Molecular Basis for Vitamin A Uptake and Storage in Vertebrates

Abstract
:1. Introduction
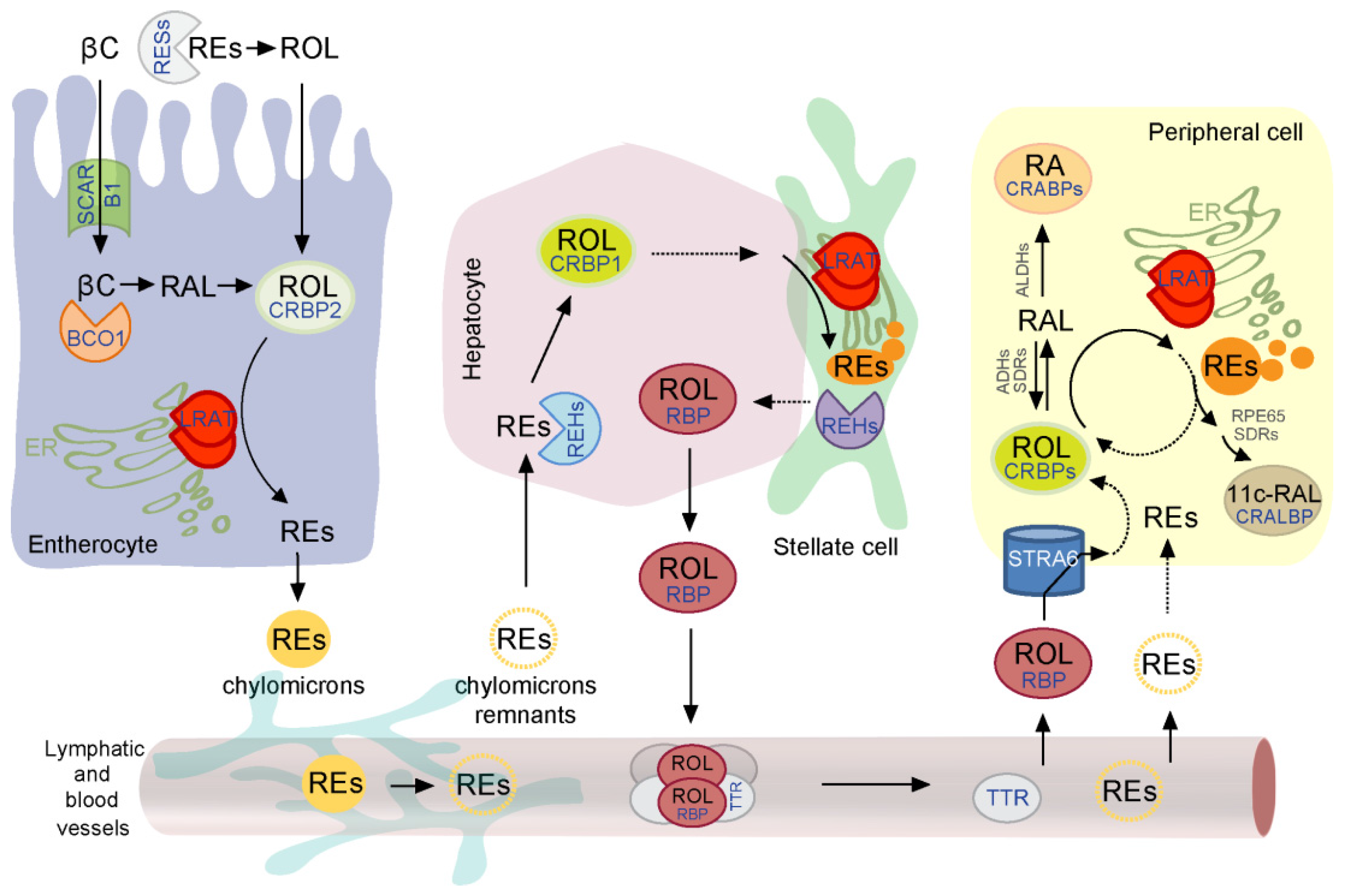
2. Overview of the Retinoid Absorption, Storage, and Transport to the Peripheral Tissues
2.1. Role of Vitamin A Esterification in the Intestinal Uptake
2.2. LRAT Activity in Hepatic Retinoid Metabolism
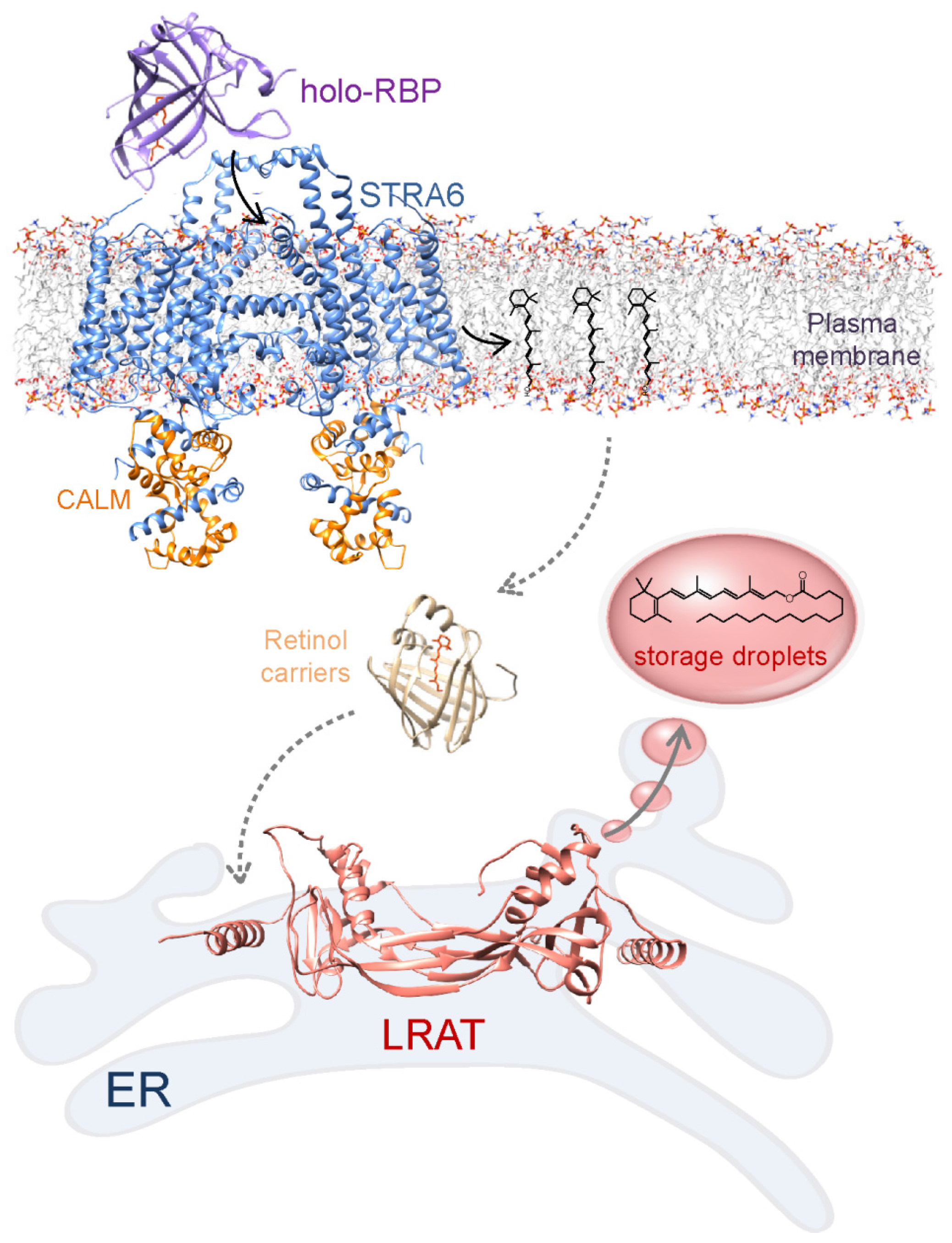
2.3. Mobilization of Retinol from the Liver Storage and LRAT-Driven Uptake by the Extrahepatic Tissue
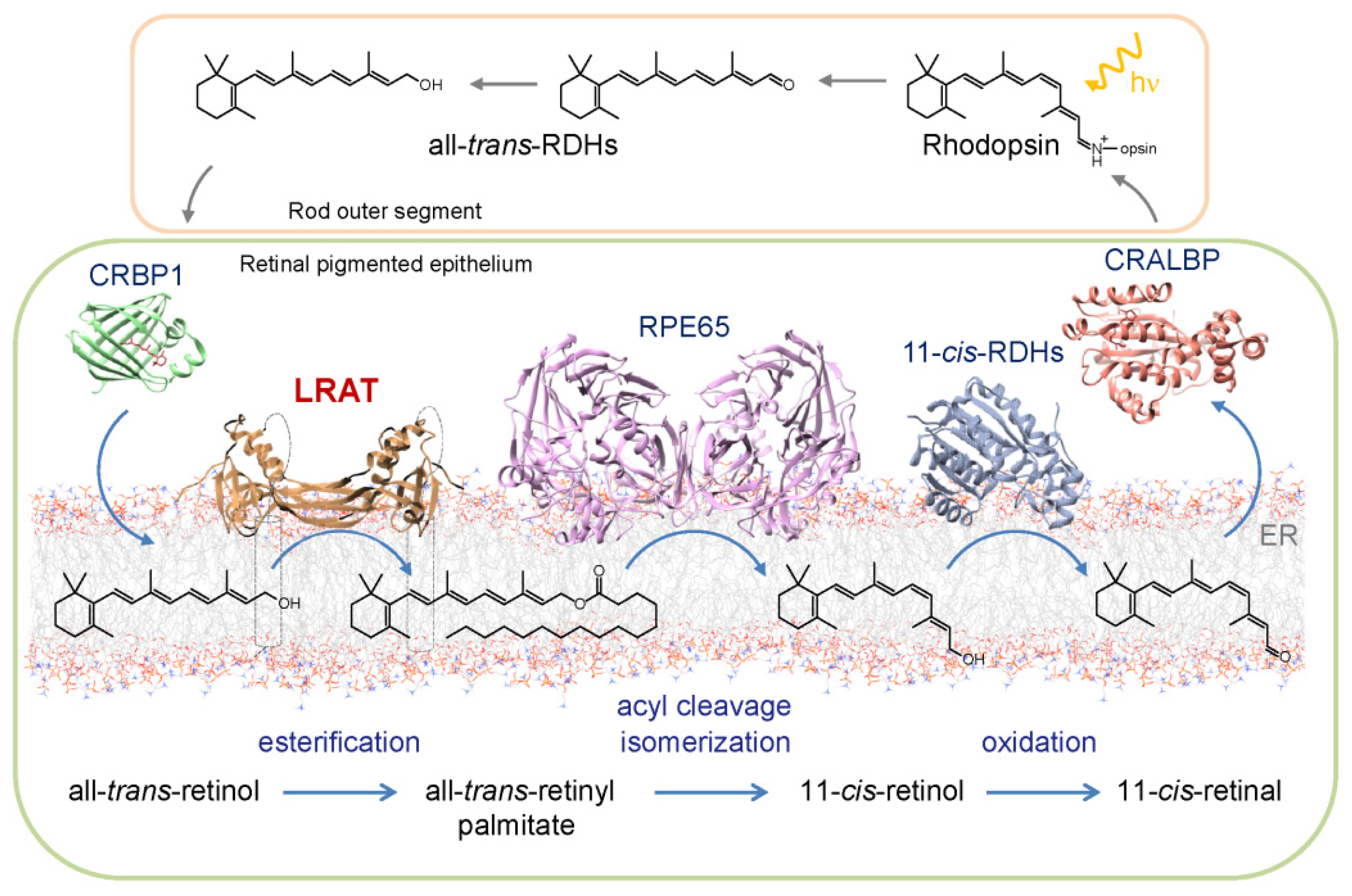
3. Role of REs in the Ocular Metabolism of Retinoids
4. Insight into Molecular Adaptations that Enable LRAT Activity in the Vertebrates
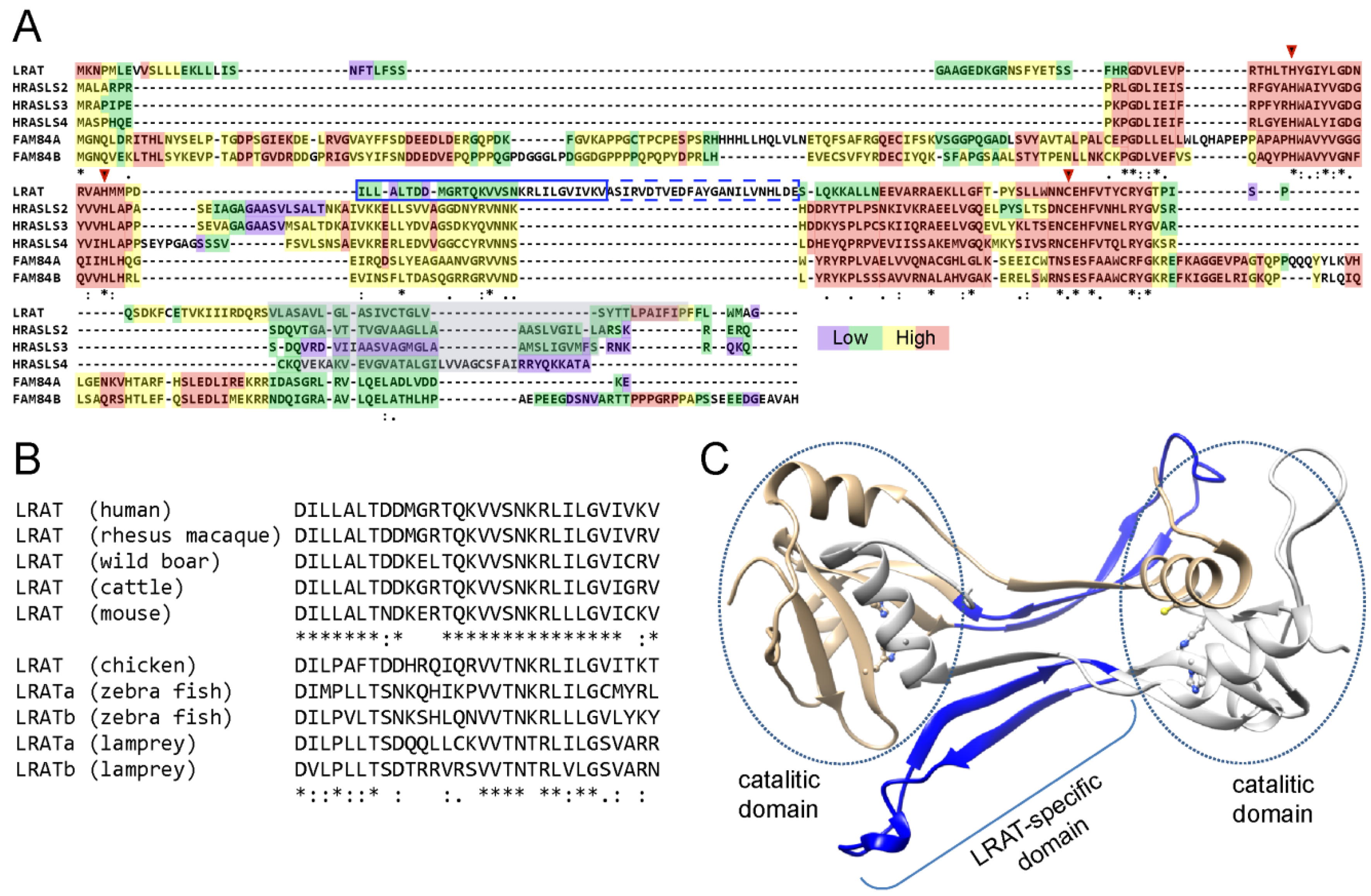
4.1. Evolutional Diversity of N1pC/P60 Protein Family
4.2. Biochemical Properties and Enzymatic Activities of N1pC/P60 Proteins
4.3. Structural Insight into Functional Adaptation of LRAT
5. Mutations in LRAT Gene and Their Clinical Manifestations
5.1. Role of LRAT in the General Retinoid Homeostasis
5.2. Synthesis of REs Incorporated into Milk
5.3. Eye Phenotype in LRAT-Deficiencies
5.4. Clinical Manifestation of Mutations in LRAT Gene
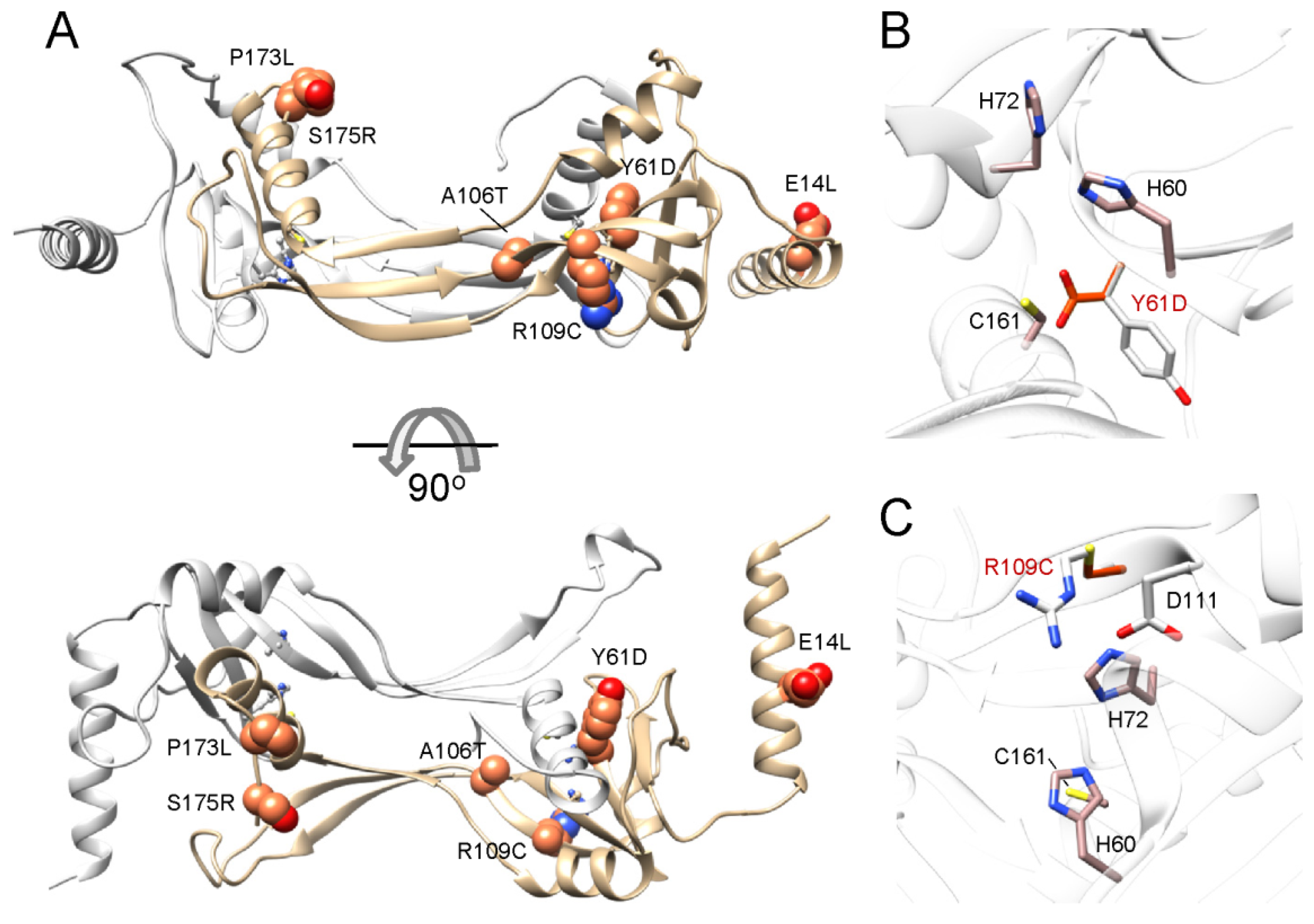
5.5. Impact of Retinal Diseases-Associated LRAT Mutations on Structure and Activity of the Enzyme
6. Conclusions and Future Directions
- (a)
- Concentration of retinol in the blood correlates with vitamin A status of the organism only in cases of depletion or over saturation of the liver storage. Between these two extremes vitamin A level is homeostatic. What is the signaling pathway in which vitamin A concentration in the blood regulates mobilization of retinoid pool in the liver? Is this process controlled on the transcription level or maybe involves allosteric regulation of enzymes involved in retinyl esters hydrolysis/formation?
- (b)
- A related unsolved problem is how retinol is trafficked from hepatocytes to lipid storage droplets in HSCs. In addition, unclear is the enzymatic machinery responsible for mobilization of retinyl esters storage and how it is regulated.
- (c)
- Uptake of vitamin A to peripheral tissue is also a controlled process. For example, excess of all-trans-retinol does not cause disproportionate accumulation of retinyl esters in the RPE. However, in vitamin A deficient states the ocular retinoid uptake is favored over other tissues [71,77]. Although the pivotal role of STRA6 and LRAT in vitamin A uptake is unquestionable, it seems that transcriptional regulation of STRA6 and LRAT expression by retinoic acid is not the only mechanism that governs “buffering” of vitamin A within the eye or other tissue. The components of this signaling pathway remain to be uncovered.
- (d)
- One of the contemplated biochemical questions in retinoid metabolism is the cone-specific visual chromophore regeneration pathway. Alterations in retinoid composition between nocturnal and diurnal animals as well as electrophysiological differences in rod and cones responses suggest the existence of an alternative to the RPE65 source of 11-cis-retinal that supports cone function. Although putative enzymes involved in this pathway have been identified [146,147], their physiological functions remain to be verified in in vivo models.
- (e)
- Despite wealth of biochemical and structural data, it is still unknown what triggers controlled release of retinol from retinol-binding proteins. Vitamin A needs to be transported from the plasma membrane or its storage sites to specific compartments within the cell. It is clear that this cannot be a stochastic event but rather a controlled process facilitated by specific interactions of retinol-binding proteins with phospholipids or protein binding partners, which are still unknown.
- (f)
- From the clinical point of view, the successful fight against vitamin A deficiency depends on reliable methods of assessing retinoid status of individuals. Unfortunately, serum retinol is not useful for this purpose. Thus, development of dependable and inexpensive diagnostic tests based on biomarkers other than serum retinol would have tremendous impact on assessing the prevalence, prevention, and treatment of vitamin A deficiencies.
Acknowledgments
Conflicts of Interest
Abbreviations
| BCO1 | β,β-carotene-15,15-dioxygenase |
| CRALBP | cellular retinaldehyde-binding protein |
| CRBP1 | cellular retinol-binding protein 1 |
| CRBP2 | cellular retinol-binding protein 2 |
| CYP26A1 | cytochrome P450 family 26 subfamily A member 1 |
| DGAT1 | diacylglycerol O-acyltransferase 1 |
| ER | endoplasmic reticulum |
| ISX | intestinal specific homeodomain transcriptional factor |
| HRASLS2 | HRAS-like tumor suppressor 2 |
| HRASLS3 | HRAS-like tumor suppressor 3 |
| HSCs | hepatic stellate cells |
| LCA | leber congenital amaurosis |
| LRAT | lecithin:retinol acyltransferase |
| RARs | retinoic acid receptors |
| RBP | serum retinol-binding protein |
| RDH | retinol dehydrogenase |
| REs | retinyl esters |
| RPE | retinal pigmented epithelium |
| RPE65 | retinal pigment epithelium-specific 65 kDa protein or retinoid isomerase |
| RXRs | retinoid X receptors |
| SCARB1 | scavenger receptor class B, type I |
| STRA6 | stimulated by retinoic acid 6 |
| TTR | transthyretin |
| WT | wild-type |
References
- Gudas, L.J. Emerging roles for retinoids in regeneration and differentiation in normal and disease states. Biochim. Biophys. Acta 2012, 1821, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Al Tanoury, Z.; Piskunov, A.; Rochette-Egly, C. Vitamin A and retinoid signaling: Genomic and nongenomic effects. J. Lipid Res. 2013, 54, 1761–1775. [Google Scholar] [CrossRef] [PubMed]
- Kiser, P.D.; Golczak, M.; Palczewski, K. Chemistry of the retinoid (visual) cycle. Chem. Rev. 2014, 114, 194–232. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.C. Vitamin A and retinoic acid in T cell-related immunity. Am. J. Clin. Nutr. 2012, 96, 1166S–1172S. [Google Scholar] [CrossRef] [PubMed]
- Mayo-Wilson, E.; Imdad, A.; Herzer, K.; Yakoob, M.Y.; Bhutta, Z.A. Vitamin A supplements for preventing mortality, illness, and blindness in children aged under 5: Systematic review and meta-analysis. BMJ 2011, 343, d5094. [Google Scholar] [CrossRef] [PubMed]
- Clagett-Dame, M.; Knutson, D. Vitamin A in reproduction and development. Nutrients 2011, 3, 385–428. [Google Scholar] [CrossRef] [PubMed]
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996, 10, 940–954. [Google Scholar] [PubMed]
- Kedishvili, N.Y. Enzymology of retinoic acid biosynthesis and degradation. J. Lipid Res. 2013, 54, 1744–1760. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, M.; Dolle, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Repa, J.J.; Evans, R.M.; Mangelsdorf, D.J. Nuclear receptors and lipid physiology: Opening the x-files. Science 2001, 294, 1866–1870. [Google Scholar] [CrossRef] [PubMed]
- Balmer, J.E.; Blomhoff, R. Gene expression regulation by retinoic acid. J. Lipid Res. 2002, 43, 1773–1808. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.H.; Gudas, L.J. Retinoids, retinoic acid receptors, and cancer. Annu. Rev. Pathol. 2011, 6, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Penniston, K.L.; Tanumihardjo, S.A. The acute and chronic toxic effects of Vitamin A. Am. J. Clin. Nutr. 2006, 83, 191–201. [Google Scholar] [PubMed]
- Mulder, G.B.; Manley, N.; Grant, J.; Schmidt, K.; Zeng, W.; Eckhoff, C.; Maggio-Price, L. Effects of excess Vitamin A on development of cranial neural crest-derived structures: A neonatal and embryologic study. Teratology 2000, 62, 214–226. [Google Scholar] [CrossRef]
- Stephensen, C.B. Vitamin A, infection, and immune function. Annu. Rev. Nutr. 2001, 21, 167–192. [Google Scholar] [CrossRef] [PubMed]
- Grotto, I.; Mimouni, M.; Gdalevich, M.; Mimouni, D. Vitamin A supplementation and childhood morbidity from diarrhea and respiratory infections: A meta-analysis. J. Pediatr. 2003, 142, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A. Vitamin A deficiency and clinical disease: An historical overview. J. Nutr. 2008, 138, 1835–1839. [Google Scholar] [PubMed]
- Bhutta, Z.A.; Haider, B.A.; Cousens, S.; Kirkwood, B.R.; Black, R.E. Neonatal Vitamin A supplementation and infant survival in Asia—Reply. Lancet 2008, 371, 1746–1748. [Google Scholar] [CrossRef]
- Black, R.E.; Allen, L.H.; Bhutta, Z.A.; Caulfield, L.E.; de Onis, M.; Ezzati, M.; Mathers, C.; Rivera, J. Maternal and child undernutrition 1—Maternal and child undernutrition: Global and regional exposures and health consequences. Lancet 2008, 371, 243–260. [Google Scholar] [CrossRef]
- Von Lintig, J. Provitamin a metabolism and functions in mammalian biology. Am. J. Clin. Nutr. 2012, 96, 1234s–1244s. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, H.H.; Poor, C.L.; Wellman, R.B.; Erdman, J.W., Jr. Concentrations of selected carotenoids and Vitamin A in human liver, kidney and lung tissue. J. Nutr. 1991, 121, 1613–1621. [Google Scholar] [PubMed]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.F.; Schwabe, R.F.; Hillman, E.M.C.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. BBA Mol. Cell Biol. Lipids 2009, 1791, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Schindler, R.; Friedrich, D.; Kramer, M.; Wacker, H.H.; Feldheim, W. Size and composition of liver vitamin A reserves of human-beings who died of various causes. Int. J. Vitam. Nutr. Res. 1988, 58, 146–154. [Google Scholar] [PubMed]
- Batten, M.L.; Imanishi, Y.; Maeda, T.; Tu, D.C.; Moise, A.R.; Bronson, D.; Possin, D.; Van Gelder, R.N.; Baehr, W.; Palczewski, K. Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J. Biol. Chem. 2004, 279, 10422–10432. [Google Scholar] [CrossRef] [PubMed]
- Goodman, D.S. Overview of current knowledge of metabolism of vitamin A and carotenoids. J. Natl. Cancer Inst. 1984, 73, 1375–1379. [Google Scholar] [PubMed]
- Weng, W.; Li, L.; van Bennekum, A.M.; Potter, S.H.; Harrison, E.H.; Blaner, W.S.; Breslow, J.L.; Fisher, E.A. Intestinal absorption of dietary cholesteryl ester is decreased but retinyl ester absorption is normal in carboxyl ester lipase knockout mice. Biochemistry 1999, 38, 4143–4149. [Google Scholar] [CrossRef] [PubMed]
- Reboul, E.; Berton, A.; Moussa, M.; Kreuzer, C.; Crenon, I.; Borel, P. Pancreatic lipase and pancreatic lipase-related protein 2, but not pancreatic lipase-related protein 1, hydrolyze retinyl palmitate in physiological conditions. BBA Mol. Cell Biol. Lipids 2006, 1761, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Van Bennekum, A.M.; Fisher, E.A.; Blaner, W.S.; Harrison, E.H. Hydrolysis of retinyl esters by pancreatic triglyceride lipase. Biochemistry 2000, 39, 4900–4906. [Google Scholar] [CrossRef] [PubMed]
- Voolstra, O.; Kiefer, C.; Hoehne, M.; Welsch, R.; Vogt, K.; von Lintig, J. The drosophila class B scavenger receptor NinaD-I is a cell surface receptor mediating carotenoid transport for visual chromophore synthesis. Biochemistry 2006, 45, 13429–13437. [Google Scholar] [CrossRef] [PubMed]
- Van Bennekum, A.; Werder, M.; Thuahnai, S.T.; Han, C.H.; Duong, P.; Williams, D.L.; Wettstein, P.; Schulthess, G.; Phillips, M.C.; Hauser, H. Class B scavenger receptor-mediated intestinal absorption of dietary β-carotene and cholesterol. Biochemistry 2005, 44, 4517–4525. [Google Scholar] [CrossRef] [PubMed]
- Lobo, G.P.; Hessel, S.; Eichinger, A.; Noy, N.; Moise, A.R.; Wyss, A.; Palczewski, K.; von Lintig, J. ISX is a retinoic acid-sensitive gatekeeper that controls intestinal β,β-carotene absorption and Vitamin A production. FASEB J. 2010, 24, 1656–1666. [Google Scholar] [CrossRef] [PubMed]
- Widjaja-Adhi, M.A.K.; Lobo, G.P.; Golczak, M.; Von Lintig, J. A genetic dissection of intestinal fat-soluble vitamin and carotenoid absorption. Hum. Mol. Genet. 2015, 24, 3206–3219. [Google Scholar] [CrossRef] [PubMed]
- Lobo, G.P.; Amengual, J.; Baus, D.; Shivdasani, R.A.; Taylor, D.; von Lintig, J. Genetics and diet regulate Vitamin A production via the homeobox transcription factor ISX. J. Biol. Chem. 2013, 288, 9017–9027. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Demmer, L.A.; Sweetser, D.A.; Ong, D.E.; Gordon, J.I. Rat cellular retinol-binding protein II—Use of a cloned cDNA to define its primary structure, tissue-specific expression, and developmental regulation. Proc. Natl. Acad. Sci. USA 1986, 83, 5779–5783. [Google Scholar] [CrossRef] [PubMed]
- Crow, J.A.; Ong, D.E. Cell-specific immunohistochemical localization of a cellular retinol-binding protein (type-2) in the small-intestine of rat. Proc. Natl. Acad. Sci. USA 1985, 82, 4707–4711. [Google Scholar] [CrossRef] [PubMed]
- Wongsiriroj, N.; Piantedosi, R.; Palczewski, K.; Goldberg, I.J.; Johnston, T.P.; Li, E.; Blaner, W.S. The molecular basis of retinoid absorption—A genetic dissection. J. Biol. Chem. 2008, 283, 13510–13519. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, P.N.; Ong, D.E. A lecithin—Retinol acyltransferase activity in human and rat-liver. Biochem. Biophys. Res. Commun. 1988, 156, 157–163. [Google Scholar] [CrossRef]
- Ruiz, A.; Winston, A.; Lim, Y.H.; Gilbert, B.A.; Rando, R.R.; Bok, D. Molecular and biochemical characterization of lecithin retinol acyltransferase. J. Biol. Chem. 1999, 274, 3834–3841. [Google Scholar] [CrossRef] [PubMed]
- Golczak, M.; Palczewski, K. An acyl-covalent enzyme intermediate of lecithin:retinol acyltransferase. J. Biol. Chem. 2010, 285, 29217–29222. [Google Scholar] [CrossRef] [PubMed]
- Orland, M.D.; Anwar, K.; Cromley, D.; Chu, C.H.; Chen, L.P.; Billheimer, J.T.; Hussain, M.M.; Cheng, D. Acyl coenzyme A dependent retinol esterification by acyl coenzyme A: Diacylglycerol acyltransferase 1. BBA Mol. Cell Biol. Lipids 2005, 1737, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Golczak, M.; Imanishi, Y.; Kuksa, V.; Maeda, T.; Kubota, R.; Palczewski, K. Lecithin:retinol acyltransferase is responsible for amidation of retinylamine, a potent inhibitor of the retinoid cycle. J. Biol. Chem. 2005, 280, 42263–42273. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Tang, H.; Seibel, W.; Papoian, R.; Oh, K.; Li, X.Y.; Zhang, J.Y.; Golczak, M.; Palczewski, K.; Kiser, P.D. Identification and characterization of novel inhibitors of mammalian aspartyl aminopeptidase. Mol. Pharmacol. 2014, 86, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.L.E.; Monetti, M.; Burri, B.J.; Farese, R.V. The triacylglycerol synthesis enzyme DGAT1 also catalyzes the synthesis of diacylglycerols, waxes, and retinyl esters. J. Lipid Res. 2005, 46, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.; Goodman, D.S.; Karmen, A. Fatty acid esterification and chylomicron formation during fat absorption in rat: III. Positional relations in triglycerides and lecithin. J. Lipid Res. 1965, 6, 233–240. [Google Scholar] [PubMed]
- Blomhoff, R.; Helgerud, P.; Rasmussen, M.; Berg, T.; Norum, K.R. Invivo uptake of chylomicron (h-3)-labeled retinyl ester by rat-liver—Evidence for retinol transfer from parenchymal to non-parenchymal cells. Proc. Natl. Acad. Sci. Biol. 1982, 79, 7326–7330. [Google Scholar] [CrossRef]
- Rohlmann, A.; Gotthardt, M.; Hammer, R.E.; Herz, J. Inducible inactivation of hepatic LRP gene by cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J. Clin. Investig. 1998, 101, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Harrison, E.H.; Gad, M.Z.; Ross, A.C. Hepatic-uptake and metabolism of chylomicron retinyl esters—Probable role of plasma-membrane endosomal retinyl ester hydrolases. J. Lipid Res. 1995, 36, 1498–1506. [Google Scholar] [PubMed]
- Blaner, W.S.; Prystowsky, J.H.; Smith, J.E.; Goodman, D.S. Rat-liver retinyl palmitate hydrolase activity—Relationship to cholesteryl oleate and triolein hydrolase activities. Biochim. Biophys. Acta 1984, 794, 419–427. [Google Scholar] [CrossRef]
- van Bennekum, A.M.; Li, L.; Piantedosi, R.; Shamir, R.; Vogel, S.; Fisher, E.A.; Blaner, W.S.; Harrison, E.H. Carboxyl ester lipase overexpression in rat hepatoma cells and CEL deficiency in mice have no impact on hepatic uptake or metabolism of chylomicron-retinyl ester. Biochemistry 1999, 38, 4150–4156. [Google Scholar] [CrossRef] [PubMed]
- Harrison, E.H. Lipases and carboxylesterases: Possible roles in the hepatic utilization of Vitamin A. J. Nutr. 2000, 130, 340s–344s. [Google Scholar] [PubMed]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A metabolism: An update. Nutrients 2011, 3, 63–103. [Google Scholar] [CrossRef] [PubMed]
- Batres, R.O.; Olson, J.A. Distribution of vitamin A among parenchymal and stellate cells of livers of rats with low vitamin A stores. Fed. Proc. 1987, 46, 1191–1191. [Google Scholar]
- Moriwaki, H.; Blaner, W.S.; Piantedosi, R.; Goodman, D.S. Effects of dietary retinoid and triglyceride on the lipid-composition of rat-liver stellate cells and stellate cell lipid droplets. J. Lipid Res. 1988, 29, 1523–1534. [Google Scholar] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Blaner, W.S.; Hendriks, H.F.J.; Brouwer, A.; Deleeuw, A.M.; Knook, D.L.; Goodman, D.S. Retinoids, retinoid-binding proteins, and retinyl palmitate hydrolase distributions in different types of rat-liver cells. J. Lipid Res. 1985, 26, 1241–1251. [Google Scholar] [PubMed]
- Nagy, N.E.; Holven, K.B.; Roos, N.; Senoo, H.; Kojima, N.; Norum, K.R.; Blomhoff, R. Storage of Vitamin A in extrahepatic stellate cells in normal rats. J. Lipid Res. 1997, 38, 645–658. [Google Scholar] [PubMed]
- Senoo, H.; Kojima, N.; Sato, M. Vitamin A-storing cells (stellate cells). Vitam. Horm. 2007, 75, 131–159. [Google Scholar] [PubMed]
- Liu, L.M.; Gudas, L.J. Disruption of the lecithin:retinol acyltransferase gene makes mice more susceptible to Vitamin A deficiency. J. Biol. Chem. 2005, 280, 40226–40234. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, S.M.; Wongsiriroj, N.; Libien, J.; Vogel, S.; Goldberg, I.J.; Baehr, W.; Palczewski, K.; Blaner, W.S. Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT). J. Biol. Chem. 2005, 280, 35647–35657. [Google Scholar] [CrossRef] [PubMed]
- Alexson, S.E.H.; Mentlein, R.; Wernstedt, C.; Hellman, U. Isolation and characterization of microsomal acyl-CoA thioesterase—A member of the rat-liver microsomal carboxylesterase multigene family. Eur. J. Biochem. 1993, 214, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Mentlein, R.; Heymann, E. Hydrolysis of retinyl esters by nonspecific carboxylesterases from rat-liver endoplasmic-reticulum. Biochem. J. 1987, 245, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.S.; Alexson, S.E.H.; Harrison, E.H. Purification and characterization of a neutral, bile salt-independent retinyl ester hydrolase from rat liver microsomes—Relationship to rat carboxylesterase es-2. J. Biol. Chem. 1997, 272, 24488–24493. [Google Scholar] [CrossRef] [PubMed]
- Episkopou, V.; Maeda, S.; Nishiguchi, S.; Shimada, K.; Gaitanaris, G.A.; Gottesman, M.E.; Robertson, E.J. Disruption of the transthyretin gene results in mice with depressed levels of plasma retinol and thyroid-hormone. Proc. Natl. Acad. Sci. USA 1993, 90, 2375–2379. [Google Scholar] [CrossRef] [PubMed]
- Quadro, L.; Blaner, W.S.; Salchow, D.J.; Vogel, S.; Piantedosi, R.; Gouras, P.; Freeman, S.; Cosma, M.P.; Colantuoni, V.; Gottesman, M.E. Impaired retinal function and Vitamin A availability in mice lacking retinol-binding protein. EMBO J. 1999, 18, 4633–4644. [Google Scholar] [CrossRef] [PubMed]
- Biesalski, H.K.; Frank, J.; Beck, S.C.; Heinrich, F.; Illek, B.; Reifen, R.; Gollnick, H.; Seeliger, M.W.; Wissinger, B.; Zrenner, E. Biochemical but not clinical Vitamin A deficiency results from mutations in the gene for retinol binding protein. Am. J. Clin. Nutr. 1999, 69, 931–936. [Google Scholar] [PubMed]
- Wei, S.H.; Episkopou, V.; Piantedosi, R.; Maeda, S.; Shimada, K.; Gottesman, M.E.; Blaner, W.S. Studies on the metabolism of retinol and retinol-binding protein in transthyretin-deficient mice produced by homologous recombination. J. Biol. Chem. 1995, 270, 866–870. [Google Scholar] [CrossRef] [PubMed]
- Monaco, H.L.; Rizzi, M.; Coda, A. Structure of a complex of 2 plasma-proteins—Transthyretin and retinol-binding protein. Science 1995, 268, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, R.; Yu, J.M.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of Vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Isken, A.; Golczak, M.; Oberhauser, V.; Hunzelmann, S.; Driever, W.; Imanishi, Y.; Palczewski, K.; von Lintig, J. RBP4 disrupts Vitamin A uptake homeostasis in a STRA6-deficient animal model for Matthew-Wood syndrome. Cell Metab. 2008, 7, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Amengual, J.; Zhang, N.; Kemerer, M.; Maeda, T.; Palczewski, K.; Von Lintig, J. STRA6 is critical for cellular Vitamin A uptake and homeostasis. Hum. Mol. Genet. 2014, 23, 5402–5417. [Google Scholar] [CrossRef] [PubMed]
- Bouillet, P.; Sapin, V.; Chazaud, C.; Messaddeq, N.; Decimo, D.; Dolle, P.; Chambon, P. Developmental expression pattern of STRA6, a retinoic acid-responsive gene encoding a new type of membrane protein. Mech. Dev. 1997, 63, 173–186. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Yu, J.M.; Wiita, P.; Honda, J.; Sun, H. An essential ligand-binding domain in the membrane receptor for retinol-binding protein revealed by large-scale mutagenesis and a human polymorphism. J. Biol. Chem. 2008, 283, 15160–15168. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, R.; Yu, J.; Wiita, P.; Ter-Stepanian, M.; Sun, H. Mapping the membrane topology and extracellular ligand binding domains of the retinol binding protein receptor. Biochemistry 2008, 47, 5387–5395. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Clarke, O.B.; Kim, J.; Stowe, S.; Kim, Y.K.; Assur, Z.; Cavalier, M.; Godoy-Ruiz, R.; von Alpen, D.C.; Manzini, C.; et al. Structure of the STRA6 receptor for retinol uptake. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, R.; Yu, J.M.; Ter-Stepanian, M.; Zhong, M.; Cheng, G.; Yuan, Q.; Jin, M.H.; Travis, G.H.; Ong, D.; Sun, H. Receptor-mediated cellular uptake mechanism that couples to intracellular storage. ACS Chem. Biol. 2011, 6, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Amengual, J.; Golczak, M.; Palczewski, K.; von Lintig, J. Lecithin:retinol acyltransferase is critical for cellular uptake of Vitamin A from serum retinol-binding protein. J. Biol. Chem. 2012, 287, 24216–24227. [Google Scholar] [CrossRef] [PubMed]
- Cowan, S.W.; Newcomer, M.E.; Jones, T.A. Crystallographic refinement of human serum retinol binding protein at 2A resolution. Proteins 1990, 8, 44–61. [Google Scholar] [CrossRef] [PubMed]
- Silvaroli, J.A.; Arne, J.M.; Chelstowska, S.; Kiser, P.D.; Banerjee, S.; Golczak, M. Ligand binding induces conformational changes in human cellular retinol-binding protein 1 (CRBP1) revealed by atomic resolution crystal structures. J. Biol. Chem. 2016, 291, 8528–8540. [Google Scholar] [CrossRef] [PubMed]
- Golczak, M.; Sears, A.E.; Kiser, P.D.; Palczewski, K. Lrat-specific domain facilitates Vitamin A metabolism by domain swapping in HRASLS3. Nat. Chem. Biol. 2015, 11, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.; Wald, G. Cis-trans isomers of vitamin A and retinene in vision. Science 1952, 115, 60–63. [Google Scholar] [CrossRef]
- Koyanagi, M.; Kawano, E.; Kinugawa, Y.; Oishi, T.; Shichida, Y.; Tamotsu, S.; Terakita, A. Bistable UV pigment in the lamprey pineal. Proc. Natl. Acad. Sci. USA 2004, 101, 6687–6691. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Terakita, A. Diversity and functional properties of bistable pigments. Photochem. Photobiol. Sci. 2010, 9, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.E. Chemistry of visual adaptation in the rat. Nature 1960, 188, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Wald, G. Molecular basis of visual excitation. Science 1968, 162, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: AG protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.O.; Crouch, R.K. Retinol dehydrogenases (RDHs) in the visual cycle. Exp. Eye Res. 2010, 91, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Rattner, A.; Smallwood, P.M.; Nathans, J. Identification and characterization of all-trans-retinol dehydrogenase from photoreceptor outer segments, the visual cycle enzyme that reduces all-trans-retinal to all-trans-retinol. J. Biol. Chem. 2000, 275, 11034–11043. [Google Scholar] [CrossRef] [PubMed]
- Haeseleer, F.; Jang, G.F.; Imanishi, Y.; Driessen, C.A.G.G.; Matsumura, M.; Nelson, P.S.; Palczewski, K. Dual-substrate specificity short chain retinol dehydrogenases from the vertebrate retina. J. Biol. Chem. 2002, 277, 45537–45546. [Google Scholar] [CrossRef] [PubMed]
- Gollapalli, D.R.; Rando, R.R. All-trans-retinyl esters are the substrates for isomerization in the vertebrate visual cycle. Biochemistry 2003, 42, 5809–5818. [Google Scholar] [CrossRef] [PubMed]
- Kiser, P.D.; Zhang, J.Y.; Badiee, M.; Li, Q.J.; Shi, W.X.; Sui, X.W.; Golczak, M.; Tochtrop, G.P.; Palczewski, K. Catalytic mechanism of a retinoid isomerase essential for vertebrate vision. Nat. Chem. Biol. 2015, 11, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Kiser, P.D.; Golczak, M.; Lodowski, D.T.; Chance, M.R.; Palczewski, K. Crystal structure of native RPE65, the retinoid isomerase of the visual cycle. Proc. Natl. Acad. Sci. USA 2009, 106, 17325–17330. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.; Hellman, U.; Wernstedt, C.; Eriksson, U. The retinal-pigment epithelial-specific 11-cis retinol dehydrogenase belongs to the family of short-chain alcohol dehydrogenases. J. Biol. Chem. 1995, 270, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Simon, A.; Eriksson, U.; Harris, E.; Berson, E.L.; Dryja, T.P. Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat. Genet. 1999, 22, 188–191. [Google Scholar] [PubMed]
- Kiser, P.D.; Golczak, M.; Maeda, A.; Palczewski, K. Key enzymes of the retinoid (visual) cycle in vertebrate retina. BBA Mol. Cell Biol. Lipids 2012, 1821, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Golczak, M.; Kiser, P.D.; Lodowski, D.T.; Maeda, A.; Palczewski, K. Importance of membrane structural integrity for RPE65 retinoid isomerization activity. J. Biol. Chem. 2010, 285, 9667–9682. [Google Scholar] [CrossRef] [PubMed]
- Bolze, C.S.; Helbling, R.E.; Owen, R.L.; Pearson, A.R.; Pompidor, G.; Dworkowski, F.; Fuchs, M.R.; Furrer, J.; Golczak, M.; Palczewski, K.; et al. Human cellular retinaldehyde-binding protein has secondary thermal 9-cis-retinal isomerase activity. J. Am. Chem. Soc. 2014, 136, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Albalat, R.; Brunet, F.; Laudet, V.; Schubert, M. Evolution of retinoid and steroid signaling: Vertebrate diversification from an amphioxus perspective. Genome Biol. Evol. 2011, 3, 985–1005. [Google Scholar] [CrossRef] [PubMed]
- Andre, A.; Ruivo, R.; Gesto, M.; Castro, L.F.C.; Santos, M.M. Retinoid metabolism in invertebrates: When evolution meets endocrine disruption. Gen. Comp. Endocrinol. 2014, 208, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Turchetto-Zolet, A.C.; Maraschin, F.S.; de Morais, G.L.; Cagliari, A.; Andrade, C.M.B.; Margis-Pinheiro, M.; Margis, R. Evolutionary view of acyl-CoA diacylglycerol acyltransferase (DGAT), a key enzyme in neutral lipid biosynthesis. BMC Evol. Biol. 2011, 11, 263. [Google Scholar] [CrossRef] [PubMed]
- Gesto, M.; Castro, L.F.C.; Reis-Henriques, M.A.; Santos, M.M. Retinol metabolism in the mollusk Osilinus lineatus indicates an ancient origin for retinyl ester storage capacity. PLoS ONE 2012, 7, e35138. [Google Scholar] [CrossRef] [PubMed]
- Biesalski, H.K.; Doepner, G.; Tzimas, G.; Gamulin, V.; Schroder, H.C.; Batel, R.; Nau, H.; Muller, W.E.G. Modulation of myb gene-expression in sponges by retinoic acid. Oncogene 1992, 7, 1765–1774. [Google Scholar] [PubMed]
- Gesto, M.; Castro, L.F.C.; Santos, M.M. Differences in retinoid levels and metabolism among gastropod lineages: Imposex-susceptible gastropods lack the ability to store retinoids in the form of retinyl esters. Aquat. Toxicol. 2013, 142, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Poliakov, E.; Gubin, A.N.; Stearn, O.; Li, Y.; Campos, M.M.; Gentleman, S.; Rogozin, I.B.; Redmond, T.M. Origin and evolution of retinoid isomerization machinery in vertebrate visual cycle: Hint from jawless vertebrates. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Theodosiou, M.; Laudet, V.; Schubert, M. From carrot to clinic: An overview of the retinoic acid signaling pathway. Cell. Mol. Life Sci. 2010, 67, 1423–1445. [Google Scholar] [CrossRef] [PubMed]
- Albalat, R. Evolution of the genetic machinery of the visual cycle: A novelty of the vertebrate eye? Mol. Biol. Evol. 2012, 29, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, V.; Aravind, L. Evolutionary history, structural features and biochemical diversity of the NlpC/P60 superfamily of enzymes. Genome Biol. 2003, 4, R11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golczak, M.; Kiser, P.D.; Sears, A.E.; Lodowski, D.T.; Blaner, W.S.; Palczewski, K. Structural basis for the acyltransferase activity of lecithin:retinol acyltransferase-like proteins. J. Biol. Chem. 2012, 287, 23790–23807. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.Y.; Cao, J.; Addington, L.; Lovell, S.; Battaile, K.P.; Zhang, N.; Rao, J.L.U.M.; Dennis, E.A.; Moise, A.R. Structure/function relationships of adipose phospholipase A(2) containing a cys-his-his catalytic triad. J. Biol. Chem. 2012, 287, 35260–35274. [Google Scholar] [CrossRef] [PubMed]
- Moise, A.R.; Golczak, M.; Imanishi, Y.; Palczewski, K. Topology and membrane association of lecithin:retinol acyltransferase. J. Biol. Chem. 2007, 282, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Uyama, T.; Morishita, J.; Jin, X.H.; Okamoto, Y.; Tsuboi, K.; Ueda, N. The tumor suppressor gene H-Rev107 functions as a novel Ca(2+)-independent cytosolic phospholipase A(1/2) of the thiol hydrolase type. J. Lipid Res. 2009, 50, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Uyama, T.; Jin, X.H.; Tsuboi, K.; Tonai, T.; Ueda, N. Characterization of the human tumor suppressors TIG3 and HRASLS2 as phospholipid-metabolizing enzymes. BBA Mol. Cell Biol. Lipids 2009, 1791, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Han, B.G.; Cho, J.W.; Cho, Y.D.; Kim, S.Y.; Yoon, H.J.; Song, H.K.; Cheong, H.K.; Jeon, Y.H.; Lee, D.K.; Lee, S.; et al. Expression, purification and biochemical characterization of the N-terminal regions of human TIG3 and HRASLS3 proteins. Protein Expr. Purif. 2010, 71, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Lin, J.; Jin, C.; Xia, B. Solution structure of the N-terminal catalytic domain of human H-REV107—A novel circular permutated NlpC/P60 domain. FEBS Lett. 2010, 584, 4222–4226. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.M.; Tang, X.H.; Gudas, L.J. Homeostasis of retinal in lecithin: Retinal acyltransferase gene knockout mice fed a high retinal diet. Biochem. Pharmacol. 2008, 75, 2316–2324. [Google Scholar] [CrossRef] [PubMed]
- Isken, A.; Holzschuh, J.; Lampert, J.M.; Fischer, L.; Oberhauser, V.; Palczewski, K.; von Lintig, J. Sequestration of retinyl esters is essential for retinoid signaling in the zebrafish embryo. J. Biol. Chem. 2007, 282, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Randolph, R.K.; Ross, A.C. Vitamin A status regulates hepatic lecithin:retinol acyltransferase activity in rats. J. Biol. Chem. 1991, 266, 16453–16457. [Google Scholar] [PubMed]
- Matsuura, T.; Ross, A.C. Regulation of hepatic lecithin:retinol acyltransferase activity by retinoic acid. Arch. Biochem. Biophys. 1993, 301, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Zolfaghari, R.; Ross, A.C. Lecithin:Retinol acyltransferase from mouse and rat liver. cDNA cloning and liver-specific regulation by dietary Vitamin A and retinoic acid. J. Lipid Res. 2000, 41, 2024–2034. [Google Scholar] [PubMed]
- Zolfaghari, R.; Ross, A.C. Lecithin:retinol acyltransferase expression is regulated by dietary Vitamin A and exogenous retinoic acid in the lung of adult rats. J. Nutr. 2002, 132, 1160–1164. [Google Scholar] [PubMed]
- Ross, A.C. Retinoid production and catabolism: Role of diet in regulating retinol esterification and retinoic acid oxidation. J. Nutr. 2003, 133, 291S–296S. [Google Scholar] [PubMed]
- Raila, J.; Stohrer, M.; Forterre, S.; Stangassinger, M.; Schweigert, F.J. Effect of exercise on the mobilization of retinol and retinyl esters in plasma of sled dogs. J. Anim. Physiol. Anim. Nutr. 2004, 88, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Raila, J.; Mathews, U.; Schweigert, F.J. Plasma transport and tissue distribution of β-carotene, Vitamin A and retinol-binding protein in domestic cats. Comp. Biochem. Physiol. 2001, 130, 849–856. [Google Scholar] [CrossRef]
- Raila, J.; Gomez, C.; Schweigert, F.J. The ferret as a model for Vitamin A metabolism in carnivores. J. Nutr. 2002, 132, 1787S–1789S. [Google Scholar] [PubMed]
- Garcia, A.L.; Raila, J.; Koebnick, C.; Eulenberger, K.; Schweigert, F.J. Great apes show highly selective plasma carotenoids and have physiologically high plasma retinyl esters compared to humans. Am. J. Phys. Anthropol. 2006, 131, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.C.; Li, N.Q. Lung retinyl ester is low in young adult rats fed a Vitamin A deficient diet after weaning, despite neonatal Vitamin A supplementation and maintenance of normal plasma retinol. J. Nutr. 2007, 137, 2213–2218. [Google Scholar] [PubMed]
- O’Byrne, S.M.; Kako, Y.; Deckelbaum, R.J.; Hansen, I.H.; Palczewski, K.; Goldberg, I.J.; Blaner, W.S. Multiple pathways ensure retinoid delivery to milk: Studies in genetically modified mice. Am. J. Physiol. 2010, 298, E862–E870. [Google Scholar] [CrossRef] [PubMed]
- Redmond, T.M.; Yu, S.; Lee, E.; Bok, D.; Hamasaki, D.; Chen, N.; Goletz, P.; Ma, J.X.; Crouch, R.K.; Pfeifer, K. RPE65 is necessary for production of 11-cis-Vitamin A in the retinal visual cycle. Nat. Genet. 1998, 20, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef] [PubMed]
- Borman, A.D.; Ocaka, L.A.; Mackay, D.S.; Ripamonti, C.; Henderson, R.H.; Moradi, P.; Hall, G.; Black, G.C.; Robson, A.G.; Holder, G.E.; et al. Early onset retinal dystrophy due to mutations in LRAT: Molecular analysis and detailed phenotypic study. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3927–3938. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.A.; Li, Y.; McHenry, C.L.; Carlson, T.J.; Ding, X.; Sieving, P.A.; Apfelstedt-Sylla, E.; Zrenner, E.; Gal, A. Mutations in lecithin retinol acyltransferase (LRAT) cause early-onset severe retinal dystrophy. Investig. Ophthalmol. Vis. Sci. 2001, 42, S647–S647. [Google Scholar]
- Senechal, A.; Humbert, G.; Surget, M.O.; Bazalgette, C.; Arnaud, B.; Arndt, C.; Laurent, E.; Brabet, P.; Hamel, C.P. Screening genes of the retinoid metabolism: Novel LRAT mutation in leber congenital amaurosis. Am. J. Ophthalmol. 2006, 142, 702–704. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Perusek, L.; Amengual, J.; Babino, D.; Palczewski, K.; von Lintig, J. Dietary 9-cis-β,β-carotene fails to rescue vision in mouse models of leber congenital amaurosis. Mol. Pharmacol. 2011, 80, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Wassef, L.; Chung, S.; Jiang, H.; Wyss, A.; Blaner, W.S.; Quadro, L. β-carotene and its cleavage enzyme β-carotene-15,15′-oxygenase (CMOI) affect retinoid metabolism in developing tissues. FASEB J. 2011, 25, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K. Retinoids for treatment of retinal diseases. Trends Pharmacol. Sci. 2010, 31, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Van Hooser, J.P.; Aleman, T.S.; He, Y.G.; Cideciyan, A.V.; Kuksa, V.; Pittler, S.J.; Stone, E.M.; Jacobson, S.G.; Palczewski, K. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc. Natl. Acad. Sci. USA 2000, 97, 8623–8628. [Google Scholar] [CrossRef] [PubMed]
- Batten, M.L.; Imanishi, Y.; Tu, D.C.; Doan, T.; Zhu, L.; Pang, J.; Glushakova, L.; Moise, A.R.; Baehr, W.; Van Gelder, R.N.; et al. Pharmacological and rAAV gene therapy rescue of visual functions in a blind mouse model of Leber congenital amaurosis. PLoS Med. 2005, 2, e333. [Google Scholar] [CrossRef] [PubMed]
- Koenekoop, R.K.; Sui, R.; Sallum, J.; van den Born, L.I.; Ajlan, R.; Khan, A.; den Hollander, A.I.; Cremers, F.P.; Mendola, J.D.; Bittner, A.K.; et al. Oral 9-cis retinoid for childhood blindness due to leber congenital amaurosis caused by RPE65 or LRAT mutations: An open-label phase 1b trial. Lancet 2014, 384, 1513–1520. [Google Scholar] [CrossRef]
- Scholl, H.P.; Moore, A.T.; Koenekoop, R.K.; Wen, Y.; Fishman, G.A.; van den Born, L.I.; Bittner, A.; Bowles, K.; Fletcher, E.C.; Collison, F.T.; et al. Safety and proof-of-concept study of oral QLT091001 in retinitis pigmentosa due to inherited deficiencies of retinal pigment epithelial 65 protein (RPE65) or lecithin:retinol acyltransferase (LRAT). PLoS ONE 2015, 10, e0143846. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.A.; Li, Y.; McHenry, C.L.; Carlson, T.J.; Ding, X.; Sieving, P.A.; Apfelstedt-Sylla, E.; Gal, A. Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat. Genet. 2001, 28, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Littink, K.W.; van Genderen, M.M.; van Schooneveld, M.J.; Visser, L.; Riemslag, F.C.; Keunen, J.E.; Bakker, B.; Zonneveld, M.N.; den Hollander, A.I.; Cremers, F.P.; et al. A homozygous frameshift mutation in LRAT causes retinitis punctata albescens. Ophthalmology 2012, 119, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Collin, R.W.; van den Born, L.I.; Klevering, B.J.; de Castro-Miro, M.; Littink, K.W.; Arimadyo, K.; Azam, M.; Yazar, V.; Zonneveld, M.N.; Paun, C.C.; et al. High-resolution homozygosity mapping is a powerful tool to detect novel mutations causative of autosomal recessive RP in the Dutch population. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2227–2239. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Sun, V.; Tuan, H.F.; Keser, V.; Wang, K.; Ren, H.; Lopez, I.; Zaneveld, J.E.; Siddiqui, S.; et al. Comprehensive molecular diagnosis of 179 leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J. Med. Genet. 2013, 50, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Bok, D.; Ruiz, A.; Yaron, O.; Jahng, W.J.; Ray, A.; Xue, L.; Rando, R.R. Purification and characterization of a transmembrane domain-deleted form of lecithin retinol acyltransferase. Biochemistry 2003, 42, 6090–6098. [Google Scholar] [CrossRef] [PubMed]
- Preising, M.N.; Paunescu, K.; Friedburg, C.; Lorenz, B. Genetic and clinical heterogeneity in LCA patients. The end of uniformity. Ophthalmologe 2007, 104, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Kaylor, J.J.; Yuan, Q.; Cook, J.; Sarfare, S.; Makshanoff, J.; Miu, A.; Kim, A.; Kim, P.; Habib, S.; Roybal, C.N.; et al. Identification of DES1 as a Vitamin A isomerase in Muller glial cells of the retina. Nat. Chem. Biol. 2013, 9, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Kaylor, J.J.; Cook, J.D.; Makshanoff, J.; Bischoff, N.; Yong, J.; Travis, G.H. Identification of the 11-cis-specific retinyl-ester synthase in retinal muller cells as multifunctional O-acyltransferase (MFAT). Proc. Natl. Acad. Sci. USA 2014, 111, 7302–7307. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide Change | AA Mutation | Exon | Reference |
|---|---|---|---|
| 12 1 del 2 C | P4fs 3 (53) | 2 | [141] |
| 40_41delGAins 4 TT | E14L | 2 | [130] |
| T181A | Y61D | 2 | [130] |
| 217_218delAT | M73fs(47) 4 | 2 | [132] |
| G316A | A106T | 2 | [130] |
| C325T | R109C | 3 | [145] |
| 397_398delAA | K134fs(11) | 3 | [131,140] |
| 427_428delCG | R143fs(2) | 3 | [139] |
| C518T | P173L | 3 | [132] |
| 519delG | I174fs(11) | 3 | [142] |
| A523T | S175R | 3 | [131,140] |
| G569A | R190H | 3 | [145] |
| 613_614delCT | S205Yfs(27) | 3 | [143] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chelstowska, S.; Widjaja-Adhi, M.A.K.; Silvaroli, J.A.; Golczak, M. Molecular Basis for Vitamin A Uptake and Storage in Vertebrates. Nutrients 2016, 8, 676. https://doi.org/10.3390/nu8110676
Chelstowska S, Widjaja-Adhi MAK, Silvaroli JA, Golczak M. Molecular Basis for Vitamin A Uptake and Storage in Vertebrates. Nutrients. 2016; 8(11):676. https://doi.org/10.3390/nu8110676
Chicago/Turabian StyleChelstowska, Sylwia, Made Airanthi K. Widjaja-Adhi, Josie A. Silvaroli, and Marcin Golczak. 2016. "Molecular Basis for Vitamin A Uptake and Storage in Vertebrates" Nutrients 8, no. 11: 676. https://doi.org/10.3390/nu8110676