The Non-Genomic Actions of Vitamin D

{kind=link}
Abstract
:1. Introduction
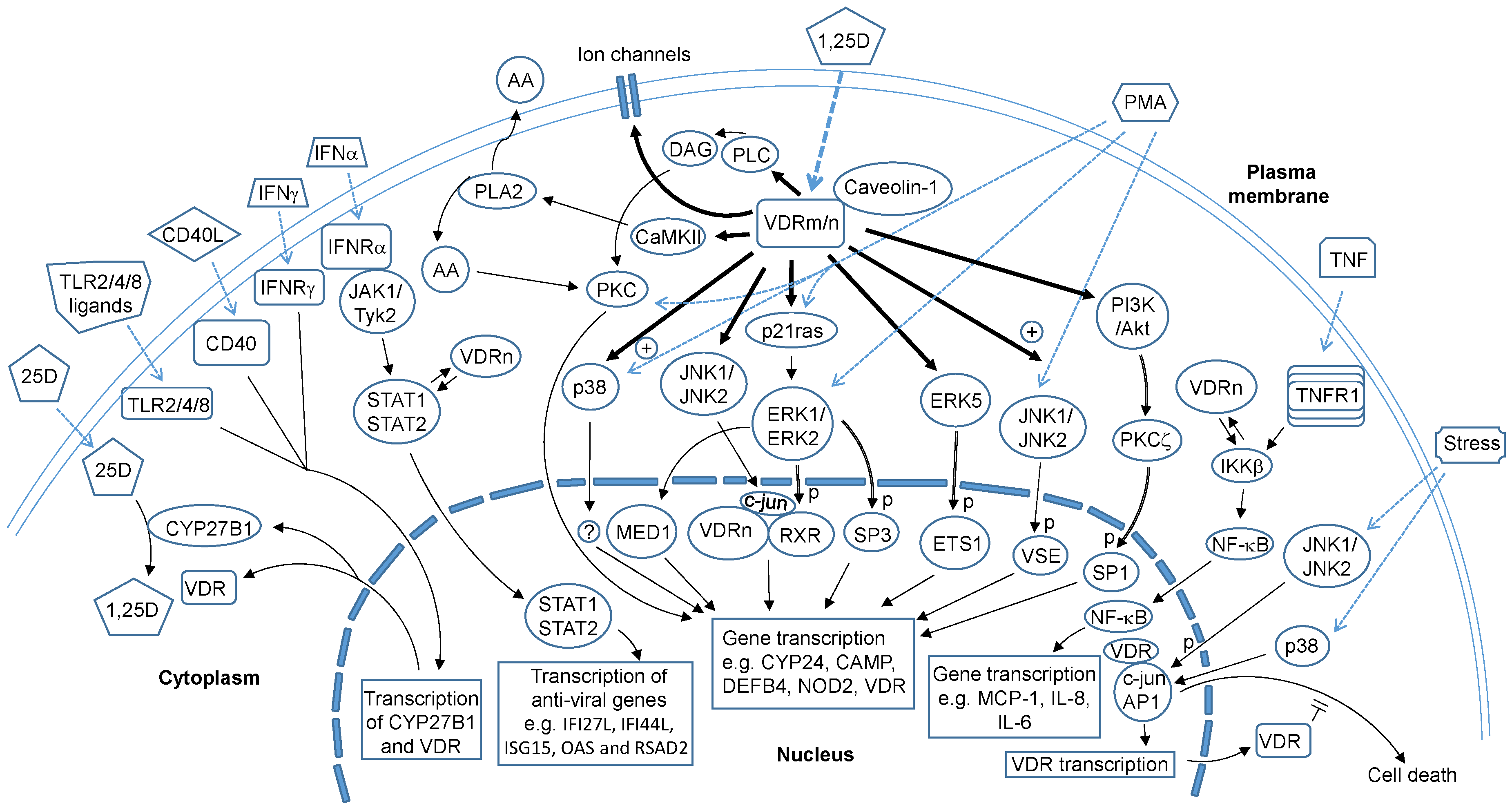
2. Vitamin D and Non-Genomic Actions
3. Vitamin D Receptors
4. VDRn and VDRm in Intracellular Signalling and Mechanism of Action
5. Regulation of 1,25D-Mediated Gene Transcription by Non-Genomic Actions
6. Non-Classical, Non-Genomic Action of the VDR that Involves Protein-Protein Interaction to Regulate Gene Transcription
7. Effects of Vitamin D Supplementation on Intracellular Signalling
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| RDA | Recommended Dietary Allowance |
| 25D | 25-hydroxyvitamin D |
| 1,25D | 1,25 dihydroxyvitamin D |
| VDRm/n | Membrane or nuclear vitamin D receptor |
| CYP27B1 | ytochrome P450C1α |
| CYP24 | Cytochrome P450C24 |
| RXR | Retinoid-X-Receptor |
| PLA2 | Phospholipase A2 |
| PI3K | Phosphatidylinositol 3-kinase |
| MAP kinase | Mitogen-activated protein kinase |
| PKC | Protein kinase C |
| MARRS | Membrane-associated rapid response steroid |
| GRP58 | Glucose responsive protein, 58 kDa |
| ERp57 and ERp60 | Endoplasmic reticulum protein 57/60 |
| Pdia3 | Protein Disulfide Isomerase Family A, Member 3 |
| RNAi | RNA interference |
| Akt | Protein kinase B |
| shRNA | Short hairpin RNA |
| ERK | Extracellular signal Regulated protein Kinase |
| JNK | c-Jun N-terminal Kinase |
| TRPV6 | Transient receptor potential cation channel, subfamily V, member 6 |
| MKK | MAP kinase ERK kinase |
| PMA | Phorbol 12-myristate 13-acetate |
| VSE | vitamin D stimulatory element |
| IL | Interleukin |
| IKK | I-κB kinase |
| Th | T-helper |
| NFAT | Nuclear factor of activated T cells |
| RunX | Runt-related transcription factor |
| AP1 | Activator protein 1 |
| IFN | Interferon |
| HCV | Hepatitis C virus |
| IFI27L | Interferon, Alpha-Inducible Protein 27 like |
| IFI44L | Interferon-Induced Protein 44-Like |
| ISG15 | Interferon-Stimulated Protein, 15 KDa |
| OAS | 2′-5′-Oligoadenylate Synthetase |
| RSAD2 | Radical S-Adenosyl Methionine Domain Containing 2 |
| AMPK | AMP-activated protein kinase |
| TLR | Toll-like receptor |
| DEFB4 | Human β-defensin 2 |
| TNF | Tumour necrosis factor |
References
- Gröber, U.; Reichrath, J.; Holick, M.F. Live longer with vitamin D? Nutrients 2015, 7, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Mazahery, H.; von Hurst, P.R. Factors Affecting 25-Hydroxyvitamin D Concentration in Response to Vitamin D Supplementation. Nutrients 2015, 7, 5111–5142. [Google Scholar] [CrossRef] [PubMed]
- Veugelers, P.J.; Ekwaru, J.P. A statistical error in the estimation of the recommended dietary allowance for vitamin D. Nutrients 2014, 6, 4472–4475. [Google Scholar] [CrossRef] [PubMed]
- Heaney, R.; Garland, C.; Baggerly, C.; French, C.; Gorham, E. Letter to Veugelers, P.J. and Ekwaru, J.P. A statistical error in the estimation of the recommended dietary allowance for vitamin D. Nutrients 2014, 6, 4472–4475, doi:10.3390/nu6104472. Nutrients 2015, 7, 1688–1690. [Google Scholar]
- Paller, C.J.; Kanaan, Y.M.; Beyene, D.A.; Naab, T.J.; Copeland, R.L.; Tsai, H.L.; Kanarek, N.F.; Hudson, T.S. Risk of prostate cancer in African-American men: Evidence of mixed effects of dietary quercetin by serum vitamin D status. Prostate 2015, 75, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Strugnell, S.A.; DeLuca, H.F. Current understanding of the molecular actions of vitamin D. Physiol. Rev. 1998, 78, 1193–1231. [Google Scholar] [PubMed]
- Christakos, S.; Pandya, M.R.; Yang, W. Genomic mechanisms involved in the pleiotropic actions of 1,25-dihydroxyvitamin D3. Biochem. J. 1996, 316, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Hewison, M.; O’Riordan, J.L.H. Immunomodulatory and cell differentiation effects of vitamin D. In Vitamin D; Feldman, D., Glorieux, F.H., Pike, J.W., Eds.; Academic Press: San Diego, CA, USA, 1997; pp. 447–462. [Google Scholar]
- Issa, L.L.; Leong, G.M.; Eisman, J.A. Molecular mechanism of vitamin D receptor action. Inflamm. Res. 1998, 47, 451–475. [Google Scholar] [CrossRef] [PubMed]
- Omdahl, J.L.; May, B.K. The 25-hydroxyvitamin D 24-hydroxylase. In Vitamin D; Feldman, D., Glorieux, F.H., Pike, J.W., Eds.; Academic Press: San Diego, CA, USA, 1997; pp. 69–85. [Google Scholar]
- Fleet, J.C. Rapid, membrane-initiated actions of 1,25 dihydroxyvitamin D: What are they and what do they mean? J. Nutr. 2004, 134, 3215–3218. [Google Scholar] [PubMed]
- Doroudi, M.; Schwartz, Z.; Boyan, B.D. Membrane-mediated actions of 1,25-dihydroxy vitamin D3: A review of the roles of phospholipase A2 activating protein and Ca(2+)/calmodulin-dependent protein kinase II. J. Steroid Biochem. Mol. Biol. 2015, 147, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, P.P.; Hii, C.S.; Ferrante, A.; Tan, J.; Der, C.J.; Omdahl, J.L.; Morris, H.A.; May, B.K. Role of MAP kinases in the 1,25-dihydroxyvitamin D3-induced transactivation of the rat cytochrome P450C24 (CYP24) promoter. Specific functions for ERK1/ERK2 and ERK5. J. Biol. Chem. 2002, 277, 29643–29653. [Google Scholar] [CrossRef] [PubMed]
- Nutchey, B.K.; Kaplan, J.S.; Dwivedi, P.P.; Omdahl, J.L.; Ferrante, A.; May, B.K.; Hii, C.S. Molecular action of 1,25-dihydroxyvitamin D3 and phorbol ester on the activation of the rat cytochrome P450C24 (CYP24) promoter: Role of MAP kinase activities and identification of an important transcription factor binding site. Biochem. J. 2005, 389, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, P.P.; Gao, X.H.; Tan, J.C.; Evdokiou, A.; Ferrante, A.; Morris, H.A.; May, B.K.; Hii, C.S. A role for the phosphatidylinositol 3-kinase—Protein kinase C zeta—Sp1 pathway in the 1,25-dihydroxyvitamin D3 induction of the 25-hydroxyvitamin D3 24-hydroxylase gene in human kidney cells. Cell. Signal. 2010, 22, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.W. Minireview: Vitamin D receptor: New assignments for an already busy receptor. Endocrinology 2006, 147, 5542–5548. [Google Scholar] [CrossRef] [PubMed]
- Dormanen, M.C.; Bishop, J.E.; Hammond, M.W.; Odamura, W.H.; Nemere, I.; Norman, A.W. Nonnuclear effects of the steroid hormone 1 alpha, 25(OH)2-vitamin D3: Analogs are able to functionally differentiate between nuclear and membrane receptors. Biochem. Biophys. Res. Commun. 1994, 201, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Nemere, I.; Dormanen, M.C.; Hammond, M.W.; Okamura, W.H.; Norman, A.W. Identification of a specific binding protein for 1,25-dihydroxyvitamin D3 in basal-lateral membranes of chick intestinal epithelium and relationship to transcaltachia. J. Biol. Chem. 1994, 269, 23750–23756. [Google Scholar] [PubMed]
- Huhtakangas, J.A.; Olivera, C.J.; Bishop, J.E.; Zanello, L.P.; Norman, A.W. The vitamin D receptor is present in caveolae-enriched plasma membranes and binds 1 alpha, 25(OH)2-vitamin D3 in vivo and in vitro. Mol. Endocrinol. 2004, 18, 2660–2671. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Doroudi, M.; Cheung, J.; Grozier, A.L.; Schwartz, Z.; Boyan, B.D. Plasma membrane Pdia3 and VDR interact to elicit rapid responses to 1α,25(OH)(2)D(3). Cell. Signal. 2013, 25, 2362–2373. [Google Scholar] [CrossRef] [PubMed]
- Boyan, B.D.; Sylvia, V.L.; McKinney, N.; Schwartz, Z. Membrane actions of vitamin D metabolites 1α,25(OH)2D3 and 24R,25(OH)2D3 are retained in growth plate cartilage cells from vitamin D receptor knockout mice. J. Cell. Biochem. 2003, 90, 1207–1223. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, J.; Sekiguchi, M.; Shirai, T.; Ishimi, Y. Vitamin D receptor is not essential for extracellular signal-related kinase phosphorylation by vitamin D(3) in human Caco-2/TC7 cells. Biosci. Biotechnol. Biochem. 2012, 76, 1588–1590. [Google Scholar] [CrossRef] [PubMed]
- Buitrago, C.; Pardo, V.G.; Boland, R. Role of VDR in 1α,25-dihydroxyvitamin D3-dependent non-genomic activation of MAPKs, Src and Akt in skeletal muscle cells. J. Steroid Biochem. Mol. Biol. 2013, 136, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Antal, C.E.; Newton, A.C. Tuning the signalling output of protein kinase C. Biochem. Soc. Trans. 2014, 42, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, V.B.; Rybchyn, M.S.; Tongkao-On, W.; Gordon-Thomson, C.; Malloy, P.J.; Nemere, I.; Norman, A.W.; Reeve, V.E.; Halliday, G.M.; Feldman, D.; et al. The role of the vitamin D receptor and ERp57 in photoprotection by 1α,25-dihydroxyvitamin D3. Mol. Endocrinol. 2012, 26, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Dwivedi, P.P.; Anderson, P.; Nutchey, B.K.; O’Loughlin, P.; Morris, H.A.; May, B.K.; Ferrante, A.; Hii, C.S. Antineoplastic agents target the 25-hydroxyvitamin D3 24-hydroxylase messenger RNA for degradation: Implications in anticancer activity. Mol. Cancer Ther. 2007, 6, 3131–3138. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pesakhov, S.; Harrison, J.S.; Danilenko, M.; Studzinski, G.P. ERK5 pathway regulates transcription factors important for monocytic differentiation of human myeloid leukemia cells. J. Cell. Physiol. 2014, 229, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, Z.; Ehland, H.; Sylvia, V.L.; Larsson, D.; Hardin, R.R.; Bingham, V.; Lopez, D.; Dean, D.D.; Boyan, B.D. 1alpha,25-dihydroxyvitamin D(3) and 24R,25-dihydroxyvitamin D(3) modulate growth plate chondrocyte physiology via protein kinase C-dependent phosphorylation of extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase. Endocrinology 2002, 143, 2775–2786. [Google Scholar] [PubMed]
- Cui, M.; Zhao, Y.; Hance, K.W.; Shao, A.; Wood, R.J.; Fleet, J.C. Effects of MAPK signalling on 1,25-dihydroxyvitamin D-mediated CYP24 gene expression in the enterocyte-like cell line, Caco-2. J. Cell. Physiol. 2009, 21, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Fleet, J.C. Phorbol esters enhance 1α,25-dihydroxyvitamin D3-regulated 25-hydroxyvitamin D-24-hydroxylase (CYP24A1) gene expression through ERK-mediated phosphorylation of specific protein 3 (Sp3) in Caco-2 cells. Mol. Cell. Endocrinol. 2012, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Simboli-Campbell, M.; Gagnon, A.; Franks, D.J.; Welsh, J. 1,25-dihydroxyvitamin D3 translocates protein kinase C beta to nucleus and enhances plasma membrane association of protein kinase C alpha in renal epithelial cells. J. Biol. Chem. 1994, 269, 3257–3264. [Google Scholar] [PubMed]
- Chen, Y.; Zhang, J.; Ge, X.; Du, J.; Deb, D.K.; Li, Y.C. Vitamin D receptor inhibits nuclear factor κB activation by interacting with IκB kinase β protein. J. Biol. Chem. 2013, 288, 19450–19458. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.M.; Gouttenoire, J.; Duong, F.H.; Morikawa, K.; Heim, M.H.; Moradpour, D. Vitamin D receptor and Jak-STAT signalling crosstalk results in calcitriol-mediated increase of hepatocellularresponse to IFN-α. J. Immunol. 2014, 192, 6037–6044. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Christakos, S. Mechanisms Underlying the Regulation of Innate and Adaptive Immunity by Vitamin D. Nutrients 2015, 7, 8251–8260. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.P.; Qi, X.; Pramanik, R.; Pohl, N.M.; Loesch, M.; Chen, G. Stress-induced c-Jun-dependent Vitamin D receptor (VDR) activation dissects the non-classical VDR pathway from the classical VDR activity. J. Biol. Chem. 2007, 282, 1544–1551. [Google Scholar] [CrossRef] [PubMed]
- Pálmer, H.G.; González-Sancho, J.M.; Espada, J.; Berciano, M.T.; Puig, I.; Baulida, J.; Quintanilla, M.; Cano, A.; de Herreros, A.G.; Lafarga, M.; et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J. Cell. Biol. 2001, 154, 369–387. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Pan, W.; Kong, J.; Zheng, W.; Szeto, F.L.; Wong, K.E.; Cohen, R.; Klopot, A.; Zhang, Z.; Li, Y.C. 1,25-Dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP AMP response element in the renin gene promoter. J. Biol. Chem. 2007, 282, 29821–29830. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Wilding, J.P.; Bing, C. 1,25-dihydroxyvitamin D3 protects against macrophage-induced activation of NFκB and MAPK signalling and chemokine release in human adipocytes. PLoS ONE 2013, 24, e61707. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kong, J.; Sun, T.; Li, G.; Szeto, F.L.; Liu, W.; Deb, D.K.; Wang, Y.; Zhao, Q.; Thadhani, R.; et al. 1,25-Dihydroxyvitamin D(3) suppresses inflammation-induced expression of plasminogen activator inhibitor-1 by blocking nuclear factor-κB activation. Arch. Biochem. Biophys. 2011, 507, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, W.; Sun, T.; Huang, Y.; Wang, Y.; Deb, D.K.; Yoon, D.; Kong, J.; Thadhani, R.; Li, Y.C. 1,25-Dihydroxyvitamin D promotes negative feedback regulation of TLR signalling via targeting microRNA-155-SOCS1 in macrophages. J. Immunol. 2013, 190, 3687–3695. [Google Scholar] [CrossRef] [PubMed]
- Deb, D.K.; Chen, Y.; Zhang, Z.; Zhang, Y.; Szeto, F.L.; Wong, K.E.; Kong, J.; Li, Y.C. 1,25-Dihydroxyvitamin D3 suppresses high glucose-induced angiotensinogen expression in kidney cells by blocking the NF-kappa B pathway. Am. J. Physiol. Ren. Physiol. 2009, 296, F1212–F1218. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, D.; Cippitelli, M.; Cocciolo, M.G.; Mazzeo, D.; Di Lucia, P.; Lang, R.; Sinigaglia, F.; Panina-Bordignon, P. Inhibition of IL-12 production by 1,25-dihydroxyvitamin D3. Involvement of NF-kappaB downregulation in transcriptional repression of the p40 gene. J. Clin. Investig. 1998, 101, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.M.; Bojunga, J.; Ramos-Lopez, E.; von Wagner, M.; Hassler, A.; Vermehren, J.; Herrmann, E.; Badenhoop, K.; Zeuzem, S.; Sarrazin, C. Vitamin D deficiency and a CYP27B1–1260 promoter polymorphism are associated with chronic hepatitis C and poor response to interferon-alfa based therapy. J. Hepatol. 2011, 54, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Gal-Tanamy, M.; Bachmetov, L.; Ravid, A.; Koren, R.; Erman, A.; Tur-Kaspa, R.; Zemel, R. Vitamin D: An innate antiviral agent suppressing hepatitis C virus in human hepatocytes. Hepatology 2011, 54, 1570–1579. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Kato, T.; Sugiyama, N.; Tasaka-Fujita, M.; Murayama, A.; Masaki, T.; Wakita, T.; Imawari, M. 25-Hydroxyvitamin D3 suppresses hepatitis C virus production. Hepatology 2012, 56, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Greiller, C.L.; Martineau, A.R. Modulation of the immune response to respiratory viruses by vitamin D. Nutrients 2015, 7, 4240–4270. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Camma`, C.; Scazzone, C.; Tripodo, C.; Di Marco, V.; Bono, A.; Cabibi, D.; Licata, G.; Porcasi, R.; Marchesini, G.; et al. Low vitamin D serum level is related to severe fibrosis and low responsiveness to interferon-based therapy in genotype 1 chronic hepatitis C. Hepatology 2010, 51, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Kroner Jde, C.; Sommer, A.; Fabri, M. Vitamin D every day to keep the infection away? Nutrients 2015, 7, 4170–4188. [Google Scholar] [CrossRef] [PubMed]
- Chun, R.F.; Liu, P.T.; Modlin, R.L.; Adams, J.S.; Hewison, M. Impact of vitamin D on immune function: Lessons learned from genome-wide analysis. Front. Physiol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Peric, M.; Koglin, S.; Dombrowski, Y.; Gross, K.; Bradac, E.; Ruzicka, T.; Schauber, J. VDR and MEK-ERK dependent induction of the antimicrobial peptide cathelicidin in keratinocytes by lithocholic acid. Mol. Immunol. 2009, 46, 3183–3187. [Google Scholar] [CrossRef] [PubMed]
- Yim, S.; Dhawan, P.; Ragunath, C.; Christakos, S.; Diamond, G. Induction of cathelicidin in normal and cf bronchial epithelial cells by 1,25-dihydroxyvitamin D(3). J. Cyst. Fibros. 2007, 6, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Autophagy induction by vitamin D inhibits both Mycobacterium tuberculosis and human immunodeficiency virus type 1. Autophagy 2012, 8, 1523–1525. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Toll-like receptor 8 ligands activate a vitamin D mediated autophagic response that inhibits human immunodeficiency virus type 1. PLoS Pathog. 2012, 8, e1003017. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Tang, J.; Pramanik, R.; Schultz, R.M.; Shirasawa, S.; Sasazuki, T.; Han, J.; Chen, G. P38 MAPK activation selectively induces cell death in K-ras-mutated human colon cancer cells through regulation of vitamin D receptor. J. Biol. Chem. 2004, 279, 22138–22144. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Pramanik, R.; Wang, J.; Schultz, R.M.; Maitra, R.K.; Han, J.; DeLuca, H.F.; Chen, G. The p38 and JNK pathways cooperate to trans-activate vitamin D receptor via c-Jun/AP-1 and sensitize human breast cancer cells to vitamin D(3)-induced growth inhibition. J. Biol. Chem. 2002, 277, 25884–25892. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Shi, D.; Zhou, X.Q.; Yin, L.; Feng, L.; Jiang, W.D.; Liu, Y.; Tang, L.; Wu, P.; Zhao, Y. Vitamin D inhibits lipopolysaccharide-induced inflammatory response potentially through the Toll-like receptor 4 signalling pathway in the intestine and enterocytes of juvenile Jian carp (Cyprinus carpio var. Jian). Br. J. Nutr. 2015, 114, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Al-Rasheed, N.M.; Al-Rasheed, N.M.; Bassiouni, Y.A.; Hasan, I.H.; Al-Amin, M.A.; Al-Ajmi, H.N.; Mohamad, R.A. Vitamin D attenuates pro-inflammatory TNF-α cytokine expression by inhibiting NF-κB/p65 signaling in hypertrophied rat hearts. J. Physiol. Biochem. 2015, 71, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Park, H.; Cho, S.; Lee, M. Vitamin D3 supplementation modulates inflammatory responses from the muscle damage induced by high-intensity exercise in SD rats. Cytokine 2013, 63, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cao, J.T.; Liang, Y.; Zhao, Y.C.; Lin, X.H.; Li, X.C.; Tan, Y.J.; Li, J.; Zhou, C.L.; Xu, H.Y.; et al. Calcitriol attenuates cardiac remodeling and dysfunction in a murine model of polycystic ovary syndrome. Endocrine 2015. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Yun, J.M.; Duncan-Staley, C.R.; Jialal, I. Low vitamin D levels correlate with the proinflammatory state in type 1 diabetic subjects with and without microvascular complications. Am. J. Clin. Pathol. 2011, 135, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.M.; Gorman, S.; Geldenhuys, S.; Hart, P.H. Vitamin D and immunity. F1000Prime Rep. 2014, 6, 118. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hii, C.S.; Ferrante, A. The Non-Genomic Actions of Vitamin D. Nutrients 2016, 8, 135. https://doi.org/10.3390/nu8030135
Hii CS, Ferrante A. The Non-Genomic Actions of Vitamin D. Nutrients. 2016; 8(3):135. https://doi.org/10.3390/nu8030135
Chicago/Turabian StyleHii, Charles S, and Antonio Ferrante. 2016. "The Non-Genomic Actions of Vitamin D" Nutrients 8, no. 3: 135. https://doi.org/10.3390/nu8030135
APA StyleHii, C. S., & Ferrante, A. (2016). The Non-Genomic Actions of Vitamin D. Nutrients, 8(3), 135. https://doi.org/10.3390/nu8030135