Ellagic Acid Inhibits Bladder Cancer Invasiveness and In Vivo Tumor Growth

 , , ,
, , ,
Abstract
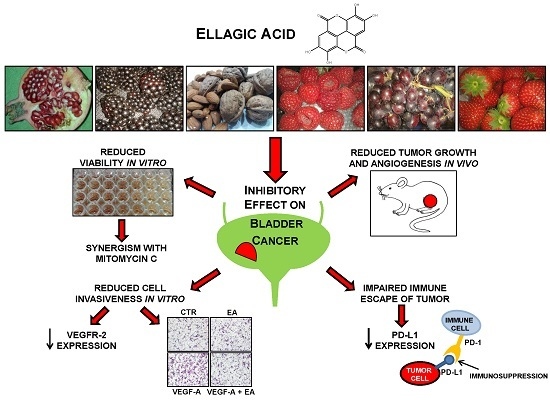
:
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Culture Conditions and Drugs
2.2. Cell Proliferation Assays
2.3. Apoptosis Analysis by Flow Cytometry
2.4. Invasion and Migration Assays
2.5. Western Blot Analysis
2.6. Evaluation of VEGF-A Secretion and Analysis of VEGFR-2 Phosphorylation
2.7. In Vivo Study
- (a)
- Percentage of tumor volume inhibition (TVI%) in treated vs. control mice, calculated as:TVI% = 100 − [(TV treated/TV control) × 100]
- (b)
- Tumor growth quadrupling time, calculated as the time required for tumor volume to increase 4-fold over initial volume at indicated times, using the formula:where tq is the interpolated quadrupling time; t1 and t2 are the lower and upper observation times bracketing the quadrupling tumor volume; Vq = 4V0, where V0 is the initial tumor volume; V1 and V2 are tumor volumes at the times t1 and t2, respectively;tq = t1 + (t2 − t1)log(Vq/V1)/log(V2/V1)
- (c)
- Tumor growth delay index, calculated as the mean treated/control tumor growth quadrupling time ratio.
2.8. Animal Care and Ethics Statement
2.9. Statistical Analysis
3. Results
3.1. Anti-Proliferative and Apoptotic Effects of EA against Bladder Cancer Cells
3.2. EA Enhances the Anti-Proliferative Effects of Mitomycin C
3.3. Ellagic Acid Reduces VEGFR-2 Expression in Human Bladder Cancer Cells
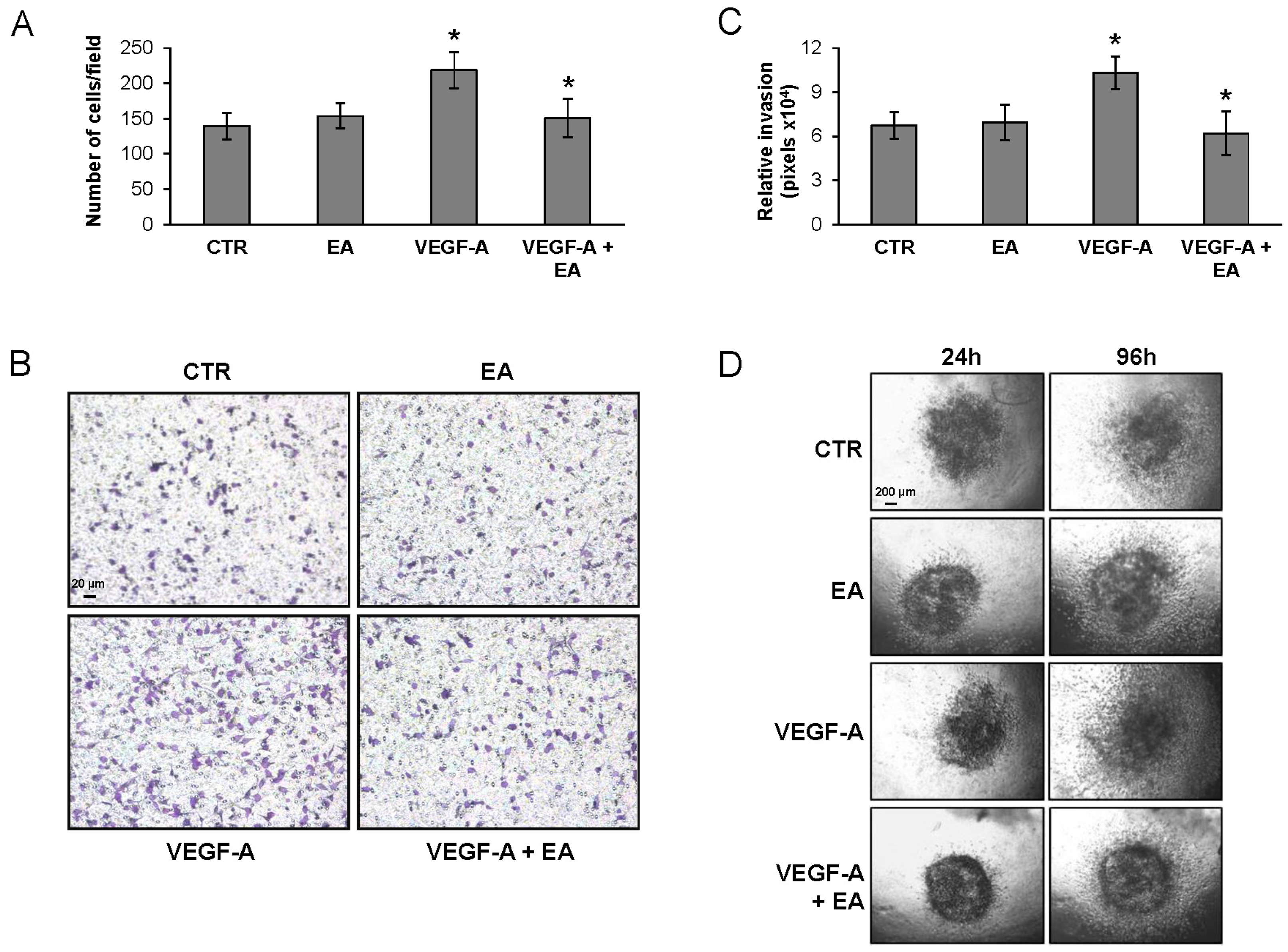
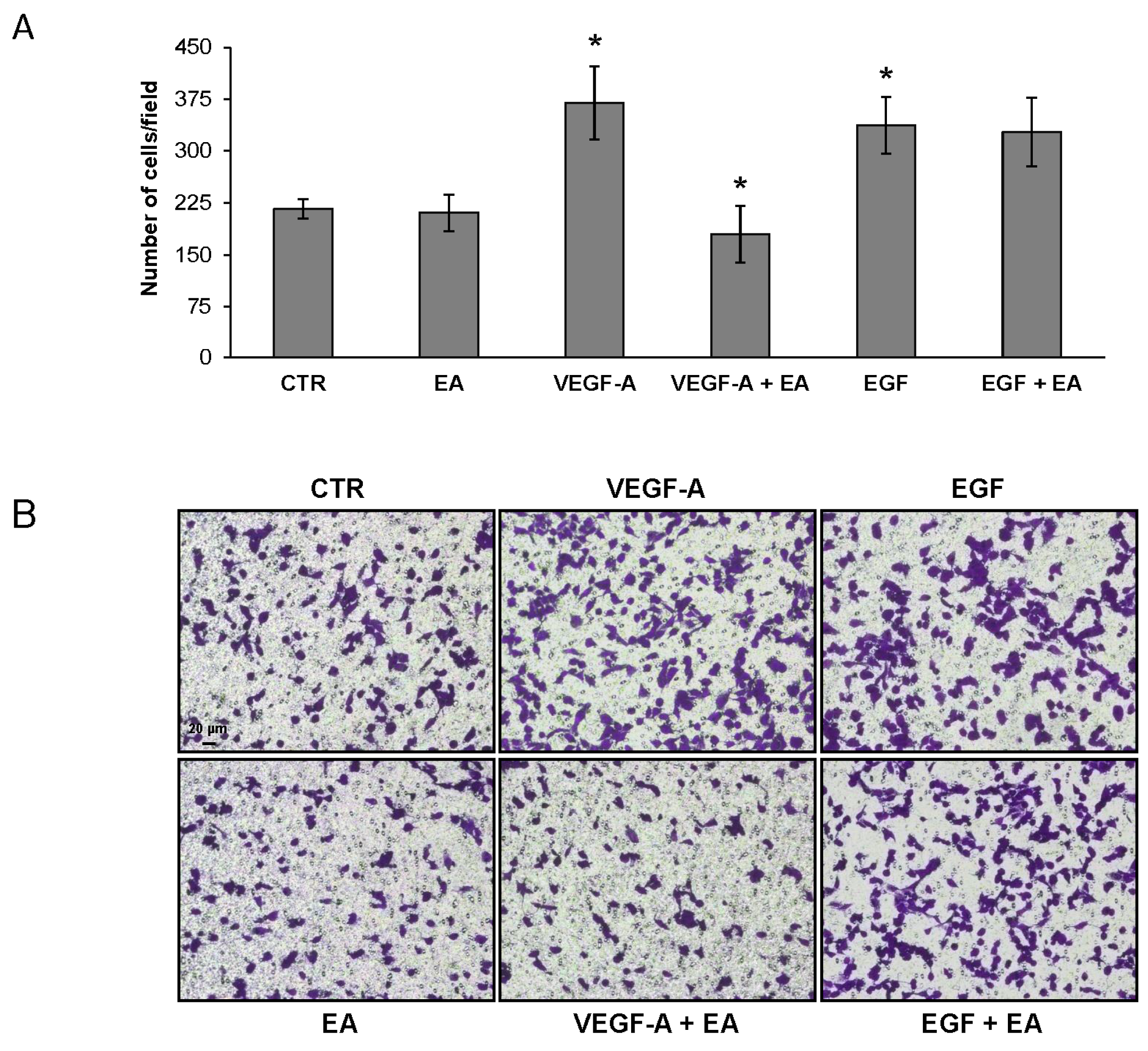
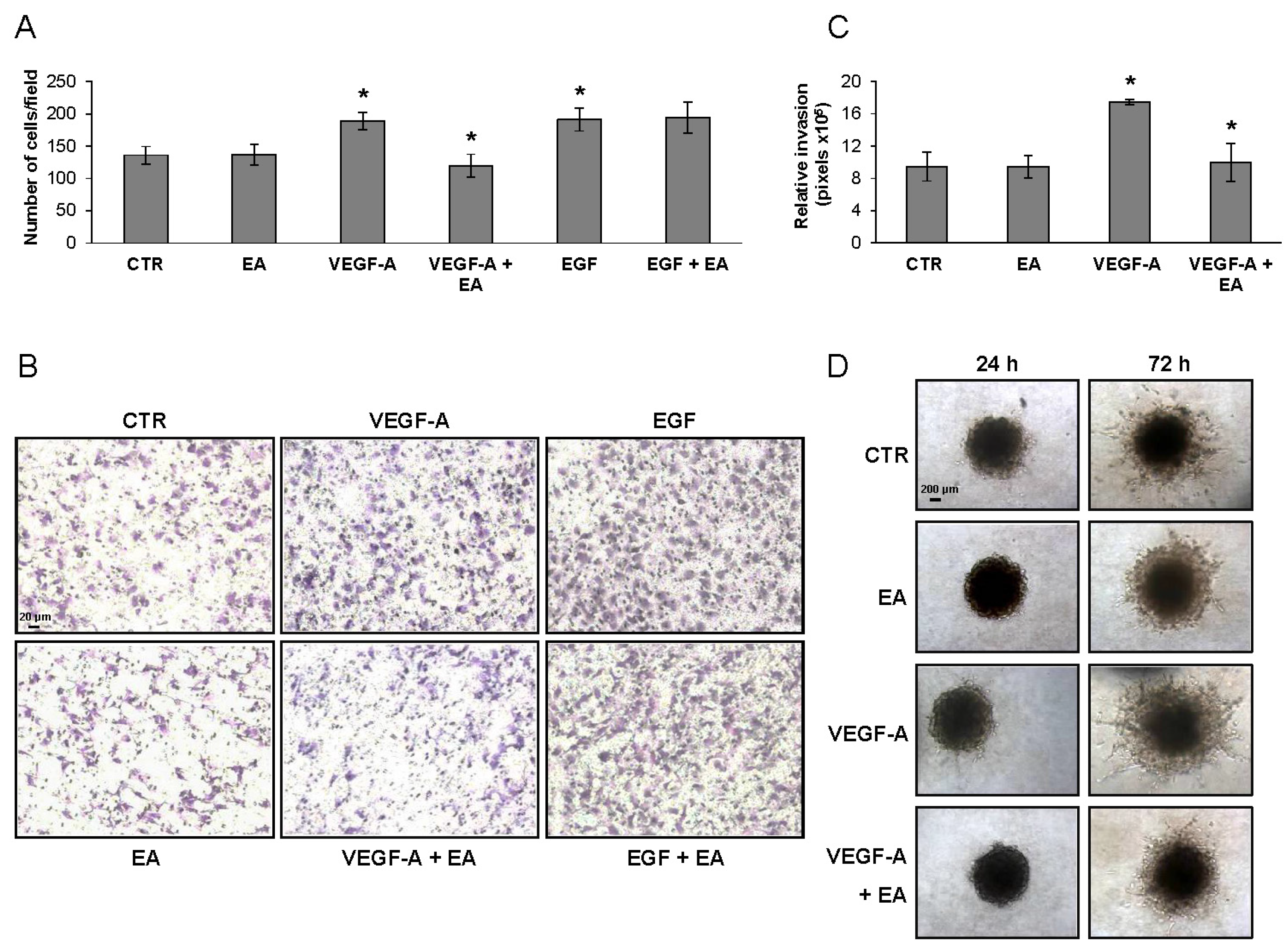
3.4. EA Inhibits Extracellular Matrix Invasion and Migration of Human Bladder Cancer Cells in Response to VEGF-A
3.5. Treatment of Bladder Cancer Cells Decrease the Expression of PD-L1
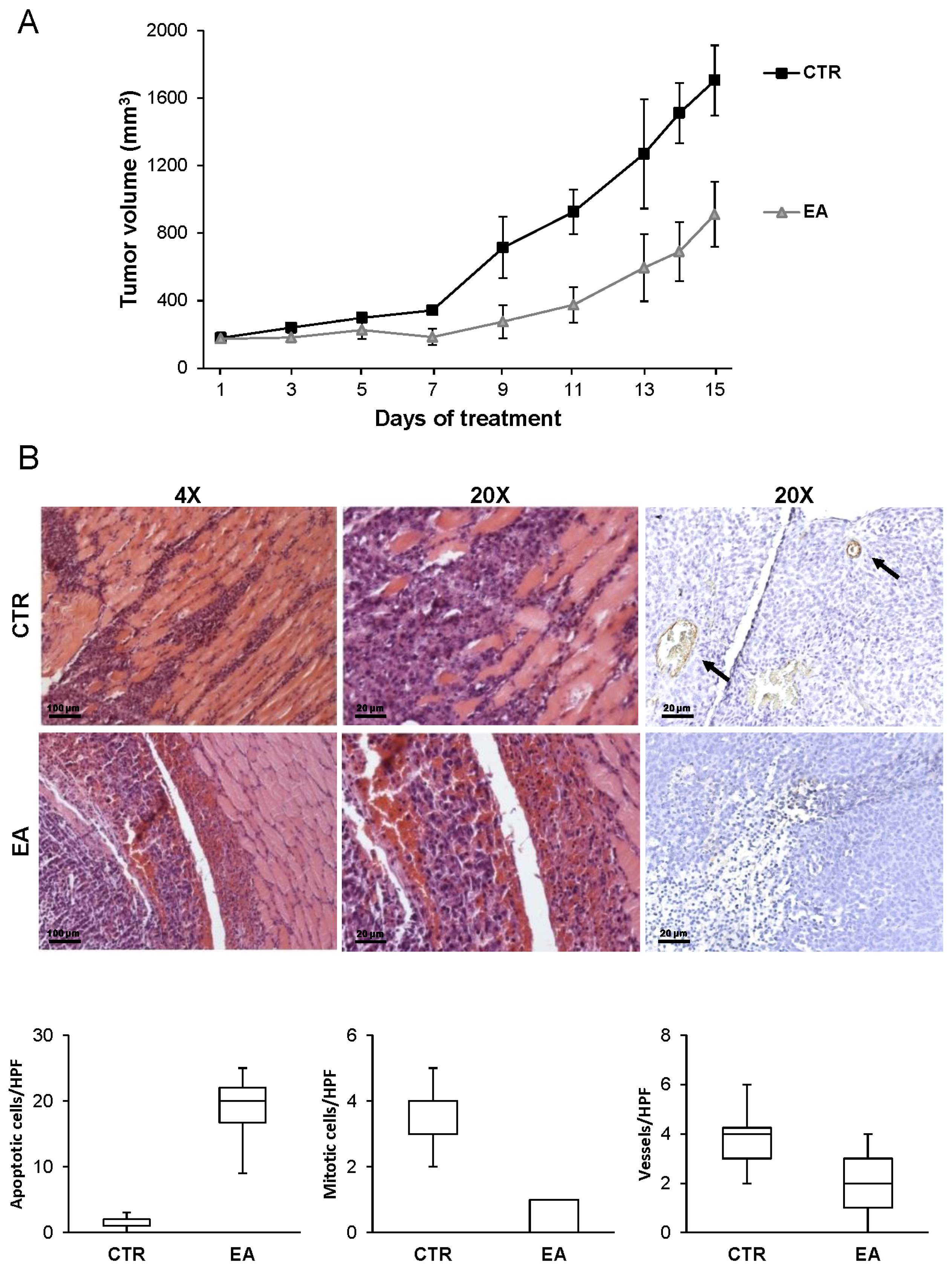
3.6. EA Inhibits In Vivo Bladder Cancer Growth
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A



References
- Zhang, H.; Zhao, L.; Li, H.; Xu, H.; Chen, W.; Tao, L. Research progress on the anticarcinogenic actions and mechanisms of ellagic acid. Cancer Biol. Med. 2014, 11, 92–100. [Google Scholar] [PubMed]
- Talcott, S.T.; Lee, J.H. Ellagic acid and flavonoid antioxidant content of muscadine wine and juice. J. Agric. Food Chem. 2002, 50, 3186–3192. [Google Scholar] [CrossRef] [PubMed]
- Vlachojannis, C.; Zimmermann, B.F.; Chrubasik-Hausmann, S. Efficacy and safety of pomegranate medicinal products for cancer. Evid. Based Complement. Alternat. Med. 2015, 2015, 258598. [Google Scholar] [CrossRef] [PubMed]
- Mertens-Talcott, S.U.; Jilma-Stohlawetz, P.; Rios, J.; Hingorani, L.; Derendorf, H. Absorption, metabolism, and antioxidant effects of pomegranate (Punica granatum I.) polyphenols after ingestion of a standardized extract in healthy human volunteers. J. Agric. Food Chem. 2006, 54, 8956–8961. [Google Scholar] [CrossRef] [PubMed]
- Núñez-Sánchez, M.Á.; Karmokar, A.; González-Sarrías, A.; García-Villalba, R.; Tomás-Barberán, F.A.; García-Conesa, M.T.; Brown, K.; Espín, J.C. In vivo relevant mixed urolithins and ellagic acid inhibit phenotypic and molecular colon cancer stem cell features: A new potentiality for ellagitannin metabolites against cancer. Food Chem. Toxicol. 2016, 92, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Heber, D. Multitargeted therapy of cancer by ellagitannins. Cancer Lett. 2008, 269, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Kasimsetty, S.G.; Bialonska, D.; Reddy, M.K.; Ma, G.; Khan, S.I.; Ferreira, D. Colon cancer chemopreventive activities of pomegranate ellagitannins and urolithins. J. Agric. Food Chem. 2010, 58, 2180–2187. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.D.; Mehta, R.; Yu, W.; Neeman, I.; Livney, T.; Amichay, A.; Poirier, D.; Nicholls, P.; Kirby, A.; Jiang, W.; et al. Chemopreventive and adjuvant therapeutic potential of pomegranate (Punica granatum) for human breast cancer. Breast Cancer Res. Treat. 2002, 71, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Kambhampati, S.; Haque, I.; Banerjee, S.K. Pomegranate sensitizes Tamoxifen action in ER-α positive breast cancer cells. J. Cell Commun. Signal 2011, 5, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.Y.; Seeram, N.P.; Heber, D. Pomegranate polyphenols down-regulate expression of androgen-synthesizing genes in human prostate cancer cells overexpressing the androgen receptor. J. Nutr. Biochem. 2008, 19, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Rettig, M.B.; Heber, D.; An, J.; Seeram, N.P.; Rao, J.Y.; Liu, H.; Klatte, T.; Belldegrun, A.; Moro, A.; Henning, S.M.; et al. Pomegranate extract inhibits androgen-independent prostate cancer growth through a nuclear factor-κB-dependent mechanism. Mol. Cancer Ther. 2008, 7, 2662–2671. [Google Scholar] [CrossRef] [PubMed]
- Adhami, V.M.; Khan, N.; Mukhtar, H. Cancer chemoprevention by pomegranate: Laboratory and clinical evidence. Nutr. Cancer 2009, 61, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Umesalma, S.; Nagendraprabhu, P.; Sudhandiran, G. Antiproliferative and apoptotic-inducing potential of ellagic acid against 1,2-dimethyl hydrazine-induced colon tumorigenesis in Wistar rats. Mol. Cell Biochem. 2014, 388, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Naiki-Ito, A.; Chewonarin, T.; Tang, M.; Pitchakarn, P.; Kuno, T.; Ogawa, K.; Asamoto, M.; Shirai, T.; Takahashi, S. Ellagic acid, a component of pomegranate fruit juice, suppresses androgen-dependent prostate carcinogenesis via induction of apoptosis. Prostate 2015, 75, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, W.; Lin, M.; Garcia, M.; Mulholland, D.; Lilly, M.; Martins-Green, M. Luteolin, ellagic acid and punicic acid are natural products that inhibit prostate cancer metastasis. Carcinogenesis 2014, 35, 2321–2330. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, Z.Y.; Mo, S.L.; Loo, T.Y.; Wang, D.M.; Luo, H.B.; Yang, D.P.; Chen, Y.L.; Shen, J.G.; Chen, J.P. Ellagic acid, a phenolic compound, exerts anti-angiogenesis effects via VEGFR-2 signaling pathway in breast cancer. Breast Cancer Res. Treat. 2012, 134, 943–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Tang, S.N.; Marsh, J.L.; Shankar, S.; Srivastava, R.K. Ellagic acid inhibits human pancreatic cancer growth in Balb c nude mice. Cancer Lett. 2013, 337, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadis, A.; de Reijke, T.M. Best practice in the treatment of nonmuscle invasive bladder cancer. Ther. Adv. Urol. 2012, 4, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; Orsola, A.; Leow, J.J.; Wiegel, T.; de Santis, M.; Horwich, A. ESMO Guidelines Working Group. Bladder cancer: ESMO Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, iii40–iii8. [Google Scholar] [CrossRef] [PubMed]
- Kim, J. Immune checkpoint blockade therapy for bladder cancer treatment. Investig. Clin. Urol. 2016, 57, S98–S105. [Google Scholar] [CrossRef] [PubMed]
- Crew, J.P.; O’Brien, T.; Bicknell, R.; Fuggle, S.; Cranston, D.; Harris, A.L. Urinary vascular endothelial growth factor and its correlation with bladder cancer recurrence rates. J. Urol. 1999, 161, 799–804. [Google Scholar] [CrossRef]
- Sidaway, P. Bladder cancer: Atezolizumab effective against advanced-stage disease. Nat. Rev. Urol. 2016, 13, 238. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, G.; Su, C.; Lin, J.; Yeh, C.; Cheng, K.; Chung, J. Ellagic acid induced p53/p21 expression, G1 arrest and apoptosis in human bladder cancer T24 cells. Anticancer Res. 2005, 25, 971–979. [Google Scholar] [PubMed]
- Ho, C.; Huang, A.; Yu, C.; Lien, J.; Wu, S.; Huang, Y.; Huang, H.; Kuo, J.; Liao, W.; Yang, J.; et al. Ellagic acid induces apoptosis in TSGH8301 human bladder cancer cells through the endoplasmic reticulum stress- and mitochondria-dependent signaling pathways. Environ. Toxicol. 2014, 29, 1262–1274. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Zhou, B.; Jin, L.; Yu, H.; Liu, L.; Liu, Y.; Qin, C.; Xie, S.; Zhu, F. In vitro antioxidant and antiproliferative effects of ellagic acid and its colonic metabolite, urolithins, on human bladder cancer T24 cells. Food Chem. Toxicol. 2013, 59, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Tentori, L.; Vergati, M.; Muzi, A.; Levati, L.; Ruffini, F.; Forini, O.; Vernole, P.; Lacal, P.M.; Graziani, G. Generation of an immortalized human endothelial cell line as a model of neovascular proliferating endothelial cells to assess chemosensitivity to anticancer drugs. Int. J. Oncol. 2005, 27, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Ruffini, F.; D’Atri, S.; Lacal, P.M. Neuropilin-1 expression promotes invasiveness of melanoma cells through vascular endothelial growth factor receptor-2-dependent and -independent mechanisms. Int. J. Oncol. 2013, 43, 297–306. [Google Scholar] [PubMed]
- Pagani, E.; Ruffini, F.; Antonini Cappellini, G.C.; Scoppola, A.; Fortes, C.; Marchetti, P.; Graziani, G.; D’Atri, S.; Lacal, P.M. Placenta growth factor and neuropilin-1 collaborate in promoting melanoma aggressiveness. Int. J. Oncol. 2016, 48, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Graziani, G.; Artuso, S.; de Luca, A.; Muzi, A.; Rotili, D.; Scimeca, M.; Atzori, M.G.; Ceci, C.; Mai, A.; Leonetti, C.; et al. A new water soluble MAPK activator exerts antitumor activity in melanoma cells resistant to the BRAF inhibitor vemurafenib. Biochem. Pharmacol. 2015, 95, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Vinci, M.; Box, C.; Eccles, S.A. Three-dimensional (3D) tumor spheroid invasion assay. J. Vis. Exp. 2015. [Google Scholar] [CrossRef] [PubMed]
- Lacal, P.M.; Failla, C.M.; Pagani, E.; Odorisio, T.; Schietroma, C.; Falcinelli, S.; Zambruno, G.; D’Atri, S. Human melanoma cells secrete and respond to placenta growth factor and vascular endothelial growth factor. J. Investg. Dermatol. 2000, 115, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Abdollah, F.; Gandaglia, G.; Thuret, R.; Schmitges, J.; Tian, Z.; Jeldres, C.; Passoni, N.M.; Briganti, A.; Shariat, S.F.; Perrotte, P.; et al. Incidence, survival and mortality rates of stage-specific bladder cancer in United States: A trend analysis. Cancer Epidemiol. 2013, 37, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Sonpavde, G.; Bellmunt, J. Bladder cancer: Angiogenesis as a therapeutic target in urothelial carcinoma. Nat. Rev. Urol. 2016, 13, 306–307. [Google Scholar] [CrossRef] [PubMed]
- Aldebasi, Y.H.; Rahmani, A.H.; Khan, A.A.; Aly, S.M. The effect of vascular endothelial growth factor in the progression of bladder cancer and diabetic retinopathy. Int. J. Clin. Exp. Med. 2013, 6, 239–251. [Google Scholar] [PubMed]
- Pinto, A.; Redondo, A.; Zamora, P.; Castelo, B.; Espinosa, E. Angiogenesis as a therapeutic target in urothelial carcinoma. Anticancer Drugs 2010, 21, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Mazzola, C.R.; Chin, J. Targeting the VEGF pathway in metastatic bladder cancer. Expert Opin. Investig. Drugs 2015, 24, 913–927. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Kim, H.; Oh, S.; Lee, J.G.; Kee, M.; Ko, H.J.; Kweon, M.N.; Won, K.J.; Baek, S.H. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 2016, 534, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Kumar, S.R.; Hawes, D.; Cai, J.; Hassanieh, L.; Groshen, S.; Zhu, S.; Masood, R.; Quinn, D.I.; Broek, D.; et al. Expression and significance of vascular endothelial growth factor receptor 2 in bladder cancer. J. Urol. 2006, 175, 1245–1252. [Google Scholar] [CrossRef]
- Kopparapu, P.K.; Boorjian, S.A.; Robinson, B.D.; Downes, M.; Gudas, L.J.; Mongan, N.P.; Persson, J.L. Expression of VEGF and its receptors VEGFR1/VEGFR2 is associated with invasiveness of bladder cancer. Anticancer Res. 2013, 33, 2381–2390. [Google Scholar] [PubMed]
- Price, D.J.; Miralem, T.; Jiang, S.; Steinberg, R.; Avraham, H. Role of vascular endothelial growth factor in the stimulation of cellular invasion and signaling of breast cancer cells. Cell Growth Differ. 2001, 12, 129–135. [Google Scholar] [PubMed]
- Lacal, P.M.; Ruffini, F.; Pagani, E.; D’Atri, S. An autocrine loop directed by the vascular endothelial growth factor promotes invasiveness of human melanoma cells. Int. J. Oncol. 2005, 27, 1625–1632. [Google Scholar] [PubMed]
- Nakanishi, R.; Oka, N.; Nakatsuji, H.; Koizumi, T.; Sakaki, M.; Takahashi, M.; Fukumori, T.; Kanayama, H.O. Effect of vascular endothelial growth factor and its receptor inhibitor on proliferation and invasion in bladder cancer. Urol. Int. 2009, 83, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Seeram, N.P.; Lee, R.; Heber, D. Bioavailability of ellagic acid in human plasma after consumption of ellagitannins from pomegranate (Punica granatum L.) juice. Clin. Chim. Acta 2004, 348, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, X.; Zu, Y.; Zhang, Y.; Ge, Y.; Zhong, C.; Wu, W. Preparation and characterization of micronized ellagic acid using antisolvent precipitation for oral delivery. Int. J. Pharm. 2015, 486, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Teply, B.A.; Kim, J.J. Systemic therapy for bladder cancer—A medical oncologist's perspective. J. Solid Tumors 2014, 4, 25–35. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | EA IC50 MTS Assay (μM) a | EA IC50 Foci Assay (μM) a |
|---|---|---|
| T24 | 21 ± 1.5 | 7.1 ± 0.5 |
| UM-UC-3 | 37.8 ± 0.7 | 7.9 ± 1.3 |
| 5637 | 26.7 ± 1.8 | 7.6 ± 1.1 |
| HT-1376 | 58.8 ± 3.4 | 19.1 ± 0.5 |
| Cell Line | IC50 Mitomycin C a (nM) | Mitomycin C (M)+ EA CI at Different FA(%) b | Dose Reduction Index (DRI) c | ||
|---|---|---|---|---|---|
| 40 | 50 | 60 | |||
| (M nM + EA µM) | |||||
| T24 | 203 ± 7.5 | 1 (92 + 10) | 1.1 (138 + 15) | 1.2 (185 + 18) | 1.9 |
| UM-UC-3 | 504.9 ± 89.5 | 0.9 (185 + 15) | 0.9 (270 + 20) | 0.9 (360 + 25) | 2.3 |
| 5637 | 451.2 ± 52.4 | 0.8 (180 + 10) | 0.8 (270 + 15) | 0.8 (360 + 20) | 2.2 |
| HT-1376 | 273 ± 34.1 | 0.9 (90 + 7.5) | 0.7 (130 + 15) | 0.7 (240 + 30) | 2.6 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceci, C.; Tentori, L.; Atzori, M.G.; Lacal, P.M.; Bonanno, E.; Scimeca, M.; Cicconi, R.; Mattei, M.; De Martino, M.G.; Vespasiani, G.; et al. Ellagic Acid Inhibits Bladder Cancer Invasiveness and In Vivo Tumor Growth. Nutrients 2016, 8, 744. https://doi.org/10.3390/nu8110744
Ceci C, Tentori L, Atzori MG, Lacal PM, Bonanno E, Scimeca M, Cicconi R, Mattei M, De Martino MG, Vespasiani G, et al. Ellagic Acid Inhibits Bladder Cancer Invasiveness and In Vivo Tumor Growth. Nutrients. 2016; 8(11):744. https://doi.org/10.3390/nu8110744
Chicago/Turabian StyleCeci, Claudia, Lucio Tentori, Maria Grazia Atzori, Pedro M. Lacal, Elena Bonanno, Manuel Scimeca, Rosella Cicconi, Maurizio Mattei, Maria Gabriella De Martino, Giuseppe Vespasiani, and et al. 2016. "Ellagic Acid Inhibits Bladder Cancer Invasiveness and In Vivo Tumor Growth" Nutrients 8, no. 11: 744. https://doi.org/10.3390/nu8110744