
Prenatal Choline Supplementation Improves Glucose Tolerance and Reduces Liver Fat Accumulation in Mouse Offspring Exposed to Ethanol during the Prenatal and Postnatal Periods

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Diets
2.2. RNA Extraction and Quantitative Real-Time PCR
2.3. Liver Triglyceride (TG) and Malondialdehyde (MDA) Measurements
2.4. Serum Measurements
2.5. Choline Measurements
2.6. Histology
2.7. Global DNA Methylation
2.8. Statistical Analysis
3. Results
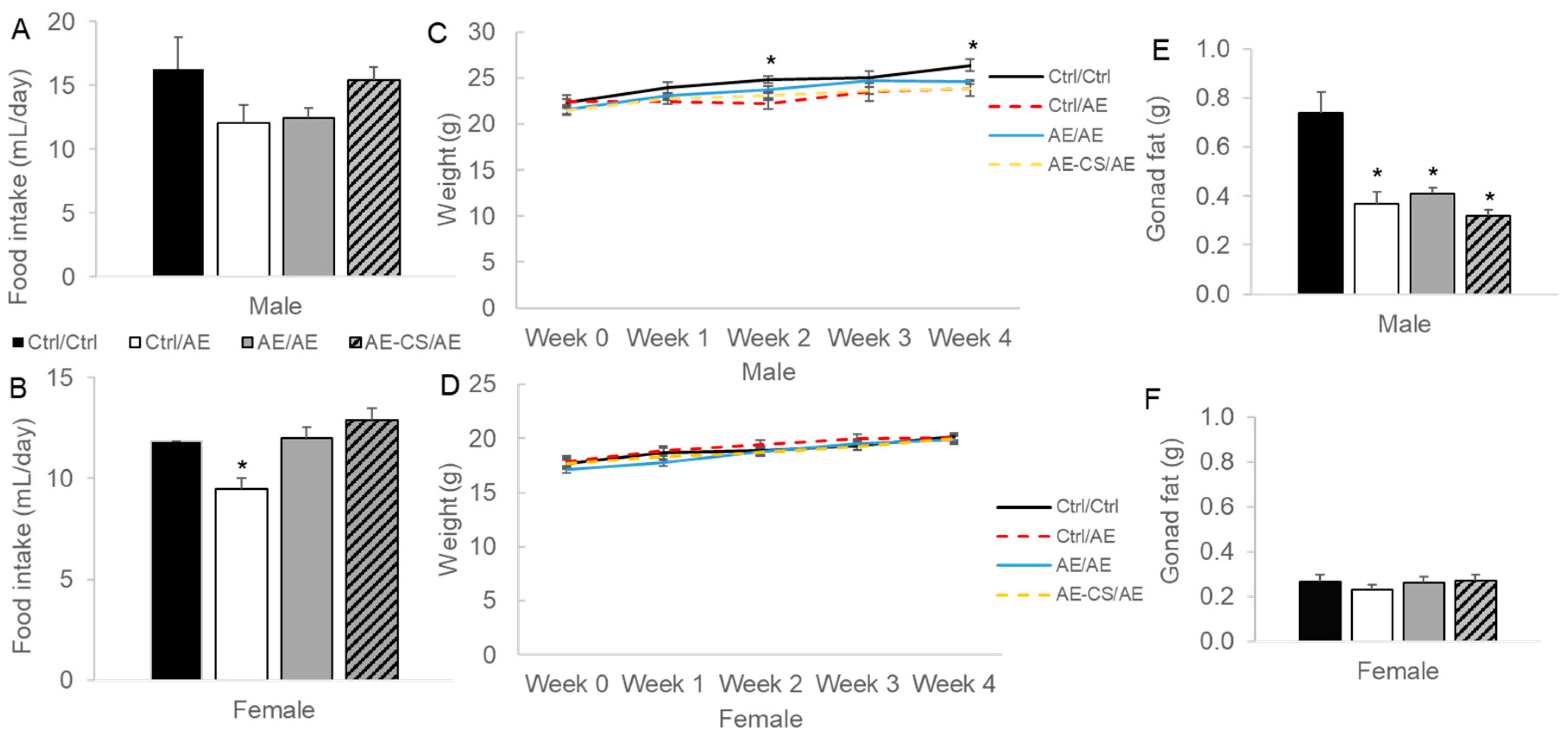
3.1. Weight Gain and Adiposity Were Lower in Male Offspring Exposed to Ethanol after Weaning
3.2. Glucose Tolerance Was Improved in Prenatal Choline-Supplemented AE Male Offspring
3.3. Liver TG Accumulation Was Exacerbated by AE and Prevented by Prenatal CS
3.4. Liver Lipid Metabolic Gene Expression
3.5. TG, Free FFA, and ApoB Levels in Serum
3.6. Hepatic Global DNA Methylation Was Not Altered by AE or CS
3.7. Choline Metabolites and Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bhuvaneswar, C.G.; Chang, G.; Epstein, L.A.; Stern, T.A. Alcohol use during pregnancy: Prevalence and impact. Prim. Care Companion J. Clin. Psychiatry 2007, 9, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Iversen, M.L.; Sørensen, N.O.; Broberg, L.; Damm, P.; Hedegaard, M.; Tabor, A.; Hegaard, H.K. Alcohol consumption and binge drinking in early pregnancy. A cross-sectional study with data from the Copenhagen Pregnancy Cohort. BMC Pregnancy Childbirth 2015, 15, 327. [Google Scholar] [CrossRef] [PubMed]
- Kesmodel, U.S.; Petersen, G.L.; Henriksen, T.B.; Strandberg-Larsen, K. Time trends in alcohol intake in early pregnancy and official recommendations in Denmark, 1998–2013. Acta Obstet Gynecol. Scand 2016, 95, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Kesmodel, U. Binge drinking in pregnancy--frequency and methodology. Am. J. Epidemiol. 2001, 154, 777–782. [Google Scholar] [CrossRef]
- Kodituwakku, P.W. Neurocognitive profile in children with fetal alcohol spectrum disorders. Dev. Disabil. Res. Rev. 2009, 15, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.C.; Jacobson, J.L.; Molteno, C.D.; Dodge, N.C.; Meintjes, E.M.; Jacobson, S.W. Fetal Alcohol Growth Restriction and Cognitive Impairment. Pediatrics 2016, 138, e20160775. [Google Scholar] [CrossRef] [PubMed]
- Feng, A.; Wang, L.; Chen, X.; Liu, X.; Li, L.; Wang, B.; Luo, H.; Mo, X.; Tobe, R.G. Developmental Origins of Health and Disease (DOHaD): Implications for health and nutritional issues among rural children in China. Biosci. Trends 2015, 9, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.C.; Mongillo, D.L.; Brien, D.C.; Stepita, R.; Poklewska-Koziell, M.; Winterborn, A.; Holloway, A.C.; Brien, J.F.; Reynolds, J.N. Chronic prenatal ethanol exposure increases adiposity and disrupts pancreatic morphology in adult guinea pig offspring. Nutr. Diabetes 2012, 2, e57. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Luo, H.; Hu, S.; Wu, Y.; Magdalou, J.; Chen, L.; Wang, H. Effects and Interactions of Prenatal Ethanol Exposure, a Post-Weaning High-Fat Diet and Gender on Adult Hypercholesterolemia Occurrence in Offspring Rats. Cell Physiol. Biochem. 2017, 44, 657–670. [Google Scholar] [CrossRef]
- Harper, K.M.; Tunc-Ozcan, E.; Graf, E.N.; Redei, E.E. Intergenerational effects of prenatal ethanol on glucose tolerance and insulin response. Physiol. Genomics 2014, 46, 159–168. [Google Scholar] [CrossRef]
- Chen, L.; Nyomba, B.L. Glucose intolerance and resistin expression in rat offspring exposed to ethanol in utero: Modulation by postnatal high-fat diet. Endocrinology 2003, 144, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Nyomba, B.L. Effects of prenatal alcohol exposure on glucose tolerance in the rat offspring. Metabolism 2003, 52, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Ponce, L.F.; Pautassi, R.M.; Spear, N.E.; Molina, J.C. Ethanol-mediated operant learning in the infant rat leads to increased ethanol intake during adolescence. Pharmacol. Biochem. Behav. 2008, 90, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Spear, N.E.; Molina, J.C. Fetal or infantile exposure to ethanol promotes ethanol ingestion in adolescence and adulthood: A theoretical review. Alcohol. Clin. Exp. Res. 2005, 29, 909–929. [Google Scholar] [CrossRef] [PubMed]
- Alati, R.; Al Mamun, A.; Williams, G.M.; O’Callaghan, M.; Najman, J.M.; Bor, W. In utero alcohol exposure and prediction of alcohol disorders in early adulthood: A birth cohort study. Arch. Gen. Psychiatry 2006, 63, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Baer, J.S.; Barr, H.M.; Bookstein, F.L.; Sampson, P.D.; Streissguth, A.P. Prenatal alcohol exposure and family history of alcoholism in the etiology of adolescent alcohol problems. J. Stud. Alcohol. 1998, 59, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Baer, J.S.; Sampson, P.D.; Barr, H.M.; Connor, P.D.; Streissguth, A.P. A 21-year longitudinal analysis of the effects of prenatal alcohol exposure on young adult drinking. Arch. Gen. Psychiatry 2003, 60, 377–385. [Google Scholar] [CrossRef]
- Dunn, W.; Shah, V.H. Pathogenesis of Alcoholic Liver Disease. Clin. Liver Dis. 2016, 20, 445–456. [Google Scholar] [CrossRef]
- da Silva, R.P.; Eudy, B.J.; Deminice, R. One-Carbon Metabolism in Fatty Liver Disease and Fibrosis: One-Carbon to Rule Them All. J. Nutr. 2020, 150, 994–1003. [Google Scholar]
- Zakhari, S. Alcohol metabolism and epigenetics changes. Alcohol. Res. 2013, 35, 6–16. [Google Scholar]
- Kharbanda, K.K.; Rogers, D.D.; Mailliard, M.E.; Siford, G.L.; Barak, A.J.; Beckenhauer, H.C.; Sorrell, M.F.; Tuma, D.J. A comparison of the effects of betaine and S-adenosylmethionine on ethanol-induced changes in methionine metabolism and steatosis in rat hepatocytes. J. Nutr. 2005, 135, 519–524. [Google Scholar] [CrossRef]
- Otero, N.K.; Thomas, J.D.; Saski, C.A.; Xia, X.; Kelly, S.J. Choline supplementation and DNA methylation in the hippocampus and prefrontal cortex of rats exposed to alcohol during development. Alcohol. Clin. Exp. Res. 2012, 36, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Bekdash, R.A.; Zhang, C.; Sarkar, D.K. Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation, and proopiomelanocortin (POMC) gene expression in β-endorphin-producing POMC neurons of the hypothalamus. Alcohol. Clin. Exp. Res. 2013, 37, 1133–1142. [Google Scholar] [CrossRef]
- Thomas, J.D.; Garrison, M.; O’Neill, T.M. Perinatal choline supplementation attenuates behavioral alterations associated with neonatal alcohol exposure in rats. Neurotoxicol. Teratol. 2004, 26, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Idrus, N.M.; Monk, B.R.; Dominguez, H.D. Prenatal choline supplementation mitigates behavioral alterations associated with prenatal alcohol exposure in rats. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Abou, E.J.; Dominguez, H.D. Prenatal choline supplementation mitigates the adverse effects of prenatal alcohol exposure on development in rats. Neurotoxicol Teratol. 2009, 31, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Tran, T.D. Choline supplementation mitigates trace, but not delay, eyeblink conditioning deficits in rats exposed to alcohol during development. Hippocampus 2012, 22, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.W.; Carter, R.C.; Molteno, C.D.; Stanton, M.E.; Herbert, J.S.; Lindinger, N.M.; Lewis, C.E.; Dodge, N.C.; Hoyme, H.E.; Zeisel, S.H.; et al. Efficacy of Maternal Choline Supplementation During Pregnancy in Mitigating Adverse Effects of Prenatal Alcohol Exposure on Growth and Cognitive Function: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Alcohol. Clin. Exp. Res. 2018, 42, 1327–1341. [Google Scholar] [CrossRef]
- Ernst, A.M.; Gimbel, B.A.; de Water, E.; Eckerle, J.K.; Radke, J.P.; Georgieff, M.K.; Wozniak, J.R. Prenatal and Postnatal Choline Supplementation in Fetal Alcohol Spectrum Disorder. Nutrients 2022, 14, 688. [Google Scholar] [CrossRef] [PubMed]
- Jack-Roberts, C.; Joselit, Y.; Nanobashvili, K.; Bretter, R.; Malysheva, O.V.; Caudill, M.A.; Saxena, A.; Axen, K.; Gomaa, A.; Jiang, X. Choline Supplementation Normalizes Fetal Adiposity and Reduces Lipogenic Gene Expression in a Mouse Model of Maternal Obesity. Nutrients 2017, 9, 899. [Google Scholar] [CrossRef]
- Korsmo, H.W.; Edwards, K.; Dave, B.; Jack-Roberts, C.; Yu, H.; Saxena, A.; Salvador, M.; Dembitzer, M.; Phagoora, J.; Jiang, X. Prenatal Choline Supplementation during High-Fat Feeding Improves Long-Term Blood Glucose Control in Male Mouse Offspring. Nutrients 2020, 12, 144. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.; Greenwald, E.; Jack-Roberts, C.; Ajeeb, T.T.; Malysheva, O.V.; Caudill, M.A.; Axen, K.; Saxena, A.; Semernina, E.; Nanobashvili, K.; et al. Choline prevents fetal overgrowth and normalizes placental fatty acid and glucose metabolism in a mouse model of maternal obesity. J. Nutr. Biochem. 2017, 49, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Jiang, X.; West, A.A.; Perry, C.A.; Malysheva, O.V.; Devapatla, S.; Pressman, E.; Vermeylen, F.; Stabler, S.P.; Allen, R.H.; et al. Maternal choline intake modulates maternal and fetal biomarkers of choline metabolism in humans. Am. J. Clin. Nutr. 2012, 95, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Janney, C.G.; Di Bisceglie, A.M.; Neuschwander-Tetri, B.A.; Bacon, B.R. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.; Yan, J.; Abratte, C.M.; Vermeylen, F.; Caudill, M.A. Choline intake exceeding current dietary recommendations preserves markers of cellular methylation in a genetic subgroup of folate-compromised men. J. Nutr. 2010, 140, 975–980. [Google Scholar] [CrossRef]
- Stewart, M.S.; Heerwagen, M.J.; Friedman, J.E. Developmental programming of pediatric nonalcoholic fatty liver disease: Redefining the “first hit”. Clin. Obstet. Gynecol. 2013, 56, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Berthier, A.; Johanns, M.; Zummo, F.P.; Lefebvre, P.; Staels, B. PPARs in liver physiology. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166097. [Google Scholar] [CrossRef]
- Zardi, E.M.; Navarini, L.; Sambataro, G.; Piccinni, P.; Sambataro, F.M.; Spina, C.; Dobrina, A. Hepatic PPARs: Their role in liver physiology, fibrosis and treatment. Curr. Med. Chem. 2013, 20, 3370–3396. [Google Scholar] [CrossRef]
- Régnier, M.; Polizzi, A.; Smati, S.; Lukowicz, C.; Fougerat, A.; Lippi, Y.; Fouché, E.; Lasserre, F.; Naylies, C.; Bétoulières, C.; et al. Hepatocyte-specific deletion of Pparα promotes NAFLD in the context of obesity. Sci. Rep. 2020, 10, 6489. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Schreyer, S.A.; Vick, C.; Lystig, T.C.; Mystkowski, P.; LeBoeuf, R.C. LDL receptor but not apolipoprotein E deficiency increases diet-induced obesity and diabetes in mice. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E207–E214. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.K.; Arjuman, A.; Chandra, N.C. Role of leptin on the expression of low density lipoprotein receptor. Indian. J. Med. Res. 2014, 140, 524–530. [Google Scholar] [PubMed]
- Korsmo, H.W.; Dave, B.; Trasino, S.; Saxena, A.; Liu, J.; Caviglia, J.M.; Edwards, K.; Dembitzer, M.; Sheeraz, S.; Khaldi, S.; et al. Maternal Choline Supplementation and High-Fat Feeding Interact to Influence DNA Methylation in Offspring in a Time-Specific Manner. Front. Nutr. 2022, 9, 841787. [Google Scholar] [CrossRef]
- Hochberg, Z.; Feil, R.; Constancia, M.; Fraga, M.; Junien, C.; Carel, J.C.; Boileau, P.; Le Bouc, Y.; Deal, C.L.; Lillycrop, K.; et al. Child health, developmental plasticity, and epigenetic programming. Endocr. Rev. 2011, 32, 159–224. [Google Scholar] [CrossRef] [PubMed]
- Linask, K.K.; Han, M. Acute alcohol exposure during mouse gastrulation alters lipid metabolism in placental and heart development: Folate prevention. Birth Defects Res. A Clin. Mol. Teratol. 2016, 106, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Korsmo, H.W.; Kadam, I.; Reaz, A.; Bretter, R.; Saxena, A.; Johnson, C.H.; Caviglia, J.M.; Jiang, X. Prenatal Choline Supplement in a Maternal Obesity Model Modulates Offspring Hepatic Lipidomes. Nutrients 2023, 15, 965. [Google Scholar] [CrossRef]
- Meck, W.H.; Williams, C.L. Metabolic imprinting of choline by its availability during gestation: Implications for memory and attentional processing across the lifespan. Neurosci. Biobehav. Rev. 2003, 27, 385–399. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ctrl/Ctrl | Ctrl/AE | AE/AE | AE-CS/AE | |
|---|---|---|---|---|
| Male | ||||
| Number of animals | n = 6 | n = 8 | n = 7 | n = 7 |
| Liver weight (g) | 1.13 ± 0.15 | 1.14 ± 0.18 | 1.20 ± 0.11 | 1.25 ± 0.22 |
| Liver NAS | 0.58 ± 0.39 | 1.69 ± 0.94 | 1.28 ± 0.33 | 2.67 ± 0.92 |
| Liver triglyceride (mg/g) | 11.5 ± 2.5 a | 16.9 ± 2.8 a,b | 22.6 ± 4.4 b | 13.9 ± 1.4 a |
| Liver MDA (nM/g) | 51.2 ± 15.0 | 44.0 ± 7.9 | 59.8 ± 10.1 | 55.5 ± 12.5 |
| Serum ALT (IU/L) | 37.3 ± 13.3 | 72.6 ± 24.4 | 34.8 ± 10.5 | 105.6 ± 17.0 |
| Serum triglyceride (mg/dL) | 45.4 ± 10.5 b | 69.0 ± 10.9 b | 60.3 ± 12.1 b | 107.3 ± 12.6 a |
| Serum FFA (nmol/L) | 0.62 ± 0.09 | 0.86 ± 0.22 | 0.69 ± 0.10 | 0.89 ± 0.10 |
| Serum ApoB (µg/mL) | 26.2 ± 16.6 | 11.6 ± 5.2 | 12.4 ± 2.2 | 19.6 ± 8.6 |
| Global DNA methylation (fold difference) | 1.00 ± 0.68 | 0.54 ± 0.18 | 0.27 ± 0.09 | 0.80 ± 0.31 |
| Female | ||||
| Number of animals | n = 6 | n = 8 | n = 6 | n = 6 |
| Liver weight (g) | 0.99 ± 0.04 | 0.94 ± 0.04 | 0.95 ± 0.03 | 0.88 ± 0.04 |
| Liver NAS | 0.25 ± 0.25 | 0.50 ± 0.19 | 0.23 ± 0.14 | 0.58 ± 0.31 |
| Liver triglyceride (mg/g) | 8.3 ± 1.4 a | 19.4 ± 2.7 b | 13.3 ± 2.0 a,b | 13.2 ± 1.6 a |
| Liver MDA (nM/g) | 58.5 ± 8.7 | 40.0 ± 5.3 | 40.6 ± 3.8 | 80.8 ± 33.5 |
| Serum ALT (IU/L) | 32.5 ± 9.5 | 64.0 ± 43.2 | 17.2 ± 3.3 | 41.8 ± 11.2 |
| Serum triglyceride (mg/dL) | 53.7 ± 5.3 | 63.9 ± 21.0 | 55.6 ± 5.5 | 45.8 ± 7.3 |
| Serum FFA (nmol/L) | 0.66 ± 0.10 | 0.91 ± 0.46 | 0.85 ± 0.30 | 0.87 ± 0.36 |
| Serum ApoB (µg/mL) | 6.6 ± 4.3 | 6.5 ± 3.3 | 10.2 ± 2.5 | 10.4 ± 1.5 |
| Global DNA methylation (fold difference) | 1.00 ± 0.22 | 0.62 ± 0.45 | 0.49 ± 0.19 | 0.56 ± 0.12 |
| Ctrl/Ctrl | Ctrl/AE | AE/AE | AE-CS/AE | |
|---|---|---|---|---|
| Male (nmol/g) | ||||
| Number of animals | n = 6 | n = 8 | n = 7 | n = 7 |
| Methionine | 191 ± 25 | 278 ± 46 | 228 ± 23 | 265 ± 21 |
| Choline | 387 ± 116 a | 301 ± 68 a | 606 ± 24 a,b | 869 ± 184 b |
| Dimethylglycine | 19.6 ± 0.8 a | 48.8 ± 10.6 b | 33.5 ± 8.8 a,b | 26.9 ± 4.3 a |
| Betaine | 205 ± 51 | 581 ± 302 | 240 ± 63 | 182 ± 48 |
| Trimethylamine-oxide | 1.3 ± 0.2 b | 0.8 ± 0.3 a,b | 0.7 ± 0.2 a,b | 0.4 ± 0.3 a |
| Glycerophosphorylcholine | 711 ± 204 b | 429 ± 91 a,b | 285 ± 37 a | 227 ± 65 a |
| Phosphorylcholine | 81 ± 23 | 105 ± 16 | 144 ± 23 | 145 ± 28 |
| Phosphatidylcholine | 16,495 ± 450 a | 20,343 ± 2728 a,b | 23,575 ± 716 b | 23,527 ± 789 b |
| Sphingomyelin | 1727 ± 80 a | 2566 ± 226 c | 1910 ± 90 a,b | 2282 ± 190 b,c |
| Lysophosphatidylcholine | 562 ± 23 a | 598 ± 63 a | 728 ± 14 b | 754 ± 28 b |
| Female (nmol/g) | ||||
| Number of animals | n = 6 | n = 8 | n = 6 | n = 6 |
| Methionine | 152 ± 13 | 182 ± 17 | 223 ± 24 | 164 ± 37 |
| Choline | 214 ± 58 | 212 ± 55 | 289 ± 124 | 194 ± 56 |
| Dimethylglycine | 39.7 ± 7.5 | 40.4 ± 10.4 | 32.9 ± 4.0 | 34.3 ± 1.6 |
| Betaine | 354 ± 103 | 467 ± 96 | 399 ± 58 | 531 ± 59 |
| Trimethylamine-oxide | 38.4 ± 17.9 | 14.1 ± 2.3 | 19.0 ± 5.0 | 13.2 ± 4.5 |
| Glycerophosphorylcholine | 372 ± 26 a | 490 ± 74 a,b | 412 ± 60 a | 721 ± 157 b |
| Phosphorylcholine | 80 ± 30 | 113 ± 32 | 115 ± 24 | 207 ± 73 |
| Phosphatidylcholine | 20,180 ± 1037 | 20,503 ± 823 | 19,698 ± 1138 | 18,393 ± 1296 |
| Sphingomyelin | 1990 ± 213 | 1998 ± 104 | 1875 ± 84 | 1792 ± 179 |
| Lysophosphatidylcholine | 646 ± 54 | 629 ± 24 | 654 ± 19 | 577 ± 44 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadam, I.; Trasino, S.E.; Korsmo, H.; Lucas, J.; Pinkas, M.; Jiang, X. Prenatal Choline Supplementation Improves Glucose Tolerance and Reduces Liver Fat Accumulation in Mouse Offspring Exposed to Ethanol during the Prenatal and Postnatal Periods. Nutrients 2024, 16, 1264. https://doi.org/10.3390/nu16091264
Kadam I, Trasino SE, Korsmo H, Lucas J, Pinkas M, Jiang X. Prenatal Choline Supplementation Improves Glucose Tolerance and Reduces Liver Fat Accumulation in Mouse Offspring Exposed to Ethanol during the Prenatal and Postnatal Periods. Nutrients. 2024; 16(9):1264. https://doi.org/10.3390/nu16091264
Chicago/Turabian StyleKadam, Isma’il, Steven E. Trasino, Hunter Korsmo, Jessica Lucas, Myriam Pinkas, and Xinyin Jiang. 2024. "Prenatal Choline Supplementation Improves Glucose Tolerance and Reduces Liver Fat Accumulation in Mouse Offspring Exposed to Ethanol during the Prenatal and Postnatal Periods" Nutrients 16, no. 9: 1264. https://doi.org/10.3390/nu16091264