Duodenal Cytochrome b (DCYTB) in Iron Metabolism: An Update on Function and Regulation

Abstract
: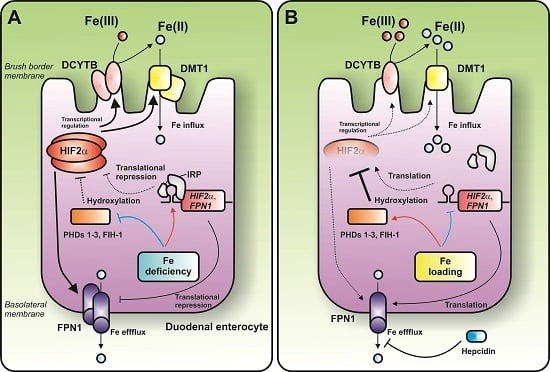
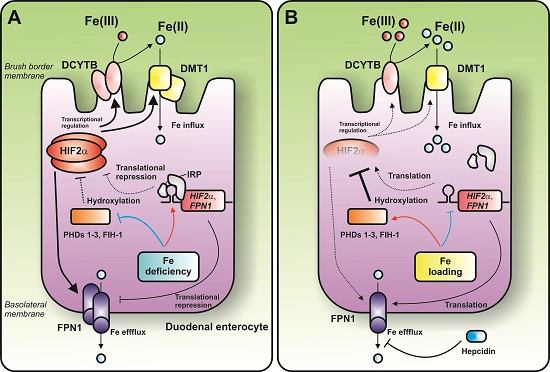{kind=link}
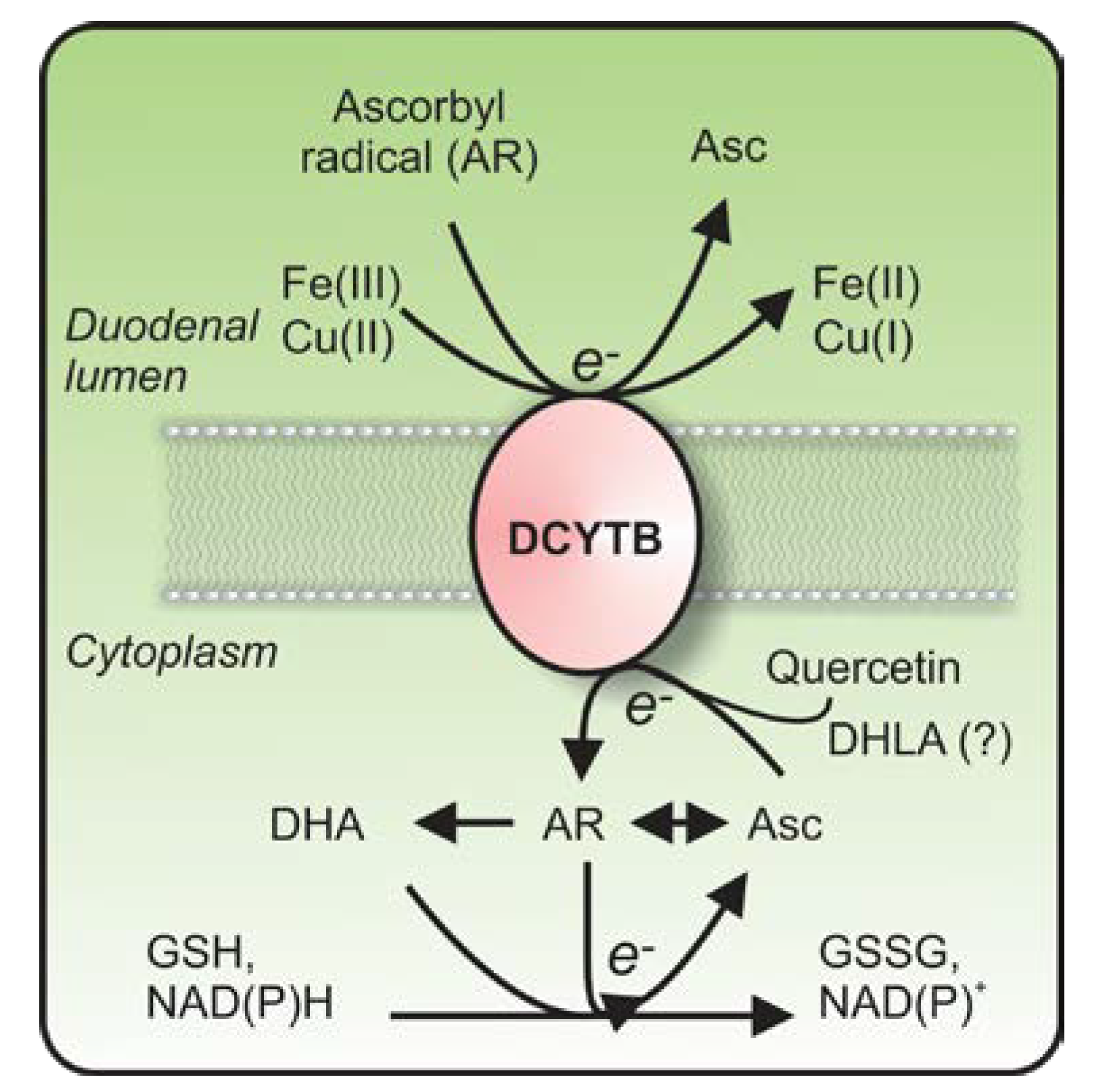{kind=link}
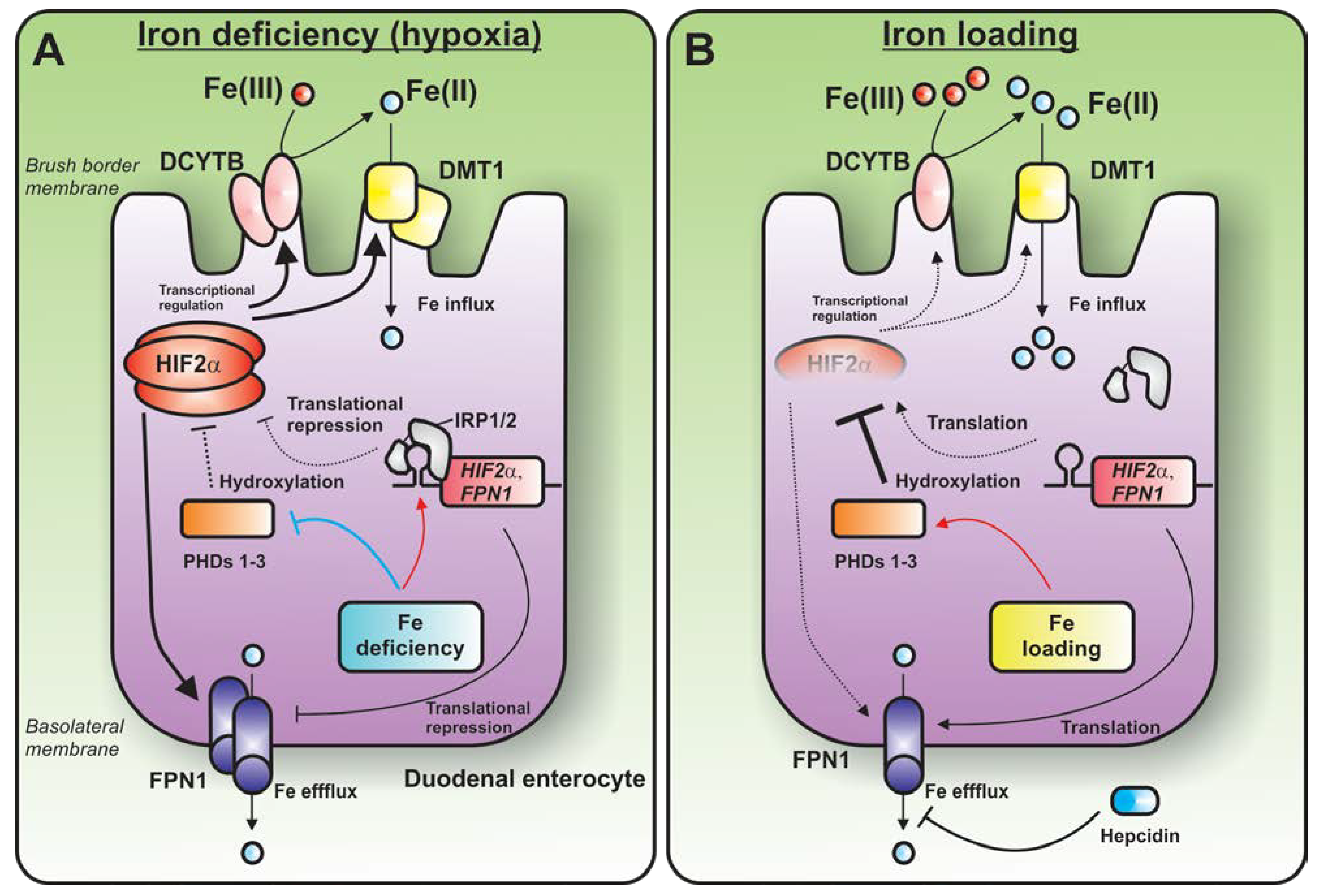{kind=link}
1. Introduction
1.1. Systemic Iron Homeostasis—An Overview
1.2. Dietary Iron Absorption—An Overview
2. DCYTB and Ascorbate: A Critical Link in Iron Metabolism?
2.1. The Cytochrome b561 Family and DCYTB
2.2. What Is the Identity of the Electron Donor for DCYTB?

2.3. DCYTB: Clarifying Its Role in Iron Metabolism
2.4. Does DCYTB Play a Role in the Reduction of Copper and Ascorbyl Radicals?
3. Coordination of Systemic and Dietary Iron Absorption: Role of the IRP1-HIF2α Axis
3.1. The IRP-IRE System
3.2. Emerging Role of the IRP1-HIF2α Axis—Regulation of Erythropoiesis
3.3. Emerging Role of the IRP1-HIF2α axis—Regulation of Duodenal Iron Absorption

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.L.; Rahmanto, Y.S.; Richardson, D.R. Iron uptake and metabolism in the new millennium. Trends Cell Biol. 2007, 17, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Lawen, A.; Lane, D.J.R. Mammalian iron homeostasis in health and disease: Uptake, storage, transport, and molecular mechanisms of action. Antioxid. Redox Signal. 2013, 18, 2473–2507. [Google Scholar] [CrossRef] [PubMed]
- Steinbicker, A.U.; Muckenthaler, M.U. Out of balance—Systemic iron homeostasis in iron-related disorders. Nutrients 2013, 5, 3034–3061. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Levi, S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta 2010, 1800, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Lill, R. Function and biogenesis of iron-sulphur proteins. Nature 2009, 460, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. Biogenesis of iron-sulfur clusters in mammalian cells: New insights and relevance to human disease. Dis. Model. Mech. 2012, 5, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Merlot, A.M.; Huang, M.L.-H.; Bae, D.-H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta. 2015, 1853, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.C.; Ryu, M.S. Special delivery: Distributing iron in the cytosol of mammalian cells. Front. Pharmacol. 2014, 5, 173. [Google Scholar] [CrossRef]
- Richardson, D.R.; Lane, D.J.R.; Becker, E.M.; Huang, M.L.; Whitnall, M.; Rahmanto, Y.S.; Sheftel, A.D.; Ponka, P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775–10782. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.; Mills, T.M.; Shafie, N.H.; Merlot, A.M.; Saleh Moussa, R.; Kalinowski, D.S.; Kovacevic, Z.; Richardson, D.R. Expanding horizons in iron chelation and the treatment of cancer: Role of iron in the regulation of ER stress and the epithelial-mesenchymal transition. Biochim. Biophys. Acta 2014, 1845, 166–181. [Google Scholar] [PubMed]
- Hentze, M.W.; Kuhn, L.C. Molecular control of vertebrate iron metabolism: MRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl. Acad. Sci. USA 1996, 93, 8175–8182. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Ponka, P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim. Biophys. Acta 1997, 1331, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Lawen, A. Ascorbate and plasma membrane electron transport—Enzymes vs. efflux. Free Radic. Biol. Med. 2009, 47, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Linster, C.L.; van Schaftingen, E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007, 274, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Atanassova, B.D.; Tzatchev, K.N. Ascorbic acid-important for iron metabolism. Folia Med. (Plovdiv) 2008, 50, 11–16. [Google Scholar]
- Toth, I.; Rogers, J.T.; McPhee, J.A.; Elliott, S.M.; Abramson, S.L.; Bridges, K.R. Ascorbic acid enhances iron-induced ferritin translation in human leukemia and hepatoma cells. J. Biol. Chem. 1995, 270, 2846–2852. [Google Scholar] [CrossRef] [PubMed]
- Bridges, K.R. Ascorbic acid inhibits lysosomal autophagy of ferritin. J. Biol. Chem. 1987, 262, 14773–14778. [Google Scholar] [PubMed]
- Hoffman, K.E.; Yanelli, K.; Bridges, K.R. Ascorbic acid and iron metabolism: Alterations in lysosomal function. Am. J. Clin. Nutr. 1991, 54, 1188S–1192S. [Google Scholar] [PubMed]
- Richardson, D.R. Role of ceruloplasmin and ascorbate in cellular iron release. J. Lab. Clin. Med. 1999, 134, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Lawen, A. Non-transferrin iron reduction and uptake are regulated by transmembrane ascorbate cycling in K562 cells. J. Biol. Chem. 2008, 283, 12701–12708. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Robinson, S.R.; Czerwinska, H.; Bishop, G.M.; Lawen, A. Two routes of iron accumulation in astrocytes: Ascorbate-dependent ferrous iron uptake via the divalent metal transporter (DMT1) plus an independent route for ferric iron. Biochem. J. 2010, 432, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Chua, A.C.; Graham, R.M.; Trinder, D.; Olynyk, J.K. The regulation of cellular iron metabolism. Crit. Rev. Clin. Lab. Sci. 2007, 44, 413–459. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: Expression and regulation. Biochim. Biophys. Acta 2012, 1823, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- De Domenico, I.; Ward, D.M.; Langelier, C.; Vaughn, M.B.; Nemeth, E.; Sundquist, W.I.; Ganz, T.; Musci, G.; Kaplan, J. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol. Biol. Cell 2007, 18, 2569–2578. [Google Scholar]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Peslova, G.; Petrak, J.; Kuzelova, K.; Hrdy, I.; Halada, P.; Kuchel, P.W.; Soe-Lin, S.; Ponka, P.; Sutak, R.; Becker, E.; et al. Hepcidin, the hormone of iron metabolism, is bound specifically to α-2-macroglobulin in blood. Blood 2009, 113, 6225–6236. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Austin, C.J.; Sari, M.A.; Suryo Rahmanto, Y.; Ponka, P.; Vyoral, D.; Richardson, D.R. Hepcidin bound to α2-macroglobulin reduces ferroportin-1 expression and enhances its activity at reducing serum iron levels. J. Biol. Chem. 2013, 288, 25450–25465. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Nemeth, E. Molecular liaisons between erythropoiesis and iron metabolism. Blood 2014, 124, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Gulec, S.; Anderson, G.J.; Collins, J.F. Mechanistic and regulatory aspects of intestinal iron absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G397–G409. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.J.; Darshan, D.; Wilkins, S.J.; Frazer, D.M. Regulation of systemic iron homeostasis: How the body responds to changes in iron demand. Biometals 2007, 20, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.A. Intestinal iron absorption: Regulation by dietary & systemic factors. Int. J. Vitam. Nutr. Res. 2010, 80, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Pollack, S.; Kaufman, R.; Crosby, W.H.; Butkiewicz, J.E. Reducing agents and absorption of iron. Nature 1963, 199, 384. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Asard, H.; Barbaro, R.; Trost, P.; Berczi, A. Cytochromes b561: Ascorbate-mediated trans-membrane electron transport. Antioxid. Redox Signal. 2013, 19, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T. The role of Dcytb in iron metabolism: An update. Biochem. Soc. Trans. 2008, 36, 1239–1241. [Google Scholar] [CrossRef] [PubMed]
- May, J.M.; Qu, Z.-C.; Mendiratta, S. Role of ascorbic acid in transferrin-independent reduction and uptake of iron by U-937 cells. Biochem. Pharmacol. 1999, 57, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Cakmak, I.; van de Wetering, D.A.M.; Marschner, H.; Bienfait, H.F. Involvement of superoxide radical in extracellular ferric reduction by iron-deficient bean roots. Plant Physiol. 1987, 85, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Ghio, A.J.; Nozik-Grayck, E.; Turi, J.; Jaspers, I.; Mercatante, D.R.; Kole, R.; Piantadosi, C.A. Superoxide-dependent iron uptake: A new role for anion exchange protein 2. Am. J. Respir. Cell Mol. Biol. 2003, 29, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Fleming, M.D.; Trenor, C.C., 3rd; Su, M.A.; Foernzler, D.; Beier, D.R.; Dietrich, W.F.; Andrews, N.C. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat. Genet. 1997, 16, 383–386. [Google Scholar] [PubMed]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Vulpe, C.D.; Kuo, Y.M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 1999, 21, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Attieh, Z.K.; Su, T.; Syed, B.A.; Gao, H.; Alaeddine, R.M.; Fox, T.C.; Usta, J.; Naylor, C.E.; Evans, R.W.; et al. Hephaestin is a ferroxidase that maintains partial activity in sex-linked anemia mice. Blood 2004, 103, 3933–3939. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.; Richardson, D.R. The active role of vitamin C in mammalian iron metabolism: Much more than just enhanced iron absorption! Biol. Med. 2014, 75C, 69–83. [Google Scholar]
- Lane, D.J.R.; Chikhani, S.; Richardson, V.; Richardson, D.R. Transferrin iron uptake is stimulated by ascorbate via an intracellular reductive mechanism. Biochim. Biophys. Acta 2013, 1833, 1527–1541. [Google Scholar] [CrossRef] [PubMed]
- Toth, I.; Bridges, K.R. Ascorbic acid enhances ferritin mRNA translation by an IRP/aconitase switch. J. Biol. Chem. 1995, 270, 19540–19544. [Google Scholar] [CrossRef] [PubMed]
- Scheers, N.; Sandberg, A.S. Iron transport through ferroportin is induced by intracellular ascorbate and involves IRP2 and HIF2α. Nutrients 2014, 6, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Asard, H. Three mammalian cytochromes b561 are ascorbate-dependent ferrireductases. FEBS J. 2006, 273, 3722–3734. [Google Scholar] [CrossRef] [PubMed]
- Fleming, P.J.; Kent, U.M. Cytochrome b561, ascorbic acid, and transmembrane electron transfer. Am. J. Clin. Nutr. 1991, 54, 1173S–1178S. [Google Scholar] [PubMed]
- Spiro, M.J.; Ball, E.G. Studies on the respiratory enzymes of the adrenal gland. I. The medulla. J. Biol. Chem. 1961, 236, 225–230. [Google Scholar] [PubMed]
- Flatmark, T.; Terland, O. Cytochrome b-561 of the bovine adrenal chromaffin granules. A high potential b-type cytochrome. Biochim. Biophys. Acta 1971, 253, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Terland, O.; Flatmark, T. Oxidoreductase activities of chromaffin granule ghosts isolated from the bovine adrenal medulla. Biochim. Biophys. Acta 1980, 597, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Yamano, T. Cytochrome 559 in the microsomes of the adrenal medulla. Biochem. Biophys. Res. Commun. 1965, 20, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Njus, D.; Knoth, J.; Cook, C.; Kelly, P.M. Electron transfer across the chromaffin granule membrane. J. Biol. Chem. 1983, 258, 27–30. [Google Scholar] [PubMed]
- Kelley, P.M.; Njus, D. Cytochrome b561 spectral changes associated with electron transfer in chromaffin-vesicle ghosts. J. Biol. Chem. 1986, 261, 6429–6432. [Google Scholar] [PubMed]
- Wakefield, L.M.; Cass, A.E.; Radda, G.K. Electron transfer across the chromaffin granule membrane. Use of EPR to demonstrate reduction of intravesicular ascorbate radical by the extravesicular mitochondrial NADH: Ascorbate radical oxidoreductase. J. Biol. Chem. 1986, 261, 9746–9752. [Google Scholar] [PubMed]
- Njus, D.; Kelley, P.M. The secretory-vesicle ascorbate-regenerating system: A chain of concerted H+/e−-transfer reactions. Biochim. Biophys. Acta 1993, 1144, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Jalukar, V.; Kelley, P.M.; Njus, D. Reaction of ascorbic acid with cytochrome b561. Concerted electron and proton transfer. J. Biol. Chem. 1991, 266, 6878–6882. [Google Scholar] [PubMed]
- Tsubaki, M.; Takeuchi, F.; Nakanishi, N. Cytochrome b561 protein family: Expanding roles and versatile transmembrane electron transfer abilities as predicted by a new classification system and protein sequence motif analyses. Biochim. Biophys. Acta 2005, 1753, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, N.; Takeuchi, F.; Tsubaki, M. Histidine cycle mechanism for the concerted proton/electron transfer from ascorbate to the cytosolic haem b centre of cytochrome b561: A unique machinery for the biological transmembrane electron transfer. J. Biochem. 2007, 142, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Ma, D.; Yan, C.; Gong, X.; Du, M.; Shi, Y. Structure and mechanism of a eukaryotic transmembrane ascorbate-dependent oxidoreductase. Proc. Natl. Acad. Sci. USA. 2014, 111, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-L.; Su, D.; Bérczi, A.; Vargas, A.; Asard, H. An ascorbate-reducible cytochrome b561 is localized in macrophage lysosomes. Biochim. Biophys. Acta 2006, 1760, 1903–1913. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.D.; Herpers, B.; McKie, A.T.; Gledhill, S.; McDonnell, J.; van den Heuvel, M.; Davies, K.E.; Ponting, C.P. Stromal cell-derived receptor 2 and cytochrome b561 are functional ferric reductases. Biochim. Biophys. Acta 2003, 1651, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Picco, C.; Scholz-Starke, J.; Naso, A.; Preger, V.; Sparla, F.; Trost, P.; Carpaneto, A. How are cytochrome B561 electron currents controlled by membrane voltage and substrate availability? Antioxid. Redox Signal. 2014, 21, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, A.; Sanuki, R.; Kakimoto, K.; Kojo, S.; Taketani, S. Involvement of 101F6, a homologue of cytochrome b561, in the reduction of ferric ions. J. Biochem. 2007, 142, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Recuenco, M.C.; Rahman, M.M.; Takeuchi, F.; Kobayashi, K.; Tsubaki, M. Electron transfer reactions of candidate tumor suppressor 101F6 protein, a cytochrome b561 homologue, with ascorbate and monodehydroascorbate radical. Biochemistry 2013, 52, 3660–3668. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.; Simpson, R.J.; McKie, A.T.; Sharp, P.A. Dcytb (Cybrd1) functions as both a ferric and a cupric reductase in vitro. FEBS Lett. 2008, 582, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Turi, J.L.; Wang, X.; McKie, A.T.; Nozik-Grayck, E.; Mamo, L.B.; Crissman, K.; Piantadosi, C.A.; Ghio, A.J. Duodenal cytochrome B: A novel ferrireductase in airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L272–L280. [Google Scholar] [CrossRef] [PubMed]
- Berczi, A.; Zimanyi, L.; Asard, H. Dihydrolipoic acid reduces cytochrome b561 proteins. Eur. Biophys. J. 2013, 42, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, J.M.; Qu, Z.-C.; Nelson, D.J. Uptake and reduction of α-lipoic acid by human erythrocytes. Clin. Biochem. 2007, 40, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.; Li, X.; Qu, Z.-C.; Perriott, L.; Whitesell, R.R.; May, J.M. Uptake, recycling, and antioxidant actions of α-lipoic acid in endothelial cells. Free Radic. Biol. Med. 2002, 33, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Latunde-Dada, G.O.; van der Westhuizen, J.; Vulpe, C.D.; Anderson, G.J.; Simpson, R.J.; McKie, A.T. Molecular and functional roles of duodenal cytochrome B (Dcytb) in iron metabolism. Blood Cells Mol. Dis. 2002, 29, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Oakhill, J.S.; Marritt, S.J.; Gareta, E.G.; Cammack, R.; McKie, A.T. Functional characterization of human duodenal cytochrome b (Cybrd1): Redox properties in relation to iron and ascorbate metabolism. Biochim. Biophys. Acta. 2008, 1777, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Latunde-Dada, G.O.; Simpson, R.J.; McKie, A.T. Duodenal cytochrome b expression stimulates iron uptake by human intestinal epithelial cells. J. Nutr. 2008, 138, 991–995. [Google Scholar] [PubMed]
- Vlachodimitropoulou, E.; Naftalin, R.J.; Sharp, P.A. Quercetin is a substrate for the transmembrane oxidoreductase Dcytb. Free Radic. Biol. Med. 2010, 48, 1366–1369. [Google Scholar] [CrossRef] [PubMed]
- Hertog, M.G.; Hollman, P.C.; Katan, M.B.; Kromhout, D. Intake of potentially anticarcinogenic flavonoids and their determinants in adults in The Netherlands. Nutr. Cancer 1993, 20, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Strobel, P.; Allard, C.; Perez-Acle, T.; Calderon, R.; Aldunate, R.; Leighton, F. Myricetin, quercetin and catechin-gallate inhibit glucose uptake in isolated rat adipocytes. Biochem. J. 2005, 386, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B.; Levine, M. Intracellular accumulation of ascorbic acid is inhibited by flavonoids via blocking of dehydroascorbic acid and ascorbic acid uptakes in HL-60, U937 and Jurkat cells. J. Nutr. 2000, 130, 1297–1302. [Google Scholar] [PubMed]
- Cunningham, P.; Afzal-Ahmed, I.; Naftalin, R.J. Docking studies show that l-glucose and quercetin slide through the transporter GLUT1. J. Biol. Chem. 2006, 281, 5797–5803. [Google Scholar] [CrossRef] [PubMed]
- Vlachodimitropoulou, E.; Sharp, P.A.; Naftalin, R.J. Quercetin-iron chelates are transported via glucose transporters. Free Radic. Biol. Med. 2011, 50, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Ludwiczek, S.; Rosell, F.I.; Ludwiczek, M.L.; Mauk, A.G. Recombinant expression and initial characterization of the putative human enteric ferric reductase Dcytb. Biochemistry 2008, 47, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Constantine, C.C.; Anderson, G.J.; Vulpe, C.D.; McLaren, C.E.; Bahlo, M.; Yeap, H.L.; Gertig, D.M.; Osborne, N.J.; Bertalli, N.A.; Beckman, K.B.; et al. A novel association between a SNP in CYBRD1 and serum ferritin levels in a cohort study of HFE hereditary haemochromatosis. Br. J. Haematol. 2009, 147, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Gunshin, H.; Starr, C.N.; Direnzo, C.; Fleming, M.D.; Jin, J.; Greer, E.L.; Sellers, V.M.; Galica, S.M.; Andrews, N.C. Cybrd1 (duodenal cytochrome b) is not necessary for dietary iron absorption in mice. Blood 2005, 106, 2879–2883. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, B.; Mudway, I.S.; Laftah, A.H.; Latunde-Dada, G.O.; McKie, A.T.; Peters, T.J.; Tzatchev, K.N.; Simpson, R.J. Duodenal ascorbate levels are changed in mice with altered iron metabolism. J. Nutr. 2004, 134, 501–505. [Google Scholar] [PubMed]
- Rathbone, B.J.; Johnson, A.W.; Wyatt, J.I.; Kelleher, J.; Heatley, R.V.; Losowsky, M.S. Ascorbic acid: A factor concentrated in human gastric juice. Clin. Sci. (Lond.) 1989, 76, 237–241. [Google Scholar]
- Atanasova, B.D.; Li, A.C.; Bjarnason, I.; Tzatchev, K.N.; Simpson, R.J. Duodenal ascorbate and ferric reductase in human iron deficiency. Am. J. Clin. Nutr. 2005, 81, 130–133. [Google Scholar] [PubMed]
- Frazer, D.M.; Wilkins, S.J.; Vulpe, C.D.; Anderson, G.J. The role of duodenal cytochrome b in intestinal iron absorption remains unclear. Blood 2005, 106, 4413. [Google Scholar] [CrossRef]
- Choi, J.; Masaratana, P.; Latunde-Dada, G.O.; Arno, M.; Simpson, R.J.; McKie, A.T. Duodenal reductase activity and spleen iron stores are reduced and erythropoiesis is abnormal in Dcytb knockout mice exposed to hypoxic conditions. J. Nutr. 2012, 142, 1929–1934. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; May, J.M.; Koury, M.J.; Asard, H. Human erythrocyte membranes contain a cytochrome b561 that may be involved in extracellular ascorbate recycling. J. Biol. Chem. 2006, 281, 39852–39859. [Google Scholar] [CrossRef] [PubMed]
- Kovar, J.; Neubauerova, J.; Cimburova, M.; Truksa, J.; Balusikova, K.; Horak, J. Stimulation of non-transferrin iron uptake by iron deprivation in K562 cells. Blood Cells Mol. Dis. 2006, 37, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Balusikova, K.; Neubauerova, J.; Dostalikova-Cimburova, M.; Horak, J.; Kovar, J. Differing expression of genes involved in non-transferrin iron transport across plasma membrane in various cell types under iron deficiency and excess. Mol. Cell. Biochem. 2009, 321, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Robinson, S.R.; Czerwinska, H.; Lawen, A. A role for Na+/H+ exchangers and intracellular pH in regulating vitamin C-driven electron transport across the plasma membrane. Biochem. J. 2010, 428, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; David, S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J. Biol. Chem. 2003, 278, 27144–27148. [Google Scholar] [CrossRef] [PubMed]
- Tulpule, K.; Robinson, S.R.; Bishop, G.M.; Dringen, R. Uptake of ferrous iron by cultured rat astrocytes. J. Neurosci. Res. 2010, 88, 563–571. [Google Scholar] [PubMed]
- Knöpfel, M.; Solioz, M. Characterization of a cytochrome b(558) ferric/cupric reductase from rabbit duodenal brush border membranes. Biochem. Biophys. Res. Commun. 2002, 291, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P. The molecular basis of copper and iron interactions. Proc. Nutr. Soc. 2004, 63, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Nose, Y.; Wood, L.K.; Kim, B.E.; Prohaska, J.R.; Fry, R.S.; Spears, J.W.; Thiele, D.J. Ctr1 is an apical copper transporter in mammalian intestinal epithelial cells in vivo that is controlled at the level of protein stability. J. Biol. Chem. 2010, 285, 32385–32392. [Google Scholar] [CrossRef] [PubMed]
- Arredondo, M.; Mendiburo, M.J.; Flores, S.; Singleton, S.T.; Garrick, M.D. Mouse divalent metal transporter 1 is a copper transporter in HEK293 cells. Biometals 2014, 27, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Garrick, M.D.; Garrick, L.M.; Zhao, L.; Collins, J.F. Divalent metal transporter 1 (Dmt1) mediates copper transport in the duodenum of iron-deficient rats and when overexpressed in iron-deprived HEK-293 cells. J. Nutr. 2013, 143, 1927–1933. [Google Scholar] [CrossRef] [PubMed]
- Matak, P.; Zumerle, S.; Mastrogiannaki, M.; el Balkhi, S.; Delga, S.; Mathieu, J.R.; Canonne-Hergaux, F.; Poupon, J.; Sharp, P.A.; Vaulont, S.; et al. Copper deficiency leads to anemia, duodenal hypoxia, upregulation of HIF-2α and altered expression of iron absorption genes in mice. PLoS ONE 2013, 8, e59538. [Google Scholar] [CrossRef]
- Xie, L.; Collins, J.F. Transcription factors Sp1 and Hif2α mediate induction of the copper-transporting ATPase (Atp7a) gene in intestinal epithelial cells during hypoxia. J. Biol. Chem. 2013, 288, 23943–23952. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Collins, J.F. Transcriptional regulation of the Menkes copper ATPase (Atp7a) gene by hypoxia-inducible factor (HIF2α) in intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 2011, 300, C1298–C1305. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P.; Beaumont, C.; Richardson, D.R. Function and regulation of transferrin and ferritin. Semin. Hematol. 1998, 35, 35–54. [Google Scholar] [PubMed]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Stehling, O.; Mascarenhas, J.; Vashisht, A.A.; Sheftel, A.D.; Niggemeyer, B.; Rosser, R.; Pierik, A.J.; Wohlschlegel, J.A.; Lill, R. Human CIA2A-FAM96A and CIA2B-FAM96B integrate iron homeostasis and maturation of different subsets of cytosolic-nuclear iron-sulfur proteins. Cell Metab. 2013, 18, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Phillips, J.D.; Yu, Y.; Leibold, E.A. Iron regulates the intracellular degradation of iron regulatory protein 2 by the proteasome. J. Biol. Chem. 1995, 270, 21645–21651. [Google Scholar] [CrossRef] [PubMed]
- Salahudeen, A.A.; Thompson, J.W.; Ruiz, J.C.; Ma, H.W.; Kinch, L.N.; Li, Q.; Grishin, N.V.; Bruick, R.K. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 2009, 326, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, A.A.; Zumbrennen, K.B.; Huang, X.; Powers, D.N.; Durazo, A.; Sun, D.; Bhaskaran, N.; Persson, A.; Uhlen, M.; Sangfelt, O.; et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 2009, 326, 718–721. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 2012, 1823, 1468–1483. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef]
- Zhang, D.L.; Ghosh, M.C.; Rouault, T.A. The physiological functions of iron regulatory proteins in iron homeostasis—An update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef]
- Meyron-Holtz, E.G.; Ghosh, M.C.; Iwai, K.; LaVaute, T.; Brazzolotto, X.; Berger, U.V.; Land, W.; Ollivierre-Wilson, H.; Grinberg, A.; Love, P.; et al. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004, 23, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Ferring-Appel, D.; Kaden, S.; Grone, H.J.; Hentze, M.W. Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell Metab. 2008, 7, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Ferring-Appel, D.; Sauer, S.W.; Kaden, S.; Lyoumi, S.; Puy, H.; Kolker, S.; Grone, H.J.; Hentze, M.W. Iron regulatory proteins secure mitochondrial iron sufficiency and function. Cell Metab. 2010, 12, 194–201. [Google Scholar] [CrossRef] [PubMed]
- LaVaute, T.; Smith, S.; Cooperman, S.; Iwai, K.; Land, W.; Meyron-Holtz, E.; Drake, S.K.; Miller, G.; Abu-Asab, M.; Tsokos, M.; et al. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat. Genet. 2001, 27, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Cooperman, S.S.; Meyron-Holtz, E.G.; Olivierre-Wilson, H.; Ghosh, M.C.; McConnell, J.P.; Rouault, T.A. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood 2005, 106, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Ferring, D.; Minana, B.; Bell, O.; Janser, H.G.; Muckenthaler, M.; Schumann, K.; Hentze, M.W. Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2). Blood 2005, 106, 2580–2589. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Pantopoulos, K. IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2α mRNA translation. Blood 2013, 122, 1658–1668. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.C.; Zhang, D.L.; Jeong, S.Y.; Kovtunovych, G.; Ollivierre-Wilson, H.; Noguchi, A.; Tu, T.; Senecal, T.; Robinson, G.; Crooks, D.R.; et al. Deletion of iron regulatory protein 1 causes polycythemia and pulmonary hypertension in mice through translational derepression of HIF2α. Cell Metab. 2013, 17, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.A.; Nizzi, C.P.; Chang, Y.I.; Deck, K.M.; Schmidt, P.J.; Galy, B.; Damnernsawad, A.; Broman, A.T.; Kendziorski, C.; Hentze, M.W.; et al. The IRP1-HIF-2α axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab. 2013, 17, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Cassavaugh, J.; Lounsbury, K.M. Hypoxia-mediated biological control. J. Cell Biochem. 2011, 112, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Mole, D.R. Iron homeostasis and its interaction with prolyl hydroxylases. Antioxid. Redox Signal. 2010, 12, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Bartz, S.; Mao, M.; Li, L.; Kaelin, W.G., Jr. The hypoxia-inducible factor 2α N-terminal and C-terminal transactivation domains cooperate to promote renal tumorigenesis in vivo. Mol. Cell. Biol. 2007, 27, 2092–2102. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood 2009, 114, 2015–2019. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, M.; Ebert, B.L.; Neil, C.; Brenner, K.; Papaioannou, I.; Melas, A.; Tolliday, N.; Lamb, J.; Pantopoulos, K.; Golub, T.; et al. Small-molecule inhibitors of HIF-2α translation link its 5ʼUTR iron-responsive element to oxygen sensing. Mol. Cell 2008, 32, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.; Galy, B.; Muckenthaler, M.U.; Hentze, M.W. Iron-regulatory proteins limit hypoxia-inducible factor-2alpha expression in iron deficiency. Nat. Struct. Mol. Biol. 2007, 14, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Shah, Y.M.; Matsubara, T.; Ito, S.; Yim, S.H.; Gonzalez, F.J. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. 2009, 9, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Mastrogiannaki, M.; Matak, P.; Keith, B.; Simon, M.C.; Vaulont, S.; Peyssonnaux, C. HIF-2α, but not HIF-1α, promotes iron absorption in mice. J. Clin. Invest. 2009, 119, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Qu, A.; Anderson, E.R.; Matsubara, T.; Martin, A.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-inducible factor-2α mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology 2011, 140, 2044–2055. [Google Scholar] [CrossRef] [PubMed]
- Frazer, D.M.; Wilkins, S.J.; Becker, E.M.; Murphy, T.L.; Vulpe, C.D.; McKie, A.T.; Anderson, G.J. A rapid decrease in the expression of DMT1 and Dcytb but not Ireg1 or hephaestin explains the mucosal block phenomenon of iron absorption. Gut 2003, 52, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Hahn, P.F.; Bale, W.F.; Ross, J.F.; Balfour, W.M.; Whipple, G.H. Radioactive iron absorption by gastro-intestinal tract: Influence of Anemia, Anoxia, and Antecedent Feeding Distribution in Growing Dogs. J. Exp. Med. 1943, 78, 169–188. [Google Scholar] [CrossRef]
- Stewart, W.B.; Yuile, C.L.; Claiborne, H.A.; Snowman, R.T.; Whipple, G.H. Radioiron absorption in anemic dogs; fluctuations in the mucosal block and evidence for a gradient of absorption in the gastrointestinal tract. J. Exp. Med. 1950, 92, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Mastrogiannaki, M.; Matak, P.; Peyssonnaux, C. The gut in iron homeostasis: Role of HIF-2 under normal and pathological conditions. Blood 2013, 122, 885–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, R.J.; McKie, A.T. Regulation of intestinal iron absorption: The mucosa takes control? Cell Metab. 2009, 10, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Latunde-Dada, G.O.; Xiang, L.; Simpson, R.J.; McKie, A.T. Duodenal cytochrome b (Cybrd 1) and HIF-2α expression during acute hypoxic exposure in mice. Eur. J. Nutr. 2011, 50, 699–704. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lane, D.J.R.; Bae, D.-H.; Merlot, A.M.; Sahni, S.; Richardson, D.R. Duodenal Cytochrome b (DCYTB) in Iron Metabolism: An Update on Function and Regulation. Nutrients 2015, 7, 2274-2296. https://doi.org/10.3390/nu7042274
Lane DJR, Bae D-H, Merlot AM, Sahni S, Richardson DR. Duodenal Cytochrome b (DCYTB) in Iron Metabolism: An Update on Function and Regulation. Nutrients. 2015; 7(4):2274-2296. https://doi.org/10.3390/nu7042274
Chicago/Turabian StyleLane, Darius J. R., Dong-Hun Bae, Angelica M. Merlot, Sumit Sahni, and Des R. Richardson. 2015. "Duodenal Cytochrome b (DCYTB) in Iron Metabolism: An Update on Function and Regulation" Nutrients 7, no. 4: 2274-2296. https://doi.org/10.3390/nu7042274