCLC Cells. Arch. Biochem. Biophys. 2021, 697, 108700. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Yan, C.; Zhou, Q.; Zhen, L. Galangin Potentiates Human Breast Cancer to Apoptosis Induced by TRAIL through Activating AMPK. Biomed. Pharmacother. 2017, 89, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; You, P.; Luo, Y.; Yang, M.; Liu, Y. Galangin Induces Apoptosis in MCF-7 Human Breast Cancer Cells through Mitochondrial Pathway and Phosphatidylinositol 3-Kinase/Akt Inhibition. Pharmacology 2018, 102, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Rao, Q.; Zhang, X.; Zhou, X. Galangin Induced Antitumor Effects in Human Kidney Tumor Cells Mediated via Mitochondrial Mediated Apoptosis, Inhi- Bition of Cell Migration and Invasion and Targeting PI3K/AKT/mTOR Signalling Pathway. J. Buon 2018, 23, 795–799. [Google Scholar] [PubMed]
- Lee, C.C.; Lin, M.L.; Meng, M.; Chen, S.S. Galangin Induces P53-Independent S-Phase Arrest and Apoptosis in Human Nasopharyngeal Carcinoma Cells through Inhibiting PI3K−AKT Signaling Pathway. Anticancer Res. 2018, 38, 1377–1389. [Google Scholar] [CrossRef]
- Liang, X.; Wang, P.; Yang, C.; Huang, F.; Wu, H.; Shi, H.; Wu, X. Galangin Inhibits Gastric Cancer Growth through Enhancing STAT3 Mediated ROS Production. Front. Pharmacol. 2021, 12, 646628. [Google Scholar] [CrossRef]
- Zou, W.-W.; Xu, S.-P. Galangin Inhibits the Cell Progression and Induces Cell Apoptosis through Activating PTEN and Caspase-3 Pathways in Retinoblastoma. Biomed. Pharmacother. 2018, 97, 851–863. [Google Scholar] [CrossRef]
- Cao, J.; Wang, H.; Chen, F.; Fang, J.; Xu, A.; Xi, W.; Zhang, S.; Wu, G.; Wang, Z. Galangin Inhibits Cell Invasion by Suppressing the Epithelial-Mesenchymal Transition and Inducing Apoptosis in Renal Cell Carcinoma. Mol. Med. Rep. 2016, 13, 4238–4244. [Google Scholar] [CrossRef]
- Han, S.-H.; Lee, J.-H.; Woo, J.-S.; Jung, G.-H.; Jung, S.-H.; Han, E.-J.; Park, Y.-S.; Kim, B.-S.; Kim, S.-K.; Park, B.-K.; et al. Myricetin Induces Apoptosis through the MAPK Pathway and Regulates JNK-mediated Autophagy in SK-BR-3 Cells. Int. J. Mol. Med. 2022, 49, 54. [Google Scholar] [CrossRef]
- Han, S.-H.; Lee, J.-H.; Woo, J.-S.; Jung, G.-H.; Jung, S.-H.; Han, E.-J.; Kim, B.; Cho, S.D.; Nam, J.S.; Che, J.H.; et al. Myricetin Induces Apoptosis and Autophagy in Human Gastric Cancer Cells through Inhibition of the PI3K/Akt/mTOR Pathway. Heliyon 2022, 8, e09309. [Google Scholar] [CrossRef]
- Ha, T.K.; Jung, I.; Kim, M.E.; Bae, S.K.; Lee, J.S. Anti-Cancer Activity of Myricetin against Human Papillary Thyroid Cancer Cells Involves Mitochondrial Dysfunction–Mediated Apoptosis. Biomed. Pharmacother. 2017, 91, 378–384. [Google Scholar] [CrossRef]
- Jo, S.; Ha, T.K.; Han, S.H.; Kim, M.E.; Jung, I.; Lee, H.W.; Bae, S.K.; Lee, J.S. Myricetin Induces Apoptosis of Human Anaplastic Thyroid Cancer Cells via Mitochondria Dysfunction. Anticancer Res. 2017, 37, 1705–1710. [Google Scholar] [CrossRef]
- Huang, H.; Chen, A.Y.; Ye, X.; Li, B.; Rojanasakul, Y.; Rankin, G.O.; Chen, Y.C. Myricetin Inhibits Proliferation of Cisplatin-Resistant Cancer Cells through a P53-Dependent Apoptotic Pathway. Int. J. Oncol. 2015, 47, 1494–1502. [Google Scholar] [CrossRef]
- Soleimani, M.; Sajedi, N. Myricetin Apoptotic Effects on T47D Breast Cancer Cells Is a P53-Independent Approach. Asian Pac. J. Cancer Prev. 2020, 21, 3697–3704. [Google Scholar] [CrossRef]
- Zhu, M.; Zhang, P.; Jiang, M.; Yu, S.; Wang, L. Myricetin Induces Apoptosis and Autophagy by Inhibiting PI3K/Akt/mTOR Signalling in Human Colon Cancer Cells. BMC Complement. Med. Ther. 2020, 20, 209. [Google Scholar] [CrossRef]
- Rajendran, P.; Maheshwari, U.; Muthukrishnan, A.; Muthuswamy, R.; Anand, K.; Ravindran, B.; Dhanaraj, P.; Balamuralikrishnan, B.; Chang, S.W.; Chung, W.J. Myricetin: Versatile Plant Based Flavonoid for Cancer Treatment by Inducing Cell Cycle Arrest and ROS–Reliant Mitochondria-Facilitated Apoptosis in A549 Lung Cancer Cells and in Silico Prediction. Mol. Cell Biochem. 2021, 476, 57–68. [Google Scholar] [CrossRef]
- Duan, R.; Liang, X.; Chai, B.; Zhou, Y.; Du, H.; Suo, Y.; Chen, Z.; Li, Q.; Huang, X. Isorhamnetin Induces Melanoma Cell Apoptosis via the PI3K/Akt and NF-κB Pathways. BioMed Res. Int. 2020, 2020, 1057943. [Google Scholar] [CrossRef]
- Choi, Y.H. Isorhamnetin Induces ROS-Dependent Cycle Arrest at G2/M Phase and Apoptosis in Human Hepatocarcinoma Hep3B Cells. Gen. Physiol. Biophys. 2019, 38, 473–484. [Google Scholar] [CrossRef]
- Li, Y.; Fan, B.; Pu, N.; Ran, X.; Lian, T.; Cai, Y.; Xing, W.; Sun, K. Isorhamnetin Suppresses Human Gastric Cancer Cell Proliferation through Mitochondria-Dependent Apoptosis. Molecules 2022, 27, 5191. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, Y.; Jiang, X.; Zhang, H.; Gao, Z.; Li, Y.; Fu, R.; Li, L.; Li, J.; Cui, H.; et al. ROS-Mediated Activation and Mitochondrial Translocation of CaMKII Contributes to Drp1-Dependent Mitochondrial Fission and Apoptosis in Triple-Negative Breast Cancer Cells by Isorhamnetin and Chloroquine. J. Exp. Clin. Cancer Res. 2019, 38, 225. [Google Scholar] [CrossRef]
- Park, C.; Cha, H.J.; Choi, E.O.; Lee, H.; Hwang-Bo, H.; Ji, S.Y.; Kim, M.Y.; Kim, S.Y.; Hong, S.H.; Cheong, J.; et al. Isorhamnetin Induces Cell Cycle Arrest and Apoptosis Via Reactive Oxygen Species-Mediated AMP-Activated Protein Kinase Signaling Pathway Activation in Human Bladder Cancer Cells. Cancers 2019, 11, 1494. [Google Scholar] [CrossRef]
- Li, C.; Li, J.; Li, Y.; Li, L.; Luo, Y.; Li, J.; Zhang, Y.; Wang, Y.; Liu, X.; Zhou, X.; et al. Isorhamnetin Promotes MKN-45 Gastric Cancer Cell Apoptosis by Inhibiting PI3K-Mediated Adaptive Autophagy in a Hypoxic Environment. J. Agric. Food Chem. 2021, 69, 8130–8143. [Google Scholar] [CrossRef]
- Ruan, Y.; Hu, K.; Chen, H. Autophagy Inhibition Enhances Isorhamnetin-Induced Mitochondria-Dependent Apoptosis in Non-Small Cell Lung Cancer Cells. Mol. Med. Rep. 2015, 12, 5796–5806. [Google Scholar] [CrossRef]
- Cai, F.; Zhang, Y.; Li, J.; Huang, S.; Gao, R. Isorhamnetin Inhibited the Proliferation and Metastasis of Androgen-Independent Prostate Cancer Cells by Targeting the Mitochondrion-Dependent Intrinsic Apoptotic and PI3K/Akt/mTOR Pathway. Biosci. Rep. 2020, 40, BSR20192826. [Google Scholar] [CrossRef]
- Shih, Y.L.; Hung, F.M.; Lee, C.H.; Yeh, M.Y.; Lee, M.H.; Lu, H.F.; Chen, Y.L.; Liu, J.Y.; Chung, J.G. Fisetin Induces Apoptosis of HSC3 Human Oral Cancer Cells through Endoplasmic Reticulum Stress and Dysfunction of Mitochondria-Mediated Signaling Pathways. In Vivo 2017, 31, 1103–1114. [Google Scholar] [CrossRef]
- Wang, K.; Hu, D.; Lin, H.; Yang, W.; Hsieh, Y.; Chien, H.; Yang, S. F Isetin Induces Apoptosis through Mitochondrial Apoptosis Pathway in Human Uveal Melanoma Cells. Environ. Toxicol. 2018, 33, 527–534. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Hyun, J.W. Fisetin Induces Apoptosis in Human Nonsmall Lung Cancer Cells via a Mitochondria-Mediated Pathway. Vitr. Cell. Dev. Biol. Anim. 2015, 51, 300–309. [Google Scholar] [CrossRef]
- Sun, X.; Ma, X.; Li, Q.; Yang, Y.; Xu, X.; Sun, J.; Yu, M.; Cao, K.; Yang, L.; Yang, G.; et al. Anti-cancer effects of fisetin on mammary carcinoma cells via regulation of the PI3K/Akt/mTOR pathway: In vitro and in vivo studies. Int. J. Mol. Med. 2018, 42, 811–820. [Google Scholar] [CrossRef]
- Sabarwal, A.; Agarwal, R.; Singh, R.P. Fisetin Inhibits Cellular Proliferation and Induces Mitochondria-Dependent Apoptosis in Human Gastric Cancer Cells. Mol. Carcinog. 2017, 56, 499–514. [Google Scholar] [CrossRef]
- Yan, W.; Chen, S.; Zhao, Y.; Ye, X. Fisetin Inhibits the Proliferation of Gastric Cancer Cells and Induces Apoptosis through Suppression of ERK 1/2 Activation. Oncol. Lett. 2018, 15, 8442–8446. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, Y.; Gao, Z.; Li, X.; Weng, M.; Shi, C.; Wang, C.; Sun, L. Fisetin Inhibits the Proliferation, Migration and Invasion of Pancreatic Cancer by Targeting PI3K/AKT/mTOR Signaling. Aging 2021, 13, 24753–24767. [Google Scholar] [CrossRef]
- Kim, T.W. Fisetin, an Anti-Inflammatory Agent, Overcomes Radioresistance by Activating the PERK-ATF4-CHOP Axis in Liver Cancer. Int. J. Mol. Sci. 2023, 24, 9076. [Google Scholar] [CrossRef]
- Pandey, A.; Trigun, S.K. Fisetin Induces Apoptosis in Colorectal Cancer Cells by Suppressing Autophagy and Down-regulating Nuclear Factor Erythroid 2-related Factor 2 (Nrf2). J. Cell. Biochem. 2023, 124, 1289–1308. [Google Scholar] [CrossRef]
- Guo, G.; Zhang, W.; Dang, M.; Yan, M.; Chen, Z. Fisetin Induces Apoptosis in Breast Cancer MDA-MB-453 Cells through Degradation of HER2/Neu and via the PI3K/Akt Pathway. J. Biochem. Mol. Toxicol. 2019, 33, e22268. [Google Scholar] [CrossRef]
- Nie, Z.-Y.; Yang, L.; Liu, X.-J.; Yang, Z.; Yang, G.-S.; Zhou, J.; Qin, Y.; Yu, J.; Jiang, L.-L.; Wen, J.-K.; et al. Morin Inhibits Proliferation and Induces Apoptosis by Modulating the miR-188-5p/PTEN/AKT Regulatory Pathway in CML Cells. Mol. Cancer Ther. 2019, 18, 2296–2307. [Google Scholar] [CrossRef]
- Hyun, H.-B.; Lee, W.S.; Go, S.-I.; Nagappan, A.; Park, C.; Han, M.H.; Hong, S.H.; Kim, G.; Kim, G.Y.; Cheong, J.; et al. The Flavonoid Morin from Moraceae Induces Apoptosis by Modulation of Bcl-2 Family Members and Fas Receptor in HCT 116 Cells. Int. J. Oncol. 2015, 46, 2670–2678. [Google Scholar] [CrossRef]
- Sithara, T.; Arun, K.B.; Syama, H.P.; Reshmitha, T.R.; Nisha, P. Morin Inhibits Proliferation of SW480 Colorectal Cancer Cells by Inducing Apoptosis Mediated by Reactive Oxygen Species Formation and Uncoupling of Warburg Effect. Front. Pharmacol. 2017, 8, 640. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kim, W.I.; Kim, S.Y.; Cho, S.W.; Nam, H.S.; Lee, S.H.; Cho, M.K. Flavonoid Morin Inhibits Proliferation and Induces Apoptosis of Melanoma Cells by Regulating Reactive Oxygen Species, Sp1 and Mcl-1. Arch. Pharm. Res. 2019, 42, 531–542. [Google Scholar] [CrossRef]
- Tezerji, S.; Nazari Robati, F.; Abdolazimi, H.; Fallah, A.; Talaei, B. Quercetin’s Effects on Colon Cancer Cells Apoptosis and Proliferation in a Rat Model of Disease. Clin. Nutr. ESPEN 2022, 48, 441–445. [Google Scholar] [CrossRef]
- Keshavarz, F.; Dorfaki, M.; Bardania, H.; Khosravani, F.; Nazari, P.; Ghalamfarsa, G. Quercetin-Loaded Liposomes Effectively Induced Apoptosis and Decreased the Epidermal Growth Factor Receptor Expression in Colorectal Cancer Cells: An In Vitro Study. Iran. J. Med. Sci. 2023, 48, 321. [Google Scholar] [CrossRef] [PubMed]
- Chai, R.; Xu, C.; Lu, L.; Liu, X.; Ma, Z. Quercetin Inhibits Proliferation of and Induces Apoptosis in Non-Small-Cell Lung Carcinoma via the lncRNA SNHG7/miR-34a-5p Pathway. Immunopharmacol. Immunotoxicol. 2021, 43, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Hashemzaei, M.; Far, A.D.; Yari, A.; Heravi, R.E.; Tabrizian, K.; Taghdisi, S.M.; Sadegh, S.E.; Tsarouhas, K.; Kouretas, D.; Tzanakakis, G.; et al. Anticancer and Apoptosis-Inducing Effects of Quercetin In Vitro and In Vivo. Oncol. Rep. 2017, 38, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Farias, M.; Carrasco-Pozo, C. The Anti-Cancer Effect of Quercetin: Molecular Implications in Cancer Metabolism. Int. J. Mol. Sci. 2019, 20, 3177. [Google Scholar] [CrossRef] [PubMed]
- Azizi, E.; Fouladdel, S.; Komeili Movahhed, T.; Modaresi, F.; Barzegar, E.; Ghahremani, M.H.; Ostad, S.N.; Atashpour, S. Quercetin Effects on Cell Cycle Arrest and Apoptosis and Doxorubicin Activity in T47D Cancer Stem Cells. Asian Pac. J. Cancer Prev. 2022, 23, 4145–4154. [Google Scholar] [CrossRef] [PubMed]
- Saraei, R.; Rahman, H.S.; Soleimani, M.; Asghari-Jafarabadi, M.; Naimi, A.; Hassanzadeh, A.; Solali, S. Kaempferol Sensitizes Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Resistance Chronic Myelogenous Leukemia Cells to Apoptosis. Mol. Biol. Rep. 2022, 49, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Da, J.; Xu, M.; Wang, Y.; Li, W.; Lu, M.; Wang, Z. Kaempferol Promotes Apoptosis While Inhibiting Cell Proliferation via Androgen-Dependent Pathway and Suppressing Vasculogenic Mimicry and Invasion in Prostate Cancer. Anal. Cell. Pathol. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dang, Q.; Song, W.; Xu, D.; Ma, Y.; Li, F.; Zeng, J.; Zhu, G.; Wang, X.; Chang, L.S.; He, D.; et al. Kaempferol Suppresses Bladder Cancer Tumor Growth by Inhibiting Cell Proliferation and Inducing Apoptosis: Kaepmferol can Inhibit Bladder Cancer Growth and Metastasis. Mol. Carcinog. 2015, 54, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Feng, Z.; Chen, A.; Qi, Q.; Han, M.; Wang, S.; Zhang, Y.; Zhang, X.; Yang, N.; Wang, J.; et al. The Natural Flavonoid Galangin Elicits Apoptosis, Pyroptosis, and Autophagy in Glioblastoma. Front. Oncol. 2019, 9, 942. [Google Scholar] [CrossRef]
- Zhong, X.; Huang, S.; Liu, D.; Jiang, Z.; Jin, Q.; Li, C.; Da, L.; Yao, Q.; Wang, D. Galangin Promotes Cell Apoptosis through Suppression of H19 Expression in Hepatocellular Carcinoma Cells. Cancer Med. 2020, 9, 5546–5557. [Google Scholar] [CrossRef]
- Huang, H.; Chen, A.Y.; Ye, X.; Guan, R.; Rankin, G.O.; Chen, Y.C. Galangin, a Flavonoid from Lesser Galangal, Induced Apoptosis via P53-Dependent Pathway in Ovarian Cancer Cells. Molecules 2020, 25, 1579. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Gong, L.; Li, N.; Pan, Y.; Zhang, L. Galangin (GG) Combined with Cisplatin (DDP) to Suppress Human Lung Cancer by Inhibition of STAT3-Regulated NF-κB and Bcl-2/Bax Signaling Pathways. Biomed. Pharmacother. 2018, 97, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Han, M.A.; Lee, D.H.; Woo, S.M.; Seo, B.R.; Min, K.; Kim, S.; Park, J.-W.; Kim, S.H.; Choi, Y.H.; Kwon, T.K. Galangin Sensitizes TRAIL-Induced Apoptosis through down-Regulation of Anti-Apoptotic Proteins in Renal Carcinoma Caki Cells. Sci. Rep. 2016, 6, 18642. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Khan, M.A.; Najmi, A.K.; Chaturvedi, S.; Akhtar, M. Myricetin-Induced Apoptosis in Triple-Negative Breast Cancer Cells through Inhibition of the PI3K/Akt/mTOR Pathway. Med. Oncol. 2022, 39, 248. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Ma, R.-H.; Zhang, X.-X.; Thakur, K.; Zhang, J.-G.; Khan, M.R.; Busquets, R.; Wei, Z.-J. Isorhamnetin Induces Apoptosis and Suppresses Metastasis of Human Endometrial Carcinoma Ishikawa Cells via Endoplasmic Reticulum Stress Promotion and Matrix Metalloproteinase-2/9 Inhibition In Vitro and In Vivo. Foods 2022, 11, 3415. [Google Scholar] [CrossRef] [PubMed]
- Zhai, T.; Zhang, X.; Hei, Z.; Jin, L.; Han, C.; Ko, A.T.; Yu, X.; Wang, J. Isorhamnetin Inhibits Human Gallbladder Cancer Cell Proliferation and Metastasis via PI3K/AKT Signaling Pathway Inactivation. Front. Pharmacol. 2021, 12, 628621. [Google Scholar] [CrossRef] [PubMed]
- Su, C.H.; Kuo, C.L.; Lu, K.W.; Yu, F.S.; Ma, Y.S.; Yang, J.L.; Chu, Y.L.; Chueh, F.S.; Liu, K.C.; Chung, J.G. Fisetin-Induced Apoptosis of Human Oral Cancer SCC-4 Cells through Reactive Oxygen Species Production, Endoplasmic Reticulum Stress, Caspase-, and Mitochondria-Dependent Signaling Pathways. Environ. Toxicol. 2017, 32, 1725–1741. [Google Scholar] [CrossRef]
- Afroze, N.; Pramodh, S.; Shafarin, J.; Bajbouj, K.; Hamad, M.; Sundaram, M.K.; Haque, S.; Hussain, A. Fisetin Deters Cell Proliferation, Induces Apoptosis, Alleviates Oxidative Stress and Inflammation in Human Cancer Cells, HeLa. Int. J. Mol. Sci. 2022, 23, 1707. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Manghwar, H.; Hu, W.; Liu, F. Degradation Mechanism of Autophagy-Related Proteins and Research Progress. Int. J. Mol. Sci. 2022, 23, 7301. [Google Scholar] [CrossRef]
- Liu, S.; Yao, S.; Yang, H.; Liu, S.; Wang, Y. Autophagy: Regulator of Cell Death. Cell Death Dis. 2023, 14, 648. [Google Scholar] [CrossRef]
- Schuck, S. Microautophagy–Distinct Molecular Mechanisms Handle Cargoes of Many Sizes. J. Cell Sci. 2020, 133, jcs246322. [Google Scholar] [CrossRef] [PubMed]
- Rakesh, R.; PriyaDharshini, L.C.; Sakthivel, K.M.; Rasmi, R.R. Role and Regulation of Autophagy in Cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166400. [Google Scholar] [CrossRef] [PubMed]
- Sorice, M. Crosstalk of Autophagy and Apoptosis. Cells 2022, 11, 1479. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and Autophagy-Related Pathways in Cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.; Erustes, A.G.; Costa, A.J.; Nascimento, A.C.; Bincoletto, C.; Ureshino, R.P.; Pereira, G.J.S.; Smaili, S.S. Autophagy and Intermittent Fasting: The Connection for Cancer Therapy? Clinics 2018, 73, e814s. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Huang, S.; Yin, X.; Zan, Y.; Guo, Y.; Han, L. Quercetin Suppresses the Mobility of Breast Cancer by Suppressing Glycolysis through Akt-mTOR Pathway Mediated Autophagy Induction. Life Sci. 2018, 208, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Li, L.; Ma, Y.-X.; Li, W.-T.; Li, L.; Zhu, H.-Z.; Wu, M.-H.; Zhou, J.-R. Quercetin Inhibits Growth of Hepatocellular Carcinoma by Apoptosis Induction in Part via Autophagy Stimulation in Mice. J. Nutr. Biochem. 2019, 69, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhang, B.; Yin, S.; Xie, S.; Huang, K.; Wang, J.; Yang, W.; Liu, H.; Zhang, G.; Liu, X.; et al. Quercetin induces autophagy-associated death in HL-60 cells through CaMKKβ/AMPK/mTOR signal pathway. ABBS 2022, 54, 1244–1256. [Google Scholar] [CrossRef] [PubMed]
- Lou, M.; Zhang, L.; Ji, P.; Feng, F.; Liu, J.; Yang, C.; Li, B.; Wang, L. Quercetin Nanoparticles Induced Autophagy and Apoptosis through AKT/ERK/Caspase-3 Signaling Pathway in Human Neuroglioma Cells: In Vitro and In Vivo. Biomed. Pharmacother. 2016, 84, 1–9. [Google Scholar] [CrossRef]
- Guo, H.; Ding, H.; Tang, X.; Liang, M.; Li, S.; Zhang, J.; Cao, J. Quercetin Induces Pro-apoptotic Autophagy via SIRT1/AMPK Signaling Pathway in Human Lung Cancer Cell Lines A549 and H1299 in Vitro. Thorac. Cancer 2021, 12, 1415–1422. [Google Scholar] [CrossRef]
- Wang, R.; Deng, Z.; Zhu, Z.; Wang, J.; Yang, X.; Xu, M.; Wang, X.; Tang, Q.; Zhou, Q.; Wan, X.; et al. Kaempferol Promotes Non-Small Cell Lung Cancer Cell Autophagy via Restricting Met Pathway. Phytomedicine 2023, 121, 155090. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-W.; Tsai, S.-C.; Peng, S.-F.; Lin, M.-W.; Chiang, J.-H.; Chiu, Y.-J.; Fushiya, S.; Tseng, M.T.; Yang, J.-S. Kaempferol Induces Autophagy through AMPK and AKT Signaling Molecules and Causes G2/M Arrest via Downregulation of CDK1/Cyclin B in SK-HEP-1 Human Hepatic Cancer Cells. Int. J. Oncol. 2013, 42, 2069–2077. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.-J.; Jung, G.-H.; Choi, E.-Y.; Han, E.-J.; Lee, J.-H.; Han, S.-H.; Woo, J.-S.; Jung, S.-H.; Jung, J.-Y. Kaempferol Induces Apoptosis through the MAPK Pathway and Regulates JNK-Mediated Autophagy in MC-3 Cells. Toxicol. Res. 2024, 40, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Lee, S.Y.; Kim, M.; Cheon, C.; Ko, S.-G. Kaempferol Induces Autophagic Cell Death via IRE1-JNK-CHOP Pathway and Inhibition of G9a in Gastric Cancer Cells. Cell Death Dis. 2018, 9, 875. [Google Scholar] [CrossRef] [PubMed]
- El-Kott, A.F.; Shati, A.A.; Al-Kahtani, M.A.; Alharbi, S.A. Kaempferol Induces Cell Death in A2780 Ovarian Cancer Cells and Increases Their Sensitivity to Cisplatin by Activation of Cytotoxic Endoplasmic Reticulum-Mediated Autophagy and Inhibition of Protein Kinase B. Folia Biol. 2020, 66, 36–46. [Google Scholar] [CrossRef]
- Zhou, Q.; Fang, G.; Pang, Y.; Wang, X. Combination of Kaempferol and Docetaxel Induces Autophagy in Prostate Cancer Cells In Vitro and In Vivo. Int. J. Mol. Sci. 2023, 24, 14519. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-X.; Tang, C. Galangin Suppresses Human Laryngeal Carcinoma via Modulation of Caspase-3 and AKT Signaling Pathways. Oncol. Rep. 2017, 38, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Xiong, Y.; Wu, J.; Ding, H.; Chen, X.; Lan, L.; Zhang, H. Galangin Induces Autophagy via Deacetylation of LC3 by SIRT1 in HepG2 Cells. Sci. Rep. 2016, 6, 30496. [Google Scholar] [CrossRef]
- Wen, M.; Wu, J.; Luo, H.; Zhang, H. Galangin Induces Autophagy through Upregulation of P53 in HepG2 Cells. Pharmacology 2012, 89, 247–255. [Google Scholar] [CrossRef]
- Cao, J.; Chen, H.; Lu, W.; Wu, Y.; Wu, X.; Xia, D.; Zhu, J. Myricetin Induces Protective Autophagy by Inhibiting the Phosphorylation of mTOR in HepG2 Cells. Anat. Rec. 2018, 301, 786–795. [Google Scholar] [CrossRef]
- Ji, A.; Hu, L.; Ma, D.; Qiang, G.; Yan, D.; Zhang, G.; Jiang, C. Myricetin Induces Apoptosis and Protective Autophagy through Endoplasmic Reticulum Stress in Hepatocellular Carcinoma. Evid. Based Complement. Altern. Med. 2022, 2022, 3115312. [Google Scholar] [CrossRef]
- Yang, W.; Su, J.; Li, M.; Li, T.; Wang, X.; Zhao, M.; Hu, X. Myricetin Induces Autophagy and Cell Cycle Arrest of HCC by Inhibiting MARCH1-Regulated Stat3 and P38 MAPK Signaling Pathways. Front. Pharmacol. 2021, 12, 709526. [Google Scholar] [CrossRef]
- Suh, Y.; Afaq, F.; Khan, N.; Johnson, J.J.; Khusro, F.H.; Mukhtar, H. Fisetin Induces Autophagic Cell Death through Suppression of mTOR Signaling Pathway in Prostate Cancer Cells. Carcinogenesis 2010, 31, 1424–1433. [Google Scholar] [CrossRef]
- Park, B.-S.; Choi, N.-E.; Lee, J.H.; Kang, H.-M.; Yu, S.-B.; Kim, H.-J.; Kang, H.-K.; Kim, I.-R. Crosstalk between Fisetin-Induced Apoptosis and Autophagy in Human Oral Squamous Cell Carcinoma. J. Cancer 2019, 10, 138–146. [Google Scholar] [CrossRef]
- Jia, S.; Xu, X.; Zhou, S.; Chen, Y.; Ding, G.; Cao, L. Fisetin Induces Autophagy in Pancreatic Cancer Cells via Endoplasmic Reticulum Stress- and Mitochondrial Stress-Dependent Pathways. Cell Death Dis. 2019, 10, 142. [Google Scholar] [CrossRef]
- Granato, M.; Rizzello, C.; Gilardini Montani, M.S.; Cuomo, L.; Vitillo, M.; Santarelli, R.; Gonnella, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Quercetin Induces Apoptosis and Autophagy in Primary Effusion Lymphoma Cells by Inhibiting PI3K/AKT/mTOR and STAT3 Signaling Pathways. J. Nutr. Biochem. 2017, 41, 124–136. [Google Scholar] [CrossRef]
- Wu, R.; Zhou, T.; Xiong, J.; Zhang, Z.; Tian, S.; Wang, Y.; Chen, J.; Tian, X. Quercetin, the Ingredient of Xihuang Pills, Inhibits Hepatocellular Carcinoma by Regulating Autophagy and Macrophage Polarization. Front. Biosci. 2022, 27, 323. [Google Scholar] [CrossRef]
- Moon, J.-H.; Eo, S.K.; Lee, J.H.; Park, S.-Y. Quercetin-Induced Autophagy Flux Enhances TRAIL-Mediated Tumor Cell Death. Oncol. Rep. 2015, 34, 375–381. [Google Scholar] [CrossRef]
- Yuan, Y.; Xia, F.; Gao, R.; Chen, Y.; Zhang, Y.; Cheng, Z.; Zhao, H.; Xu, L. Kaempferol Mediated AMPK/mTOR Signal Pathway Has a Protective Effect on Cerebral Ischemic-Reperfusion Injury in Rats by Inducing Autophagy. Neurochem. Res. 2022, 47, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Pal Singh, M.; Pal Khaket, T.; Bajpai, V.K.; Alfarraj, S.; Kim, S.-G.; Chen, L.; Huh, Y.S.; Han, Y.-K.; Kang, S.C. Morin Hydrate Sensitizes Hepatoma Cells and Xenograft Tumor towards Cisplatin by Downregulating PARP-1-HMGB1 Mediated Autophagy. Int. J. Mol. Sci. 2020, 21, 8253. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Tian, S.; Pan, Y.; Li, W.; Wang, Q.; Tang, Y.; Yu, T.; Wu, X.; Shi, Y.; Ma, P.; et al. Pyroptosis: A New Frontier in Cancer. Biomed. Pharmacother. 2020, 121, 109595. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and Diseases. Sig. Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Du, T.; Gao, J.; Li, P.; Wang, Y.; Qi, Q.; Liu, X.; Li, J.; Wang, C.; Du, L. Pyroptosis, Metabolism, and Tumor Immune Microenvironment. Clin. Transl. Med. 2021, 11, e492. [Google Scholar] [CrossRef]
- Rao, Z.; Zhu, Y.; Yang, P.; Chen, Z.; Xia, Y.; Qiao, C.; Liu, W.; Deng, H.; Li, J.; Ning, P.; et al. Pyroptosis in Inflammatory Diseases and Cancer. Theranostics 2022, 12, 4310–4329. [Google Scholar] [CrossRef]
- Chen, S.; Ma, J.; Yang, L.; Teng, M.; Lai, Z.-Q.; Chen, X.; He, J. Anti-Glioblastoma Activity of Kaempferol via Programmed Cell Death Induction: Involvement of Autophagy and Pyroptosis. Front. Bioeng. Biotechnol. 2020, 8, 614419. [Google Scholar] [CrossRef]
- Han, J.; Cheng, C.; Zhang, J.; Fang, J.; Yao, W.; Zhu, Y.; Xiu, Z.; Jin, N.; Lu, H.; Li, X.; et al. Myricetin Activates the Caspase-3/GSDME Pathway via ER Stress Induction of Pyroptosis in Lung Cancer Cells. Front. Pharmacol. 2022, 13, 959938. [Google Scholar] [CrossRef]
- Li, J.; Yu, J.; Zhang, T.; Pu, X.; Li, Y.; Wu, Z. Genomic Analysis Quantifies Pyroptosis in the Immune Microenvironment of HBV-Related Hepatocellular Carcinoma. Front. Immunol. 2022, 13, 932303. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Deng, L.; He, S.; Guo, N.; Tian, W.; Zhang, W.; Luo, L. Molecular Mechanisms of Ferroptosis and Relevance to Inflammation. Inflamm. Res. 2023, 72, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Wang, X.; Zhou, Y.; Wang, X.; Yu, Y. Autophagy, Ferroptosis, Pyroptosis, and Necroptosis in Tumor Immunotherapy. Sig. Transduct. Target. Ther. 2022, 7, 196. [Google Scholar] [CrossRef]
- Zhao, L.; Zhou, X.; Xie, F.; Zhang, L.; Yan, H.; Huang, J.; Zhang, C.; Zhou, F.; Chen, J.; Zhang, L. Ferroptosis in Cancer and Cancer Immunotherapy. Cancer Commun. 2022, 42, 88–116. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a New Form of Cell Death: Opportunities and Challenges in Cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kang, R.; Tang, D. Signaling Pathways and Defense Mechanisms of Ferroptosis. FEBS J. 2022, 289, 7038–7050. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Dang, S.; Sun, M.; Zhou, D.; Sun, Y.; Li, E.; Peng, S.; Li, J.; Li, G. Quercetin Induces Ferroptosis in Gastric Cancer Cells by Targeting SLC1A5 and Regulating the P-Camk2/p-DRP1 and NRF2/GPX4 Axes. Free Radic. Biol. Med. 2024, 213, 150–163. [Google Scholar] [CrossRef]
- Wang, Z.X.; Ma, J.; Li, X.Y.; Wu, Y.; Shi, H.; Chen, Y.; Lu, G.; Shen, H.M.; Lu, G.D.; Zhou, J. Quercetin Induces P53-Independent Cancer Cell Death through Lysosome Activation by the Transcription Factor EB and Reactive Oxygen Species-Dependent Ferroptosis. Br. J. Pharmacol. 2021, 178, 1133–1148. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, Q.; Ma, M.; Guo, H. Quercetin Inhibits the Progression of Endometrial HEC-1-A Cells by Regulating Ferroptosis—A Preliminary Study. Eur. J. Med. Res. 2022, 27, 292. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Hu, M. Quercetin Promotes TFEB Nuclear Translocation and Activates Lysosomal Degradation of Ferritin to Induce Ferroptosis in Breast Cancer Cells. Comput. Intell. Neurosci. 2022, 2022, 5299218. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Song, Y.-R.; Kim, Y.E.; Bae, S.-J.; Lee, W.-Y.; Bak, S.-B.; Kim, Y.W. Kaempferol Stimulation of Autophagy Regulates the Ferroptosis under the Oxidative Stress as Mediated with AMP-Activated Protein Kinase. Free Radic. Biol. Med. 2023, 208, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Xue, R.; Geng, Y.; Zhang, S. Galangin Inhibited Ferroptosis through Activation of the PI3K/AKT Pathway In Vitro and In Vivo. FASEB J. 2022, 36, e22569. [Google Scholar] [CrossRef]
- Yan, J.; Wan, P.; Choksi, S.; Liu, Z.-G. Necroptosis and Tumor Progression. Trends Cancer 2022, 8, 21–27. [Google Scholar] [CrossRef]
- Khoury, M.K.; Gupta, K.; Franco, S.R.; Liu, B. Necroptosis in the Pathophysiology of Disease. Am. J. Pathol. 2020, 190, 272–285. [Google Scholar] [CrossRef]
- Seo, J.; Nam, Y.W.; Kim, S.; Oh, D.-B.; Song, J. Necroptosis Molecular Mechanisms: Recent Findings Regarding Novel Necroptosis Regulators. Exp. Mol. Med. 2021, 53, 1007–1017. [Google Scholar] [CrossRef]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The Role of Necroptosis in Cancer Biology and Therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef]
- Lomphithak, T.; Jaikla, P.; Sae-Fung, A.; Sonkaew, S.; Jitkaew, S. Natural Flavonoids Quercetin and Kaempferol Targeting G2/M Cell Cycle-Related Genes and Synergize with Smac Mimetic LCL-161 to Induce Necroptosis in Cholangiocarcinoma Cells. Nutrients 2023, 15, 3090. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, H.; Zhao, Y.; Shan, L.; Lan, S. Fisetin-Induced Cell Death in Human Ovarian Cancer Cell Lines via Zbp1-Mediated Necroptosis. J. Ovarian Res. 2022, 15, 57. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Y.; Zhang, J.; Yang, Y.; Fleishman, J.S.; Wang, Y.; Wang, J.; Chen, J.; Li, Y.; Wang, H. Cuproptosis: A Novel Therapeutic Target for Overcoming Cancer Drug Resistance. Drug Resist. Updates 2024, 72, 101018. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Yang, Y.; Gao, Y.; He, J. Cuproptosis: Mechanisms and Links with Cancers. Mol. Cancer 2023, 22, 46. [Google Scholar] [CrossRef]
- Wang, J.; Luo, L.-Z.; Liang, D.-M.; Guo, C.; Huang, Z.-H.; Sun, G.-Y.; Wen, J. Progress in the Research of Cuproptosis and Possible Targets for Cancer Therapy. World J. Clin. Oncol. 2023, 14, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, J.; Pakrashi, S.; Sarbajna, A.; Dutta, M.; Bandyopadhyay, J. Quercetin Attenuates Copper-Induced Apoptotic Cell Death and Endoplasmic Reticulum Stress in SH-SY5Y Cells by Autophagic Modulation. Biol. Trace Elem. Res. 2022, 200, 5022–5041. [Google Scholar] [CrossRef]
- Sadžak, A.; Vlašić, I.; Kiralj, Z.; Batarelo, M.; Oršolić, N.; Jazvinšćak Jembrek, M.; Kušen, I.; Šegota, S. Neurotoxic Effect of Flavonol Myricetin in the Presence of Excess Copper. Molecules 2021, 26, 845. [Google Scholar] [CrossRef]
- Huang, J.; Chen, J.; Li, J. Quercetin Promotes ATG5-Mediating Autophagy-Dependent Ferroptosis in Gastric Cancer. J. Mol. Histol. 2024, 55, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, J.; Yu, C.; Xiang, L.; Li, L.; Shi, D.; Lin, F. Quercetin Enhanced Paclitaxel Therapeutic Effects Towards PC-3 Prostate Cancer through ER Stress Induction and ROS Production. OncoTargets Ther. 2020, 13, 513–523. [Google Scholar] [CrossRef]
- Wu, L.; Li, J.; Liu, T.; Li, S.; Feng, J.; Yu, Q.; Zhang, J.; Chen, J.; Zhou, Y.; Ji, J.; et al. Quercetin Shows Anti-tumor Effect in Hepatocellular Carcinoma LM3 Cells by Abrogating JAK2/STAT3 Signaling Pathway. Cancer Med. 2019, 8, 4806–4820. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Pan, L.; Gao, C.; Xu, H.; Li, Y.; Zhang, L.; Ma, L.; Meng, L.; Sun, X.; Qin, H. Quercetin Inhibits the Proliferation of Glycolysis-Addicted HCC Cells by Reducing Hexokinase 2 and Akt-mTOR Pathway. Molecules 2019, 24, 1993. [Google Scholar] [CrossRef]
- Fernández-Palanca, P.; Fondevila, F.; Méndez-Blanco, C.; Tuñón, M.J.; González-Gallego, J.; Mauriz, J.L. Antitumor Effects of Quercetin in Hepatocarcinoma In Vitro and In Vivo Models: A Systematic Review. Nutrients 2019, 11, 2875. [Google Scholar] [CrossRef]
- Ibrahim, F.A.R.; Hussein, N.A.; Soliman, A.Y.M.; Shalaby, T.I.; Rashad, M.M.; Matar, N.A.; El-Sewedy, T.S. Chitosan-Loaded Piperlongumine Nanoparticles and Kaempferol Enhance the Anti-Cancer Action of Doxorubicin in Targeting of Ehrlich Solid Adenocarcinoma: In Vivo and In Silico Modeling Study. Med. Oncol. 2024, 41, 61. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Ma, X.; Yan, X.; Zhang, G.; Hong, S.; Ma, R.; Wang, Y.; Ma, M. Kaempferol Induces Mitochondrial Dysfunction and Mitophagy by Activating the LKB1/AMPK/MFF Pathway in Breast Precancerous Lesions. Phytother. Res. 2023, 37, 3602–3616. [Google Scholar] [CrossRef]
- Kim, S.-H.; Hwang, K.-A.; Choi, K.-C. Treatment with Kaempferol Suppresses Breast Cancer Cell Growth Caused by Estrogen and Triclosan in Cellular and Xenograft Breast Cancer Models. J. Nutr. Biochem. 2016, 28, 70–82. [Google Scholar] [CrossRef]
- Yang, H.-W.; Lan, Y.; Li, A.; Wu, H.; Song, Z.-W.; Wan, A.-L.; Wang, Y.; Li, S.-B.; Ji, S.; Wang, Z.-C.; et al. Myricetin Suppresses TGF-β-Induced Epithelial-to-Mesenchymal Transition in Ovarian Cancer. Front. Pharmacol. 2023, 14, 1288883. [Google Scholar] [CrossRef]
- Mei, C.; Zhang, X.; Zhi, Y.; Liang, Z.; Xu, H.; Liu, Z.; Liu, Y.; Lyu, Y.; Wang, H. Isorhamnetin Regulates Programmed Death Ligand-1 Expression by Suppressing the EGFR–STAT3 Signaling Pathway in Canine Mammary Tumors. Int. J. Mol. Sci. 2024, 25, 670. [Google Scholar] [CrossRef]
- Luan, Y.; Luan, Y.; Zhao, Y.; Xiong, F.; Li, Y.; Liu, L.; Cao, Y.; Dai, F. Isorhamnetin in Tsoong Blocks Hsp70 Expression to Promote Apoptosis of Colon Cancer Cells. Saudi J. Biol. Sci. 2019, 26, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Manu, K.A.; Shanmugam, M.K.; Ramachandran, L.; Li, F.; Siveen, K.S.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Arfuso, F.; Kumar, A.P.; et al. Isorhamnetin Augments the Anti-Tumor Effect of Capeciatbine through the Negative Regulation of NF-κB Signaling Cascade in Gastric Cancer. Cancer Lett. 2015, 363, 28–36. [Google Scholar] [CrossRef]
- Li, Q.; Ren, F.-Q.; Yang, C.-L.; Zhou, L.-M.; Liu, Y.-Y.; Xiao, J.; Zhu, L.; Wang, Z.-G. Anti-Proliferation Effects of Isorhamnetin on Lung Cancer Cells In Vitro and In Vivo. Asian Pac. J. Cancer Prev. 2015, 16, 3035–3042. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Gou, H.; Lin, T.; Yang, Y.; Jin, X.; Dong, T.; Zhang, Y.; Chen, X. Fisetin Nanoparticles Based on Cells Cycle and Apoptosis Intervention for the Treatment of Lymphoma and Leukemia. Int. J. Pharm. 2024, 654, 123971. [Google Scholar] [CrossRef] [PubMed]
- Talaat, S.M.; Elnaggar, Y.S.R.; Gowayed, M.A.; El-Ganainy, S.O.; Allam, M.; Abdallah, O.Y. Novel PEGylated Cholephytosomes for Targeting Fisetin to Breast Cancer: In Vitro Appraisal and In Vivo Antitumoral Studies. Drug Deliv. Transl. Res. 2024, 14, 433–454. [Google Scholar] [CrossRef]
- Aboushanab, A.R.; El-Moslemany, R.M.; El-Kamel, A.H.; Mehanna, R.A.; Bakr, B.A.; Ashour, A.A. Targeted Fisetin-Encapsulated β-Cyclodextrin Nanosponges for Breast Cancer. Pharmaceutics 2023, 15, 1480. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ding, Y.; Jin, J.; Xu, C.; Hu, W.; Wu, S.; Ding, G.; Cheng, R.; Cao, L.; Jia, S. Post-Translational Modification of CDK1–STAT3 Signaling by Fisetin Suppresses Pancreatic Cancer Stem Cell Properties. Cell Biosci. 2023, 13, 176. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, M.A.; El-Refaie, W.M.; Elnaggar, Y.S.R.; El-Mezayen, N.S.; Awaad, A.K.; Abdallah, O.Y. Fucoidan/Hyaluronic Acid Cross-Linked Zein Nanoparticles Loaded with Fisetin as a Novel Targeted Nanotherapy for Oral Cancer. Int. J. Biol. Macromol. 2023, 241, 124528. [Google Scholar] [CrossRef]
- Ravichandran, N.; Suresh, G.; Ramesh, B.; Manikandan, R.; Choi, Y.W.; Vijaiyan Siva, G. Fisetin Modulates Mitochondrial Enzymes and Apoptotic Signals in Benzo(a)Pyrene-Induced Lung Cancer. Mol. Cell Biochem. 2014, 390, 225–234. [Google Scholar] [CrossRef]
- Li, J.; Qu, W.; Cheng, Y.; Sun, Y.; Jiang, Y.; Zou, T.; Wang, Z.; Xu, Y.; Zhao, H. The Inhibitory Effect of Intravesical Fisetin against Bladder Cancer by Induction of P53 and Down-Regulation of NF -kappa B Pathways in a Rat Bladder Carcinogenesis Model. Basic Clin. Pharmacol. Toxicol. 2014, 115, 321–329. [Google Scholar] [CrossRef]





| Flavonoid | Mechanism | Cancer Type | Cell Line | Ref. |
|---|---|---|---|---|
| Quercetin |
| Breast cancer | MCF-7 | [20] |
| Leukemia | HL-60 | [21] | ||
| Breast cancer | MCF-7 | [20] | |
| Leukemia | HL-60 | [21] | ||
| Gastric cancer | AGS | [22] | ||
| Gastric cancer | AGS | [22] | |
| Kaempferol |
| Cervical cancer | HeLa | [23] |
| Submandibular gland cancer | A253 | [24] | ||
| Ovarian cancer | OVACAR-3 | [25,26] | ||
| Liver cancer | HepG2 | [27] | ||
| Pancreatic cancer | PaCa-2, PANC-1 | [28] | ||
| Bladder cancer | 5637, T24 | [29] | ||
| Cervical cancer | HeLa | [23] | |
| Submandibular gland cancer | A253 | [24] | ||
| Ovarian cancer | OVACAR-3, A2780/CP70 | [25,26] | ||
| Colorectal cancer | HT-29 | [30] | ||
| Pancreatic cancer | PaCa-2, PANC-1 | [28] | ||
| Breast cancer | MDA-MB-231 | [31] | ||
| Cervical cancer | HeLa | [23] | |
| Cervical cancer | HeLa | [23] | |
| Colorectal cancer | HCT15, HCT116 | [32] | ||
| Bladder cancer | 5637, T24 | [29] | ||
| Cervical cancer | HeLa | [23] | |
| Bladder cancer | 5637, T24 | [29] | ||
| Oral cancer | SCC-9 | [24] | |
| Submandibular gland cancer | A-253 | [24] | ||
| Ovarian cancer | OVACAR-3 | [25] | ||
| Ovarian cancer | OVACAR-3 | [25] | |
| Ovarian cancer | OVACAR-3 | [25] | |
| Colorectal cancer | HT-29 | [30] | |
| Ovarian cancer | A2780/CP70 | [26] | ||
| Pancreatic cancer | PaCa-2, PANC-1 | [28] | |
| Colorectal cancer | HCT15, HCT116 | [32] | ||
| Non-small-cell lung cancer | NSCLC | [33] | ||
| Galangin |
| Breast cancer | MCF-7, T47D | [34,35] |
| Kidney cancer | A498 | [36] | ||
| Nasopharyngeal cancer | NPC-TW039, NPC-TW076 | [37] | ||
| Gastric cancer | MGC 803 | [38] | ||
| Breast cancer | MCF-7, T47D | [34,35] | |
| Retinoblastoma | Y-79, HXO-Rb44 | [39] | ||
| Breast cancer | MCF-7, T47D | [34,35] | |
| Retinoblastoma | Y-79, HXO-Rb44 | [39] | |
| Kidney cancer | A498 | [36] | ||
| Nasopharyngeal cancer | NPC-TW039, NPC-TW076 | [37] | ||
| Breast cancer | MCF-7 | [35] | ||
| Kidney cancer | A498 | [36] | |
| Gastric cancer | MGC 803 | [38] | |
| Gastric cancer | MGC 803 | [38] | |
| Kidney cancer | Caki1, 7860 | [40] | ||
| Myricetin |
| Breast cancer | SKBR3,T47-D | [41] |
| Gastric cancer | AGS | [42] | ||
| Thyroid cancer | SNU-790 H, SNU-80 HATC | [43,44] | ||
| Ovarian cancer | A2780/CP70 OVCAR-3 | [45] | ||
| Thyroid cancer | SNU-790 H, SNU-80 HATC | [43,44] | |
| Breast cancer | T47-D | [46] | ||
| Ovarian cancer | A2780/CP70 OVCAR-3 | [45] | ||
| Gastric cancer | AGS | [42] | |
| Colorectal cancer | HCT116, SW620 | [47] | ||
| Lung cance | A549 | [48] | |
| Isorhamnetin |
| Melanoma | B16F10 | [49] |
| Liver cancer | Hep3B | [50] | ||
| Gastric cancer | AGS-1, HGC-27 | [51] | ||
| Breast cancer | MDA-MB-231, MCF-7 | [52] | ||
| Melanoma | B16F10 | [49] | |
| Bladder cancer | T24, 5637 | [53] | ||
| Liver cancer | Hep3B | [50] | ||
| Gastric cancer | AGS-1, HGC-27, MKN-45 | [51,54] | ||
| Breast cancer | MDA-MB-231, MCF-7 | [52] | ||
| Non-small lung cancer | A549 | [55] | ||
| Melanoma | B16F10 | [49] | |
| Prostate cancer | DU145, PC3 | [56] | ||
| Gastric cancer | MKN-45 | [54] | ||
| Bladder cancer | T24, 5637 | [53] | |
| Breast cancer | MDA-MB-231, MCF-7 | [52] | ||
| Gastric cancer | MKN-45 | [54] | ||
| Fisetin |
| Oral cancer | HSC3 | [57] |
| Melanoma | M17, SP6.5 | [58] | ||
| Non-small lung cancer | NCI-H460 | [59] | ||
| Breast cancer | 4T1 | [60] | ||
| Gastric cancer | AGS, SNU-1, SGC790 | [61,62] | ||
| Oral cancer | HSC3 | [57] | |
| Melanoma | M17, SP6.5 | [58] | ||
| Non-small lung cancer | NCI-H460 | [59] | ||
| Pancreatic cancer | PANC-1 | [63] | ||
| Gastric cancer | AGS, SNU-1, SGC790 | [61,62] | ||
| Liver cancer | HepG2, Hep3B | [64] | ||
| Oral cancer | HSC3 | [57] | |
| Melanoma | M17, SP6.5 | [58] | ||
| Colorectal cancer | SW-480 | [65] | |
| Non-small lung cancer | NCI-H460 | [59] | ||
| Gastric cancer | AGS, SNU-1 | [61] | ||
| Pancreatic cancer | PANC-1 | [63] | |
| Breast cancer | 4T1, MDA-MB-453 | [60,66] | ||
| Morin |
| Myeloid leukemia | K562, KCL22 | [67] |
| Colorectal cancer | HCT-116. SW480 | [68,69] | ||
| Melanoma | G361, SK-MEL-2 | [70] | ||
| Myeloid leukemia | K562, KCL22 | [67] | |
| Colorectal cancer | HCT-116, SW480 | [68,69] | ||
| Melanoma | G361, SK-MEL-2 | [70] | ||
| Myeloid leukemia | K562, KCL22 | [67] | |
| Colorectal cancer | HCT-116 | [68] | |
| Melanoma | G361, SK-MEL-2 | [70] | |
| Colorectal cancer | SW480 | [69] | ||
| Colorectal cancer | HCT-116 | [68] |
| Flavonoid | Mechanism | Cancer Type | Cell Line | Ref. |
|---|---|---|---|---|
| Quercetin |
| Breast cancer | MCF-7, MDA-MB-231 | [97] |
| Liver cancer | SMMC7721, HepG2 | [98] | ||
| Acute myeloid leukemia | HL-60 | [99] | ||
| Liver cancer | SMMC7721, HepG2 | [98] | |
| Neuroglioma | U87 | [100] | ||
| Acute myeloid leukemia | HL-60 | [99] | ||
| Lung cancer | A549, H1299 | [101] | ||
| Neuroglioma | U87 | [100] | |
| Lung cancer | A549, H1299 | [101] | ||
| Lung cancer | A549, H1299 | [101] | |
| Kaempferol |
| Lung cancer | A549, H1299 | [102] |
| Liver cancer | SK-HEP-1 | [103] | ||
| Lung cancer | A549, H1299 | [102] | |
| Liver cancer | SK-HEP-1 | [103] | ||
| Oral cancer | MC-3 | [104] | ||
| Gastric cancer | SNU-638 | [105] | ||
| Ovarian cancer | A2780 | [106] | ||
| Prostate cancer | PC-3 | [107] | ||
| Lung cancer | A549, H1299 | [102] | |
| Liver cancer | SK-HEP-1 | [103] | ||
| Oral cancer | MC-3 | [104] | ||
| Gastric cancer | SNU-638 | [105] | ||
| Ovarian cancer | A2780 | [106] | ||
| Liver cancer | SK-HEP-1 | [103] | |
| Gastric cancer | SNU-638 | [105] | ||
| Ovarian cancer | A2780 | [106] | ||
| Galangin |
| Laryngeal cancer | TU212, HEP-2 | [108] |
| Laryngeal cancer | TU212, HEP-2 | [108] | |
| Liver cancer | HepG2 | [109,110] | ||
| Laryngeal cancer | TU212, HEP-2 | [108] | |
| Liver cancer | HepG2 | [109,110] | ||
| Myricetin |
| Gastric cancer | AGS | [42] |
| Colorectal cancer | HCT116, SW620 | [47] | ||
| Liver cancer | HepG2 | [111] | ||
| Gastric cancer | AGS | [42] | |
| Colorectal cancer | HCT116, SW620 | [47] | ||
| Liver cancer | HepG2, Hep3B | [111,112,113] | ||
| Gastric cancer | AGS | [42] | |
| Isorhamnetin |
| Gastric cancer | MKN-45 | [54] |
| Fisetin |
| Prostate cancer | PC3 | [114] |
| Prostate cancer | PC3 | [114] | |
| Oral cancer | Ca9-22 | [115] | ||
| Pancreatic cancer | PANC-1, BxPC-3 | [116] | ||
| Oral cancer | Ca9-22 | [115] | |
| Oral cancer | Ca9-22 | [115] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wendlocha, D.; Kubina, R.; Krzykawski, K.; Mielczarek-Palacz, A. Selected Flavonols Targeting Cell Death Pathways in Cancer Therapy: The Latest Achievements in Research on Apoptosis, Autophagy, Necroptosis, Pyroptosis, Ferroptosis, and Cuproptosis. Nutrients 2024, 16, 1201. https://doi.org/10.3390/nu16081201
Wendlocha D, Kubina R, Krzykawski K, Mielczarek-Palacz A. Selected Flavonols Targeting Cell Death Pathways in Cancer Therapy: The Latest Achievements in Research on Apoptosis, Autophagy, Necroptosis, Pyroptosis, Ferroptosis, and Cuproptosis. Nutrients. 2024; 16(8):1201. https://doi.org/10.3390/nu16081201
Chicago/Turabian StyleWendlocha, Dominika, Robert Kubina, Kamil Krzykawski, and Aleksandra Mielczarek-Palacz. 2024. "Selected Flavonols Targeting Cell Death Pathways in Cancer Therapy: The Latest Achievements in Research on Apoptosis, Autophagy, Necroptosis, Pyroptosis, Ferroptosis, and Cuproptosis" Nutrients 16, no. 8: 1201. https://doi.org/10.3390/nu16081201