Activation and Regulation of the Pattern Recognition Receptors in Obesity-Induced Adipose Tissue Inflammation and Insulin Resistance

{kind=link}
{kind=link}
{kind=link}
Abstract
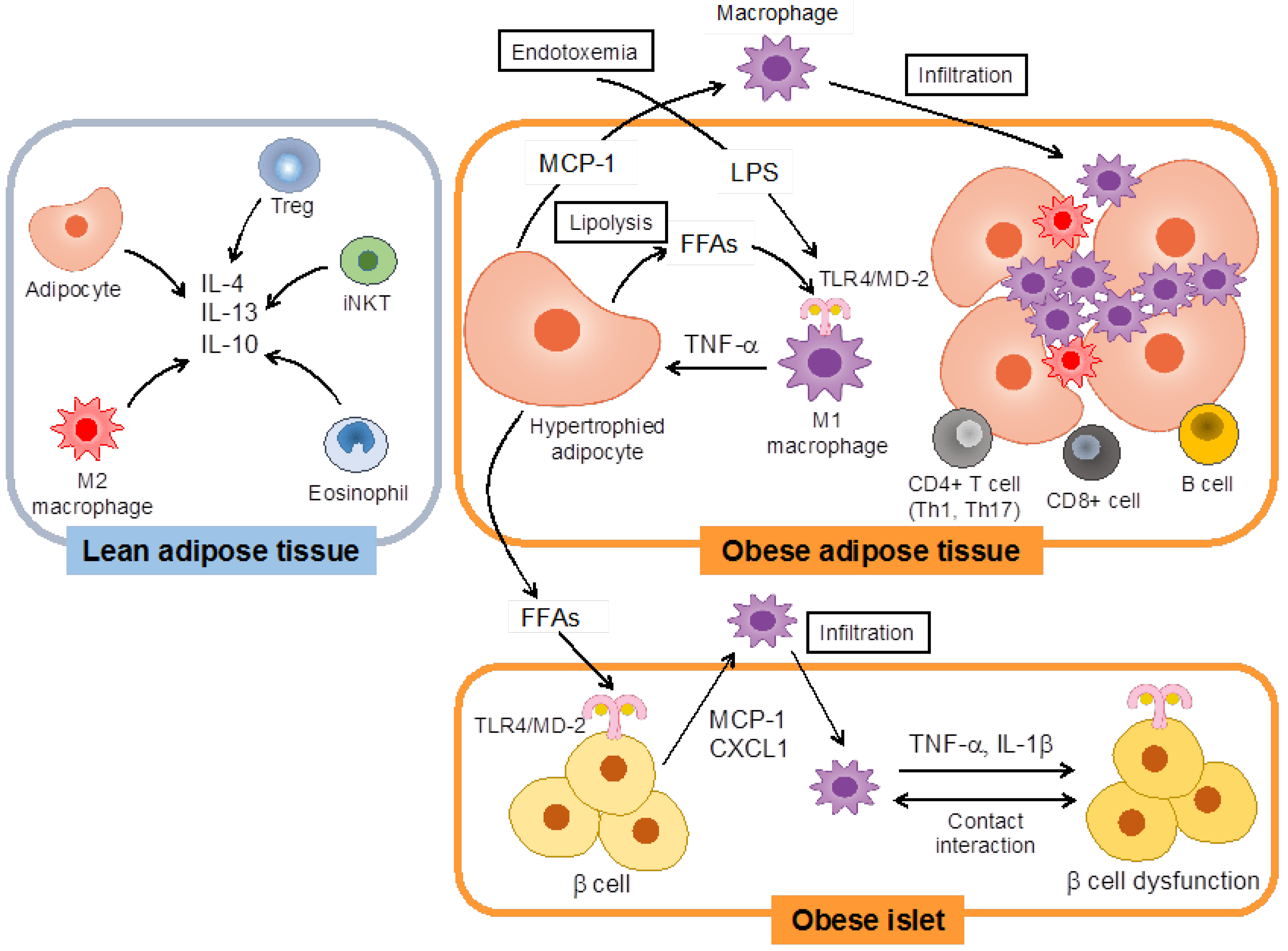
:1. Introduction

2. Toll-Like Receptors
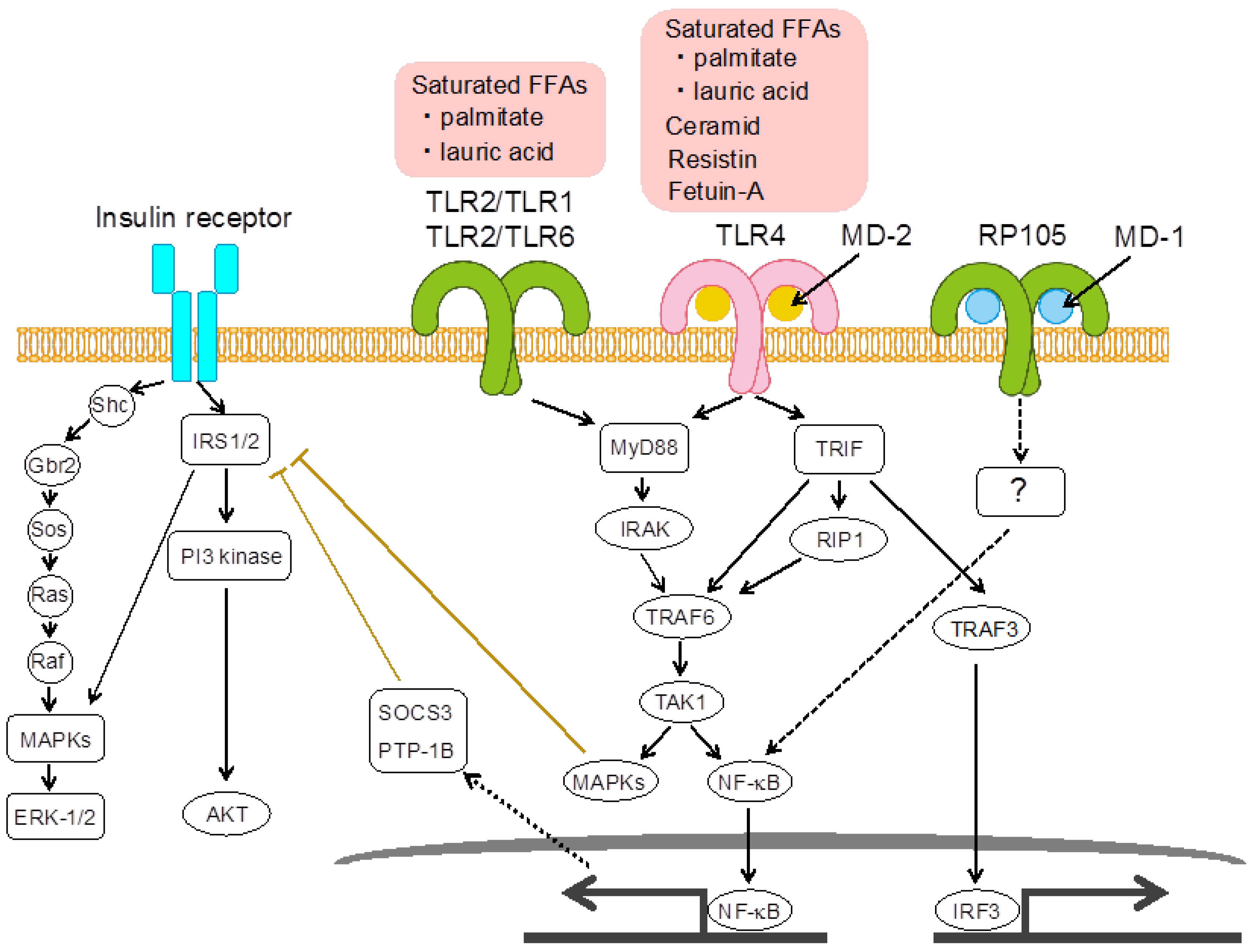
2.1. Toll-Like Receptor 4

2.2. Radioprotective 105
2.3. Toll-Like Receptor 2
2.4. Toll-Like Receptor 5
3. Nucleotide-Binding Oligomerization Domain (NOD)-Like Receptors and Inflammasomes
3.1. Nucleotide-Binding Oligomerization Domain (NOD)-Like Receptors

3.2. NOD-Like Receptor Family, Pyrin Domain Containing 3
4. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Schenk, S.; Saberi, M.; Olefsky, J. Insulin sensitivity: Modulation by nutrients and inflammation. J. Clin. Investig. 2008, 118, 2992–3002. [Google Scholar] [CrossRef]
- Kalupahana, N.S.; Moustaid-Moussa, N.; Claycombe, K.J. Immunity as a link between obesity and insulin resistance. Mol. Aspects Med. 2012, 33, 26–34. [Google Scholar] [CrossRef]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Ryan, K.K.; Woods, S.C.; Seeley, R.J. Central nervous system mechanisms linking the consumption of palatable high-fat diets to the defense of greater adiposity. Cell Metab. 2012, 15, 137–149. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Suganami, T.; Ogawa, Y. Adipose tissue macrophages: Their role in adipose tissue remodeling. J. Leukoc. Biol. 2010, 88, 33–39. [Google Scholar] [CrossRef]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef]
- Wu, D.; Molofsky, A.B.; Liang, H.E.; Ricardo-Gonzalez, R.R.; Jouihan, H.A.; Bando, J.K.; Chawla, A.; Locksley, R.M. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 2011, 332, 243–247. [Google Scholar] [CrossRef]
- Lynch, L.; Nowak, M.; Varghese, B.; Clark, J.; Hogan, A.E.; Toxavidis, V.; Balk, S.P.; O’Shea, D.; O’Farrelly, C.; Exley, M.A. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity 2012, 37, 574–587. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Cildir, G.; Akincilar, S.C.; Tergaonkar, V. Chronic adipose tissue inflammation: All immune cells on the stage. Trends Mol. Med. 2013, 19, 487–500. [Google Scholar] [CrossRef]
- Chmelar, J.; Chung, K.J.; Chavakis, T. The role of innate immune cells in obese adipose tissue inflammation and development of insulin resistance. Thromb. Haemost. 2013, 109, 399–406. [Google Scholar] [CrossRef]
- Kang, K.; Reilly, S.M.; Karabacak, V.; Gangl, M.R.; Fitzgerald, K.; Hatano, B.; Lee, C.H. Adipocyte-derived Th2 cytokines and myeloid PPARδ regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008, 7, 485–495. [Google Scholar] [CrossRef]
- Zeyda, M.; Stulnig, T.M. Adipose tissue macrophages. Immunol. Lett. 2007, 112, 61–67. [Google Scholar] [CrossRef]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef]
- Elgazar-Carmon, V.; Rudich, A.; Hadad, N.; Levy, R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J. Lipid Res. 2008, 49, 1894–1903. [Google Scholar] [CrossRef]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef]
- Fabbrini, E.; Cella, M.; McCartney, S.A.; Fuchs, A.; Abumrad, N.A.; Pietka, T.A.; Chen, Z.; Finck, B.N.; Han, D.H.; Magkos, F.; et al. Association between specific adipose tissue CD4(+) T-Cell populations and insulin resistance in obese individuals. Gastroenterology 2013, 145, 366–374. [Google Scholar] [CrossRef]
- Jagannathan-Bogdan, M.; McDonnell, M.E.; Shin, H.; Rehman, Q.; Hasturk, H.; Apovian, C.M.; Nikolajczyk, B.S. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J. Immunol. 2011, 186, 1162–1172. [Google Scholar] [CrossRef]
- Zeng, C.; Shi, X.; Zhang, B.; Liu, H.; Zhang, L.; Ding, W.; Zhao, Y. The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: Relationship with metabolic factors and complications. J. Mol. Med. 2012, 90, 175–186. [Google Scholar] [CrossRef]
- Morris, D.L.; Cho, K.W.; Delproposto, J.L.; Oatmen, K.E.; Geletka, L.M.; Martinez-Santibanez, G.; Singer, K.; Lumeng, C.N. Adipose tissue macrophages function as antigen-presenting cells and regulate adipose tissue CD4+ T cells in mice. Diabetes 2013, 62, 2762–2772. [Google Scholar] [CrossRef]
- Winer, D.A.; Winer, S.; Shen, L.; Wadia, P.P.; Yantha, J.; Paltser, G.; Tsui, H.; Wu, P.; Davidson, M.G.; Alonso, M.N.; et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 2011, 17, 610–617. [Google Scholar] [CrossRef]
- DeFuria, J.; Belkina, A.C.; Jagannathan-Bogdan, M.; Snyder-Cappione, J.; Carr, J.D.; Nersesova, Y.R.; Markham, D.; Strissel, K.J.; Watkins, A.A.; Zhu, M.; et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. USA 2013, 110, 5133–5138. [Google Scholar] [CrossRef]
- Jagannathan, M.; McDonnell, M.; Liang, Y.; Hasturk, H.; Hetzel, J.; Rubin, D.; Kantarci, A.; van Dyke, T.E.; Ganley-Leal, L.M.; Nikolajczyk, B.S. Toll-like receptors regulate B cell cytokine production in patients with diabetes. Diabetologia 2010, 53, 1461–1471. [Google Scholar] [CrossRef]
- Leahy, J.L. Pathogenesis of type 2 diabetes mellitus. Arch. Med. Res. 2005, 36, 197–209. [Google Scholar] [CrossRef]
- Beaudry, J.L.; Riddell, M.C. Effects of glucocorticoids and exercise on pancreatic β-cell function and diabetes development. Diabetes Metab. Res. Rev. 2012, 28, 560–573. [Google Scholar] [CrossRef]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Islet-associated macrophages in type 2 diabetes. Diabetologia 2009, 52, 1686–1688. [Google Scholar] [CrossRef]
- Lizarbe, B.; Benitez, A.; Pelaez-Brioso, G.A.; Sanchez-Montanes, M.; Lopez-Larrubia, P.; Ballesteros, P.; Cerdan, S. Hypothalamic metabolic compartmentation during appetite regulation as revealed by magnetic resonance imaging and spectroscopy methods. Front. Neuroenerget. 2013, 5. [Google Scholar] [CrossRef]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef]
- Reyna, S.M.; Ghosh, S.; Tantiwong, P.; Meka, C.S.; Eagan, P.; Jenkinson, C.P.; Cersosimo, E.; Defronzo, R.A.; Coletta, D.K.; Sriwijitkamol, A.; et al. Elevated Toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes 2008, 57, 2595–2602. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef]
- Piccinini, A.M.; Midwood, K.S. DAMPening inflammation by modulating TLR signalling. Mediat. Inflamm. 2010, 2010, e672395. [Google Scholar]
- De Nardo, D.; Latz, E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011, 32, 373–379. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Watanabe, Y.; Nakamura, T.; Ishikawa, S.; Fujisaka, S.; Usui, I.; Tsuneyama, K.; Ichihara, Y.; Wada, T.; Hirata, Y.; Suganami, T.; et al. The radioprotective 105/MD-1 complex contributes to diet-induced obesity and adipose tissue inflammation. Diabetes 2012, 61, 1199–1209. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Uematsu, S.; Akira, S. Toll-like receptors and innate immunity. J. Mol. Med. 2006, 84, 712–725. [Google Scholar] [CrossRef]
- Uematsu, S.; Akira, S. Toll-like receptors and type I interferons. J. Biol. Chem. 2007, 282, 15319–15323. [Google Scholar] [CrossRef]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef]
- Nagai, Y.; Akashi, S.; Nagafuku, M.; Ogata, M.; Iwakura, Y.; Akira, S.; Kitamura, T.; Kosugi, A.; Kimoto, M.; Miyake, K. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat. Immunol. 2002, 3, 667–672. [Google Scholar]
- Ohto, U.; Fukase, K.; Miyake, K.; Satow, Y. Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science 2007, 316, 1632–1634. [Google Scholar] [CrossRef]
- Ohto, U.; Fukase, K.; Miyake, K.; Shimizu, T. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc. Natl. Acad. Sci. USA 2012, 109, 7421–7426. [Google Scholar] [CrossRef]
- Kim, H.M.; Park, B.S.; Kim, J.I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Favelyukis, S.; Nguyen, A.K.; Reichart, D.; Scott, P.A.; Jenn, A.; Liu-Bryan, R.; Glass, C.K.; Neels, J.G.; Olefsky, J.M. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 2007, 282, 35279–35292. [Google Scholar] [CrossRef]
- Tsukumo, D.M.; Carvalho-Filho, M.A.; Carvalheira, J.B.; Prada, P.O.; Hirabara, S.M.; Schenka, A.A.; Araujo, E.P.; Vassallo, J.; Curi, R.; Velloso, L.A.; et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 2007, 56, 1986–1998. [Google Scholar] [CrossRef]
- Saberi, M.; Woods, N.B.; de Luca, C.; Schenk, S.; Lu, J.C.; Bandyopadhyay, G.; Verma, I.M.; Olefsky, J.M. Hematopoietic cell-specific deletion of Toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009, 10, 419–429. [Google Scholar] [CrossRef]
- Suganami, T.; Tanimoto-Koyama, K.; Nishida, J.; Itoh, M.; Yuan, X.; Mizuarai, S.; Kotani, H.; Yamaoka, S.; Miyake, K.; Aoe, S.; et al. Role of the Toll-like receptor 4/NF-κB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 84–91. [Google Scholar] [CrossRef]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M.; et al. GPR120 is an ω-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar]
- Boden, G. Obesity, insulin resistance and free fatty acids. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 139–143. [Google Scholar] [CrossRef]
- Wong, S.W.; Kwon, M.J.; Choi, A.M.; Kim, H.P.; Nakahira, K.; Hwang, D.H. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J. Biol. Chem. 2009, 284, 27384–27392. [Google Scholar] [CrossRef]
- Lee, J.Y.; Ye, J.; Gao, Z.; Youn, H.S.; Lee, W.H.; Zhao, L.; Sizemore, N.; Hwang, D.H. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J. Biol. Chem. 2003, 278, 37041–37051. [Google Scholar]
- Yamakawa, N.; Ohto, U.; Akashi-Takamura, S.; Takahashi, K.; Saitoh, S.; Tanimura, N.; Suganami, T.; Ogawa, Y.; Shibata, T.; Shimizu, T.; et al. Human TLR4 polymorphism D299G/T399I alters TLR4/MD-2 conformation and response to a weak ligand monophosphoryl lipid A. Int. Immunol. 2013, 25, 45–52. [Google Scholar] [CrossRef]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef]
- Suganami, T.; Nishida, J.; Ogawa, Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: Role of free fatty acids and tumor necrosis factor α. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2062–2068. [Google Scholar] [CrossRef]
- Erridge, C.; Samani, N.J. Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1944–1949. [Google Scholar] [CrossRef]
- Galbo, T.; Perry, R.J.; Jurczak, M.J.; Camporez, J.P.; Alves, T.C.; Kahn, M.; Guigni, B.A.; Serr, J.; Zhang, D.; Bhanot, S.; et al. Saturated and unsaturated fat induce hepatic insulin resistance independently of TLR-4 signaling and ceramide synthesis in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 12780–12785. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef]
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 2012, 18, 1279–1285. [Google Scholar] [CrossRef]
- Benomar, Y.; Gertler, A.; de Lacy, P.; Crepin, D.; Ould Hamouda, H.; Riffault, L.; Taouis, M. Central resistin overexposure induces insulin resistance through Toll-like receptor 4. Diabetes 2013, 62, 102–114. [Google Scholar] [CrossRef]
- Dasgupta, S.; Bhattacharya, S.; Biswas, A.; Majumdar, S.S.; Mukhopadhyay, S.; Ray, S. NF-κB mediates lipid-induced fetuin-A expression in hepatocytes that impairs adipocyte function effecting insulin resistance. Biochem. J. 2010, 429, 451–462. [Google Scholar] [CrossRef]
- Bluher, M.; Michael, M.D.; Peroni, O.D.; Ueki, K.; Carter, N.; Kahn, B.B.; Kahn, C.R. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev. Cell 2002, 3, 25–38. [Google Scholar] [CrossRef]
- Havekes, B.; Sauerwein, H.P. Adipocyte-myocyte crosstalk in skeletal muscle insulin resistance; is there a role for thyroid hormone? Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 641–646. [Google Scholar] [CrossRef]
- Meshkani, R.; Adeli, K. Hepatic insulin resistance, metabolic syndrome and cardiovascular disease. Clin. Biochem. 2009, 42, 1331–1346. [Google Scholar] [CrossRef]
- Stockhorst, U.; de Fries, D.; Steingrueber, H.J.; Scherbaum, W.A. Insulin and the CNS: Effects on food intake, memory, and endocrine parameters and the role of intranasal insulin administration in humans. Physiol. Behav. 2004, 83, 47–54. [Google Scholar]
- Konner, A.C.; Bruning, J.C. Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 2012, 16, 144–152. [Google Scholar] [CrossRef]
- Tanti, J.F.; Jager, J. Cellular mechanisms of insulin resistance: Role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr. Opin. Pharmacol. 2009, 9, 753–762. [Google Scholar] [CrossRef]
- Boura-Halfon, S.; Zick, Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E581–E591. [Google Scholar]
- Gonzalez-Rodriguez, A.; Mas Gutierrez, J.A.; Sanz-Gonzalez, S.; Ros, M.; Burks, D.J.; Valverde, A.M. Inhibition of PTP1B restores IRS1-mediated hepatic insulin signaling in IRS2-deficient mice. Diabetes 2010, 59, 588–599. [Google Scholar] [CrossRef]
- Miyake, K.; Shimazu, R.; Kondo, J.; Niki, T.; Akashi, S.; Ogata, H.; Yamashita, Y.; Miura, Y.; Kimoto, M. Mouse MD-1, a molecule that is physically associated with RP105 and positively regulates its expression. J. Immunol. 1998, 161, 1348–1353. [Google Scholar]
- Miyake, K.; Yamashita, Y.; Ogata, M.; Sudo, T.; Kimoto, M. RP105, a novel B cell surface molecule implicated in B cell activation, is a member of the leucine-rich repeat protein family. J. Immunol. 1995, 154, 3333–3340. [Google Scholar]
- Miyake, K.; Ogata, H.; Nagai, Y.; Akashi, S.; Kimoto, M. Innate recognition of lipopolysaccharide by Toll-like receptor 4/MD-2 and RP105/MD-1. J. Endotoxin Res. 2000, 6, 389–391. [Google Scholar]
- Yamashita, Y.; Miyake, K.; Miura, Y.; Kaneko, Y.; Yagita, H.; Suda, T.; Nagata, S.; Nomura, J.; Sakaguchi, N.; Kimoto, M. Activation mediated by RP105 but not CD40 makes normal B cells susceptible to anti-IgM-induced apoptosis: A role for Fc receptor coligation. J. Exp. Med. 1996, 184, 113–120. [Google Scholar] [CrossRef]
- Nagai, Y.; Yanagibashi, T.; Watanabe, Y.; Ikutani, M.; Kariyone, A.; Ohta, S.; Hirai, Y.; Kimoto, M.; Miyake, K.; Takatsu, K. The RP105/MD-1 complex is indispensable for TLR4/MD-2-dependent proliferation and IgM-secreting plasma cell differentiation of marginal zone B cells. Int. Immunol. 2012, 24, 389–400. [Google Scholar] [CrossRef]
- Ogata, H.; Su, I.; Miyake, K.; Nagai, Y.; Akashi, S.; Mecklenbrauker, I.; Rajewsky, K.; Kimoto, M.; Tarakhovsky, A. The Toll-like receptor protein RP105 regulates lipopolysaccharide signaling in B cells. J. Exp. Med. 2000, 192, 23–29. [Google Scholar] [CrossRef]
- Nagai, Y.; Shimazu, R.; Ogata, H.; Akashi, S.; Sudo, K.; Yamasaki, H.; Hayashi, S.; Iwakura, Y.; Kimoto, M.; Miyake, K. Requirement for MD-1 in cell surface expression of RP105/CD180 and B-cell responsiveness to lipopolysaccharide. Blood 2002, 99, 1699–1705. [Google Scholar] [CrossRef]
- Nagai, Y.; Kobayashi, T.; Motoi, Y.; Ishiguro, K.; Akashi, S.; Saitoh, S.; Kusumoto, Y.; Kaisho, T.; Akira, S.; Matsumoto, M.; et al. The radioprotective 105/MD-1 complex links TLR2 and TLR4/MD-2 in antibody response to microbial membranes. J. Immunol. 2005, 174, 7043–7049. [Google Scholar]
- Divanovic, S.; Trompette, A.; Atabani, S.F.; Madan, R.; Golenbock, D.T.; Visintin, A.; Finberg, R.W.; Tarakhovsky, A.; Vogel, S.N.; Belkaid, Y.; et al. Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat. Immunol. 2005, 6, 571–578. [Google Scholar] [CrossRef]
- Nagai, Y.; Watanabe, Y.; Takatsu, K. The TLR family protein RP105/MD-1 complex: A new player in obesity and adipose tissue inflammation. Adipocyte 2013, 2, 61–66. [Google Scholar] [CrossRef]
- Blumenthal, A.; Kobayashi, T.; Pierini, L.M.; Banaei, N.; Ernst, J.D.; Miyake, K.; Ehrt, S. RP105 facilitates macrophage activation by Mycobacterium tuberculosis lipoproteins. Cell Host Microbe 2009, 5, 35–46. [Google Scholar] [CrossRef]
- Kuo, L.H.; Tsai, P.J.; Jiang, M.J.; Chuang, Y.L.; Yu, L.; Lai, K.T.; Tsai, Y.S. Toll-like receptor 2 deficiency improves insulin sensitivity and hepatic insulin signalling in the mouse. Diabetologia 2011, 54, 168–179. [Google Scholar] [CrossRef]
- Himes, R.W.; Smith, C.W. Tlr2 is critical for diet-induced metabolic syndrome in a murine model. FASEB J. 2010, 24, 731–739. [Google Scholar] [CrossRef]
- Ozinsky, A.; Underhill, D.M.; Fontenot, J.D.; Hajjar, A.M.; Smith, K.D.; Wilson, C.B.; Schroeder, L.; Aderem, A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 13766–13771. [Google Scholar] [CrossRef]
- Takeuchi, O.; Kawai, T.; Muhlradt, P.F.; Morr, M.; Radolf, J.D.; Zychlinsky, A.; Takeda, K.; Akira, S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 2001, 13, 933–940. [Google Scholar]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef]
- Oliveira-Nascimento, L.; Massari, P.; Wetzler, L.M. The role of TLR2 in infection and immunity. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef]
- Lee, J.Y.; Zhao, L.; Youn, H.S.; Weatherill, A.R.; Tapping, R.; Feng, L.; Lee, W.H.; Fitzgerald, K.A.; Hwang, D.H. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J. Biol. Chem. 2004, 279, 16971–16979. [Google Scholar] [CrossRef]
- Senn, J.J. Toll-like receptor-2 is essential for the development of palmitate-induced insulin resistance in myotubes. J. Biol. Chem. 2006, 281, 26865–26875. [Google Scholar] [CrossRef]
- Caricilli, A.M.; Nascimento, P.H.; Pauli, J.R.; Tsukumo, D.M.; Velloso, L.A.; Carvalheira, J.B.; Saad, M.J. Inhibition of Toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. J. Endocrinol. 2008, 199, 399–406. [Google Scholar] [CrossRef]
- Caricilli, A.M.; Picardi, P.K.; de Abreu, L.L.; Ueno, M.; Prada, P.O.; Ropelle, E.R.; Hirabara, S.M.; Castoldi, A.; Vieira, P.; Camara, N.O.; et al. Gut microbiota is a key modulator of insulin resistance in TLR 2 knockout mice. PLoS Biol. 2011, 9, e1001212. [Google Scholar] [CrossRef]
- Shechter, R.; London, A.; Kuperman, Y.; Ronen, A.; Rolls, A.; Chen, A.; Schwartz, M. Hypothalamic neuronal Toll-like receptor 2 protects against age-induced obesity. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Uematsu, S.; Akira, S. Immune responses of TLR5+ lamina propria dendritic cells in enterobacterial infection. J. Gastroenterol. 2009, 44, 803–811. [Google Scholar] [CrossRef]
- Hayashi, F.; Smith, K.D.; Ozinsky, A.; Hawn, T.R.; Yi, E.C.; Goodlett, D.R.; Eng, J.K.; Akira, S.; Underhill, D.M.; Aderem, A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 2001, 410, 1099–1103. [Google Scholar] [CrossRef]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef]
- Martin, B.; Hirota, K.; Cua, D.J.; Stockinger, B.; Veldhoen, M. Interleukin-17-producing γδ T cells selectively expand in response to pathogen products and environmental signals. Immunity 2009, 31, 321–330. [Google Scholar] [CrossRef]
- Kinnebrew, M.A.; Buffie, C.G.; Diehl, G.E.; Zenewicz, L.A.; Leiner, I.; Hohl, T.M.; Flavell, R.A.; Littman, D.R.; Pamer, E.G. Interleukin 23 production by intestinal CD103+CD11b+ dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity 2012, 36, 276–287. [Google Scholar] [CrossRef]
- Vijay-Kumar, M.; Aitken, J.D.; Gewirtz, A.T. Toll like receptor-5: Protecting the gut from enteric microbes. Semin. Immunopathol. 2008, 30, 11–21. [Google Scholar] [CrossRef]
- Scarpellini, E.; Campanale, M.; Leone, D.; Purchiaroni, F.; Vitale, G.; Lauritano, E.C.; Gasbarrini, A. Gut microbiota and obesity. Intern. Emerg. Med. 2010, 5, S53–S56. [Google Scholar]
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010, 328, 228–231. [Google Scholar] [CrossRef]
- Rubino, S.J.; Selvanantham, T.; Girardin, S.E.; Philpott, D.J. Nod-like receptors in the control of intestinal inflammation. Curr. Opin. Immunol. 2012, 24, 398–404. [Google Scholar] [CrossRef]
- Kufer, T.A.; Sansonetti, P.J. NLR functions beyond pathogen recognition. Nat. Immunol. 2011, 12, 121–128. [Google Scholar] [CrossRef]
- Broz, P.; Monack, D.M. Molecular mechanisms of inflammasome activation during microbial infections. Immunol. Rev. 2011, 243, 174–190. [Google Scholar] [CrossRef]
- Proell, M.; Riedl, S.J.; Fritz, J.H.; Rojas, A.M.; Schwarzenbacher, R. The Nod-like receptor (NLR) family: A tale of similarities and differences. PLoS One 2008, 3, e2119. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Munoz-Planillo, R.; Nunez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar]
- Ye, Z.; Ting, J.P. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr. Opin. Immunol. 2008, 20, 3–9. [Google Scholar]
- Jacobs, S.R.; Damania, B. NLRs, inflammasomes, and viral infection. J. Leukoc. Biol. 2012, 92, 469–477. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Kostura, M.J.; Tocci, M.J.; Limjuco, G.; Chin, J.; Cameron, P.; Hillman, A.G.; Chartrain, N.A.; Schmidt, J.A. Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proc. Natl. Acad. Sci. USA 1989, 86, 5227–5231. [Google Scholar] [CrossRef]
- Kuida, K.; Lippke, J.A.; Ku, G.; Harding, M.W.; Livingston, D.J.; Su, M.S.; Flavell, R.A. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 1995, 267, 2000–2003. [Google Scholar]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef]
- Osborn, L. Leukocyte adhesion to endothelium in inflammation. Cell 1990, 62, 3–6. [Google Scholar] [CrossRef]
- Girardin, S.E.; Boneca, I.G.; Carneiro, L.A.; Antignac, A.; Jehanno, M.; Viala, J.; Tedin, K.; Taha, M.K.; Labigne, A.; Zahringer, U.; et al. NOD1 detects a unique muropeptide from Gram-negative bacterial peptidoglycan. Science 2003, 300, 1584–1587. [Google Scholar] [CrossRef]
- Girardin, S.E.; Boneca, I.G.; Viala, J.; Chamaillard, M.; Labigne, A.; Thomas, G.; Philpott, D.J.; Sansonetti, P.J. NOD2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 2003, 278, 8869–8872. [Google Scholar] [CrossRef]
- Inohara, N.; Ogura, Y.; Fontalba, A.; Gutierrez, O.; Pons, F.; Crespo, J.; Fukase, K.; Inamura, S.; Kusumoto, S.; Hashimoto, M.; et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J. Biol. Chem. 2003, 278, 5509–5512. [Google Scholar] [CrossRef]
- Inohara, N.; Koseki, T.; Lin, J.; del Peso, L.; Lucas, P.C.; Chen, F.F.; Ogura, Y.; Nunez, G. An induced proximity model for NF-κB activation in the NOD1/RICK and RIP signaling pathways. J. Biol. Chem. 2000, 275, 27823–27831. [Google Scholar]
- Abbott, D.W.; Wilkins, A.; Asara, J.M.; Cantley, L.C. The Crohn’s disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr. Biol. 2004, 14, 2217–2227. [Google Scholar] [CrossRef]
- Da Silva Correia, J.; Miranda, Y.; Leonard, N.; Ulevitch, R. SGT1 is essential for NOD1 activation. Proc. Natl. Acad. Sci. USA 2007, 104, 6764–6769. [Google Scholar] [CrossRef]
- Pan, Q.; Kravchenko, V.; Katz, A.; Huang, S.; Ii, M.; Mathison, J.C.; Kobayashi, K.; Flavell, R.A.; Schreiber, R.D.; Goeddel, D.; et al. NF-κB-inducing kinase regulates selected gene expression in the NOD2 signaling pathway. Infect. Immun. 2006, 74, 2121–2127. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, Y.G.; Shaw, M.; Kanneganti, T.D.; Fujimoto, Y.; Fukase, K.; Inohara, N.; Nunez, G. NOD1/RICK and TLR signaling regulate chemokine and antimicrobial innate immune responses in mesothelial cells. J. Immunol. 2007, 179, 514–521. [Google Scholar]
- Kim, J.Y.; Omori, E.; Matsumoto, K.; Nunez, G.; Ninomiya-Tsuji, J. TAK1 is a central mediator of NOD2 signaling in epidermal cells. J. Biol. Chem. 2008, 283, 137–144. [Google Scholar]
- Schertzer, J.D.; Tamrakar, A.K.; Magalhaes, J.G.; Pereira, S.; Bilan, P.J.; Fullerton, M.D.; Liu, Z.; Steinberg, G.R.; Giacca, A.; Philpott, D.J.; et al. NOD1 activators link innate immunity to insulin resistance. Diabetes 2011, 60, 2206–2215. [Google Scholar] [CrossRef]
- Correa, R.G.; Milutinovic, S.; Reed, J.C. Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in innate immunity and inflammatory diseases. Biosci. Rep. 2012, 32, 597–608. [Google Scholar] [CrossRef]
- Tamrakar, A.K.; Schertzer, J.D.; Chiu, T.T.; Foley, K.P.; Bilan, P.J.; Philpott, D.J.; Klip, A. NOD2 activation induces muscle cell-autonomous innate immune responses and insulin resistance. Endocrinology 2010, 151, 5624–5637. [Google Scholar] [CrossRef]
- Schertzer, J.D.; Klip, A. Give a NOD to insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E585–E586. [Google Scholar] [CrossRef]
- Zhao, L.; Kwon, M.J.; Huang, S.; Lee, J.Y.; Fukase, K.; Inohara, N.; Hwang, D.H. Differential modulation of NODs signaling pathways by fatty acids in human colonic epithelial HCT116 cells. J. Biol. Chem. 2007, 282, 11618–11628. [Google Scholar]
- Guarda, G.; Zenger, M.; Yazdi, A.S.; Schroder, K.; Ferrero, I.; Menu, P.; Tardivel, A.; Mattmann, C.; Tschopp, J. Differential expression of NLRP3 among hematopoietic cells. J. Immunol. 2011, 186, 2529–2534. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Kanneganti, T.D.; Lamkanfi, M.; Kim, Y.G.; Chen, G.; Park, J.H.; Franchi, L.; Vandenabeele, P.; Nunez, G. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 2007, 26, 433–443. [Google Scholar] [CrossRef]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef]
- Wen, H.; Ting, J.P.; O’Neill, L.A. A role for the NLRP3 inflammasome in metabolic diseases—Did Warburg miss inflammation? Nat. Immunol. 2012, 13, 352–357. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Stienstra, R.; Joosten, L.A.; Koenen, T.; van Tits, B.; van Diepen, J.A.; van den Berg, S.A.; Rensen, P.C.; Voshol, P.J.; Fantuzzi, G.; Hijmans, A.; et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010, 12, 593–605. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar]
- Fischer, H.; Ellstrom, P.; Ekstrom, K.; Gustafsson, L.; Gustafsson, M.; Svanborg, C. Ceramide as a TLR4 agonist; a putative signalling intermediate between sphingolipid receptors for microbial ligands and TLR4. Cell Microbiol. 2007, 9, 1239–1251. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Watanabe, Y.; Nagai, Y.; Takatsu, K. Activation and Regulation of the Pattern Recognition Receptors in Obesity-Induced Adipose Tissue Inflammation and Insulin Resistance. Nutrients 2013, 5, 3757-3778. https://doi.org/10.3390/nu5093757
Watanabe Y, Nagai Y, Takatsu K. Activation and Regulation of the Pattern Recognition Receptors in Obesity-Induced Adipose Tissue Inflammation and Insulin Resistance. Nutrients. 2013; 5(9):3757-3778. https://doi.org/10.3390/nu5093757
Chicago/Turabian StyleWatanabe, Yasuharu, Yoshinori Nagai, and Kiyoshi Takatsu. 2013. "Activation and Regulation of the Pattern Recognition Receptors in Obesity-Induced Adipose Tissue Inflammation and Insulin Resistance" Nutrients 5, no. 9: 3757-3778. https://doi.org/10.3390/nu5093757