Diagnosing and Treating Intolerance to Carbohydrates in Children

Abstract
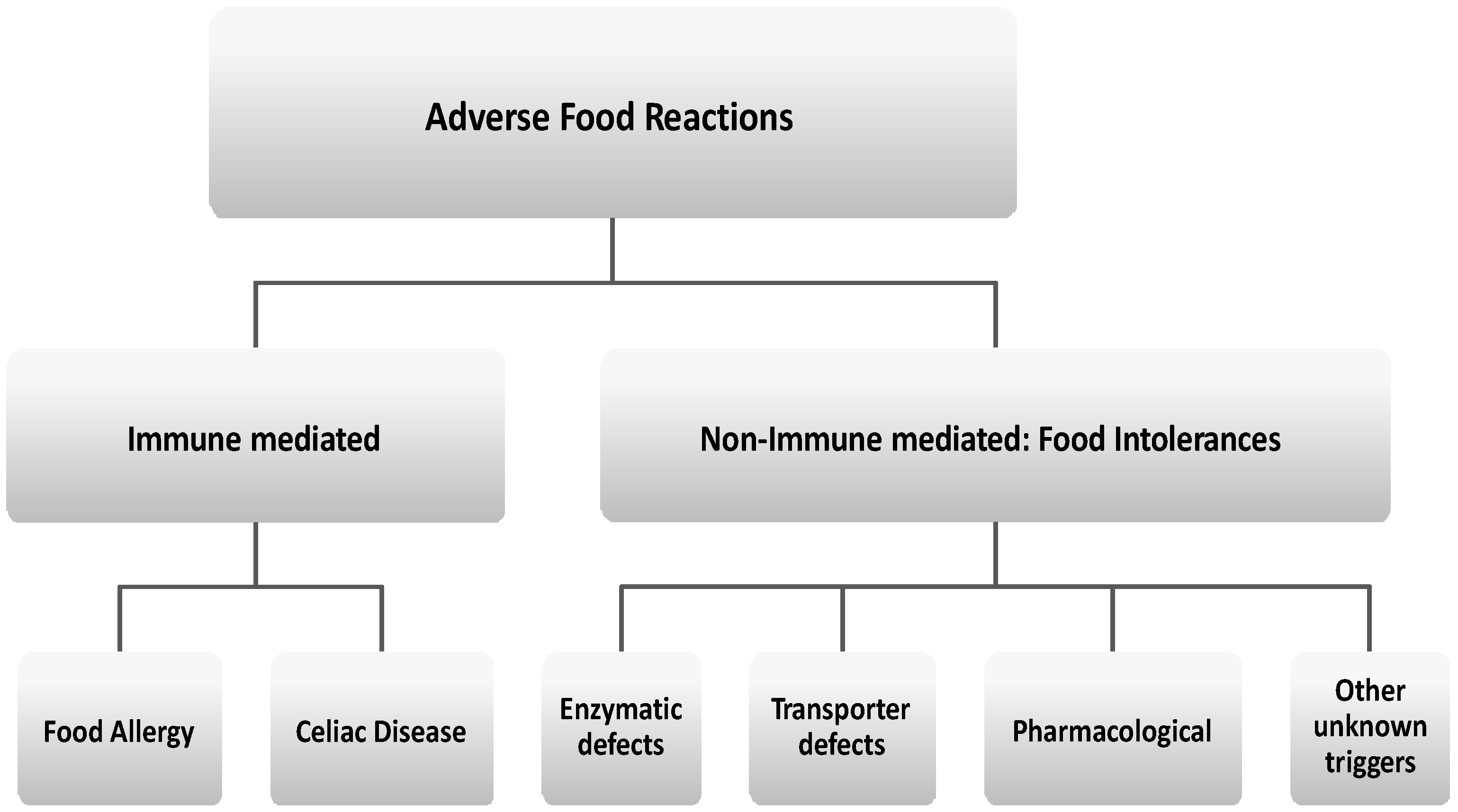
:1. Introduction
2. Genetic Etiology with Early Onset Carbohydrate Intolerances
2.1. Congenital Sucrase-Isomaltase Deficiency (CSID)
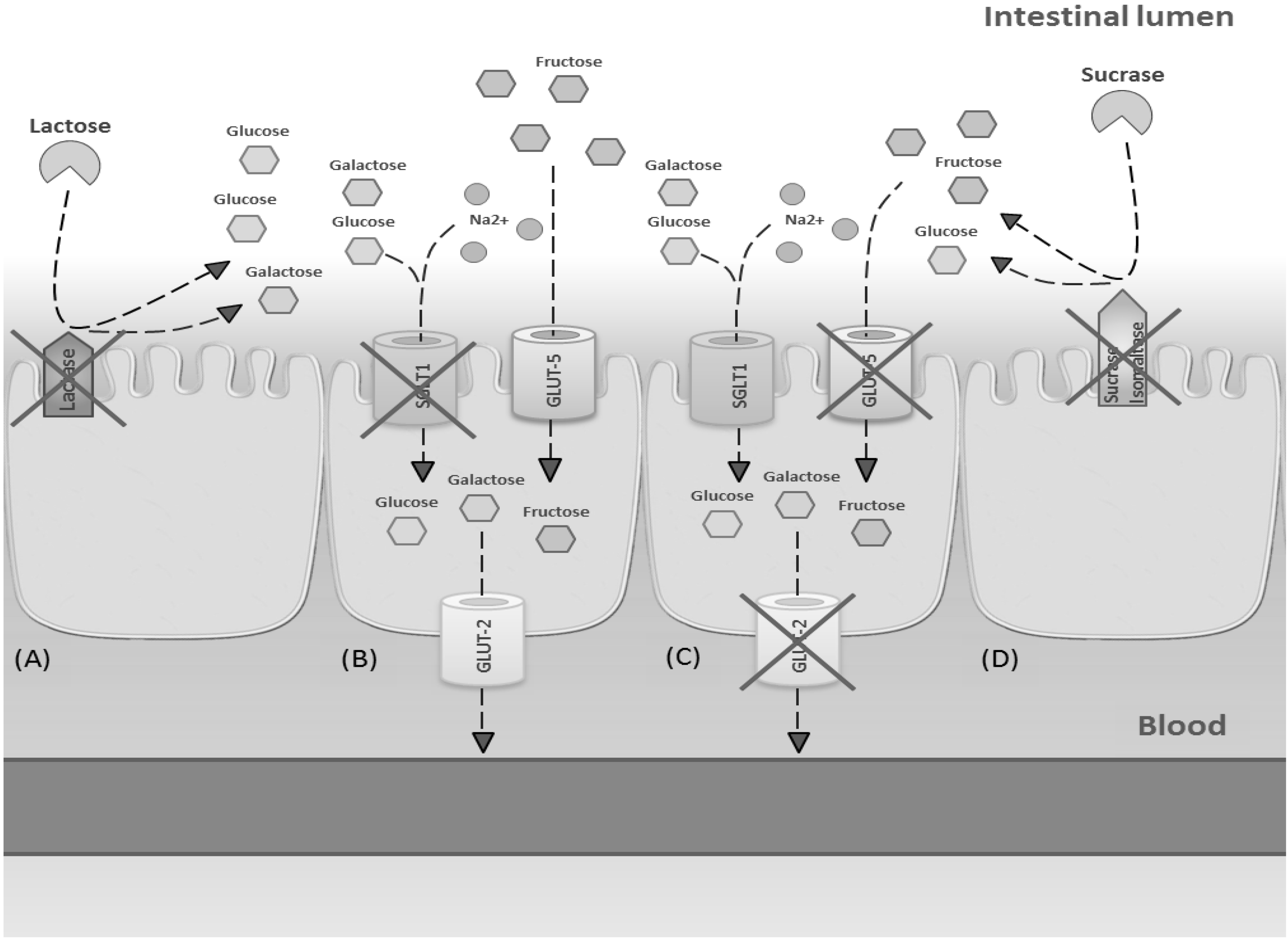
2.2. Glucose-Galactose Malabsorption
3. Genetic Etiology with Late-Onset Carbohydrate Intolerances
Lactose Intolerance
- Congenital lactase deficiency: a rare autosomal recessive disease where enzymatic activity is absent or reduced from birth;
- Secondary lactase deficiency: a transient condition deriving from intestinal damage secondary to small bowel bacterial overgrowth, infections, celiac disease, Crohn’s disease, or radiation enteritis;
- Adult type lactase deficiency: an autosomal recessive condition resulting from a developmentally regulated change of the lactase gene product, responsible for reduced synthesis of the precursor protein.
4. Non-Genetic Etiology Carbohydrate Intolerances
4.1. Fructose Malabsorption
4.2. Sorbitol Intolerance
4.3. Trehalose Intolerance
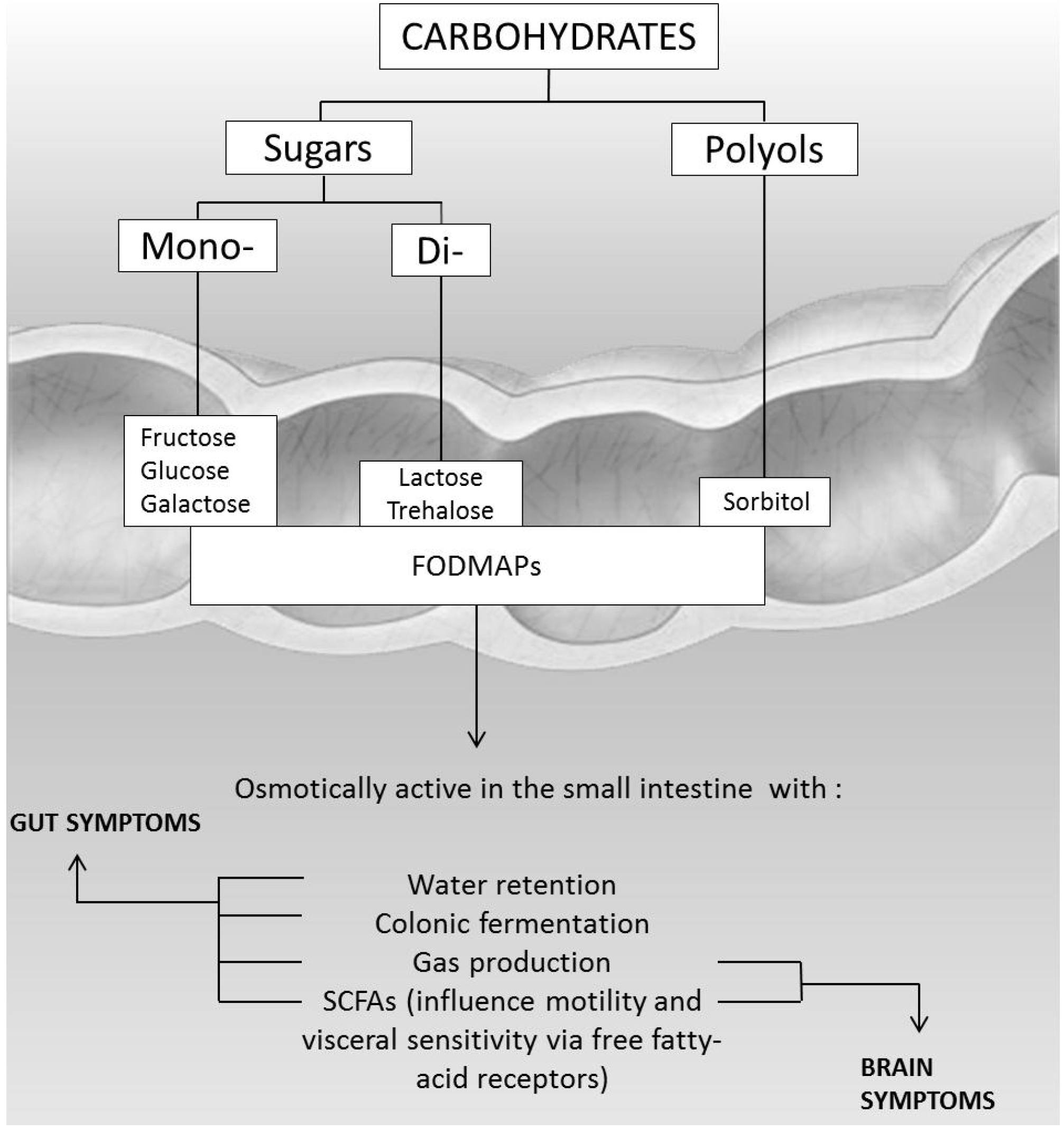
4.4. FODMAPs Intolerance
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lomer, M.C.E. The aetiology, diagnosis, mechanisms and clinical evidence for food intolerance. Aliment. Pharmacol. Ther. 2015, 41, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Hammer, H.F.; Hammer, J. Diarrhea caused by carbohydrate malabsorption. Gastroenterol. Clin. North. Am. 2012, 41, 611–627. [Google Scholar] [CrossRef] [PubMed]
- Zar, S.; Kumar, D.; Benson, M.J. Food hypersensitivity and irritable bowel syndrome. Aliment. Pharmacol. Ther. 2001, 15, 439–449. [Google Scholar] [CrossRef]
- Dupont, C. Food allergy: recent advances in pathophysiology and diagnosis. Ann. Nutr. Metab. 2011, 59. [Google Scholar] [CrossRef] [PubMed]
- Ortolani, C. Atlas on mechanisms in adverse reaction to food. Introduction. Allergy 1995, 50, 5. [Google Scholar] [CrossRef] [PubMed]
- Sicherer, S.H.; Leung, D.Y. Advances in allergic skin disease, anaphylaxis, and hypersensitivity reactions to foods, drugs, and insects in 2010. J. Allergy Clin. Immunol. 2011, 127, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Vickery, B.P.; Chin, S.; Burks, A.W. Pathophysiology of food allergy. Pediatr. Clin. North Am. 2011, 58, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Waserman, S.; Watson, W. Food allergy. Allergy Asthma Clin. Immunol. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Schulze, M.B.; Hu, F.B. Intake of sugar sweetened beverages and weight gain: A systematic review. Am. J. Clin. Nutr. 2006, 84, 274–288. [Google Scholar] [PubMed]
- Diet, M.N.; Talley, N.J. Are adverse food reactions linked to irritable bowel syndrome? Am. J. Gastroenterol. 1998, 93, 2184–2190. [Google Scholar]
- Latulippe, M.E.; Skoog, S.M. Fructose malabsorption and intolerance: Effects of fructose with and without simultaneous glucose ingestion. Clin. Rev. Food Sci. Nutr. 2011, 51, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Born, P.; Sekatcheva, M.; Rosch, T.; Classen, M. Carbohydrate malabsorption in clinical routine: A prospective observational study. Hepatogastroenterology 2006, 53, 673–677. [Google Scholar] [PubMed]
- Fernandez-Banares, F.; Rosinach, M.; Esteve, M.; Forne, M.; Espinos, J.C.; Viver, J.M. Sugar malabsorption in functional abdominal bloating: A pilot study on the long-term effect of dietary treatment. Clin. Nutr. 2006, 25, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Raithel, M.; Weidenhiller, M.; Hagel, A.F.K.; Hetterich, U.; Neurath, M.F.; Konturek, P.C. The malabsorption of commonly occurring mono and disaccharides: levels of investigation and differential diagnoses. Dtsch. Arztebl. Int. 2013, 110, 775–782. [Google Scholar] [PubMed]
- Matthews, S.B.; Waud, J.P.; Roberts, A.G.; Campbell, A.K. Systemic lactose intolerance: a new perspective on an old problem. Postgrad. Med. J. 2005, 81, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.K.; Matthews, S.B.; Vassel, N.; Cox, C.D.; Naseem, R.; Chaichi, J.; Holland, I.B.; Green, J.; Wann, K.T. Bacterial metabolic “toxins”: A new mechanism for lactose and food intolerance, and irritable bowel syndrome. Toxicology 2010, 278, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Ritz, V.; Alfalah, M.; Zimmer, K.F.; Schmitz, J.; Jacob, R.; Naim, H.Y. Congenital sucrose-isomaltase deficiency because of an accumulation of the mutant enzyme in the endoplasmic reticulum. Gastroenterology 2003, 125, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.; Zimmer, K.P.; Schmitz, J.; Naim, H.Y. Congenital sucrase-isomaltase deficiency arising from cleavage and secretion of a mutant form of the enzyme. J. Clin. Investig. 2000, 106, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Alfalah, M.; Keiser, M.; Leeb, T.; Zimmer, K.P.; Nami, H.Y. Compound heterozygous mutations affect protein folding and function in patients with congenital sucrase-isomaltase deficiency. Gastroenterology 2009, 136, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Ouwendijk, J.; Moolenaar, C.E.; Peters, W.J.; Hollenberg, C.P.; Ginsel, L.A.; Fransen, J.A.; Naim, H.Y. Congenital sucrase-isomaltase deficiency. Identification of a glutamine to proline substitution that leads to a transport block of sucrase-isomaltase in a pre-Golgi compartment. J. Clin. Investig. 1996, 97, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Treem, W.R. Clinical aspects and treatment of congenital sucrase isomaltase deficiency. J. Pediatr. Gastroenterol. Nutr. 2012, 55, S7–S13. [Google Scholar] [CrossRef] [PubMed]
- Marcadier, J.L.; Boland, M.; Scott, C.R.; Issa, K.; Wu, Z.; McIntyre, A.D.; Hegele, R.A.; Geraghty, M.T.; Lines, M.A. Congenital sucrase-isomaltase deficiency: Identification of a common Inuit founder mutation. Can. Med. Assoc. J. 2015, 187, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Li, D.-Y.; Ou, W.; Yang, Q.; Fang, T.; Chen, P.; Yang, M.; Gong, S. Congenital sucrase-isomaltase deficiency: An under-diagnosed disease in Chinese children. BMC Pediatr. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Uhrich, S.; Wu, Z.; Huang, J.-Y.; Scott, C.R. Four mutations in the SI gene are responsible for the majority of clinical symptoms of CSID. J. Pediatr. Gastroenterol. Nutr. 2012, 55, S34–S35. [Google Scholar] [CrossRef] [PubMed]
- Robayo-Torres, C.C.; Opekun, A.R.; Quezada-Calvillo, R.; Xavier, V.; Smith, E.O.; Navarrete, M.; Baker, S.S.; Nichols, B.L. 13C-breath tests for sucrose digestion in congenital sucrase isomaltase-deficient and sacrosidase-supplemented patients. J. Pediatr. Gastroenterol. Nutr. 2009, 48, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Puntis, J.W.; Zamvar, V. Congenital sucrase-isomaltase deficiency: Diagnostic challenges and response to enzyme replacement therapy. Arch. Dis. Child. 2015, 100, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Saadah, O.I.; Alghamdi, S.A.; Sindi, H.H.; Alhunaitti, H.; Bin-Taleb, Y.Y.; Alhussaini, B.H. Congenital glucose-galactose malabsorption: A descriptive study of clinical characteristics and outcome from Western Saudi Arabia. Arab J. Gastroenterol. 2014, 15, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Raja, M.; Kinne, R.K. Structural insights into genetic variants of Na (+)/glucose cotransporter SGLT1 causing glucose-galactose malabsorption: vSGLT as a model structure. Cell Biochem. Biophys. 2012, 63, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M. I. Glucose galactose malabsorption. Am. J. Physiol. 1998, 275, G879–G882. [Google Scholar] [PubMed]
- Pahari, A.; Milla, P.J.; van’t Hoff, W.G. Neonatal nephrocalcinosis in association with glucose–galactose malabsorption. Pediatr. Nephrol. 2003, 18, 700–702. [Google Scholar] [PubMed]
- Vallaeys, L.; van Biervliet, S.; de Bruyn, G.; Loeys, B.; Moring, A.S.; van Deynse, E.; Cornette, L. Congenital glucose–galactose malabsorption: A novel deletion within the SLC5A1 gene. Eur. J. Pediatr. 2013, 172, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Gok, F.; Aydin, H.I.; Kurt, I.; Gokcay, E.; Maeda, M.; Kasahara, M. A novel mutation of Na+/glucose cotransporter in a Turkish newborn with congenital glucose– galactose malabsorption. J. Pediatr. Gastroenterol. Nutr. 2005, 40, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Kianifar, H.R.; Talebi, S.; Tavakkol-Afshari, J.; Esmaili, M.; Davachi, B.; Brook, A. D28G mutation in congenital glucose–galactose malabsorption. Arch. Iran. Med. 2007, 10, 514–518. [Google Scholar] [PubMed]
- Wright, E.M.; Martin, M.G.; Turk, E. Intestinal absorption in health and disease–sugars. Best Pract. Res. Clin. Gastroenterol. 2003, 17, 943–956. [Google Scholar] [CrossRef]
- Uchida, N.; Sakamoto, O.; Irie, M.; Abukawa, D.; Takeyama, J.; Kure, S.; Tsuchiya, S. Two novel mutations in the lactase gene in a Japanese infant with congenital lactase deficiency. Tohoku J. Exp. Med. 2012, 227, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Torniainen, S.; Freddara, R.; Routi, T.; Gijsbers, C.; Catassi, C.; Hoglund, P.; Savilahti, E.; Jarvel, I. Four novel mutations in the lactase gene (LCT) underlying congenital lactase deficiency (CLD). BMC Gastroenterol. 2009, 9. [Google Scholar] [CrossRef] [PubMed]
- Kuokkanen, M.; Kokkonen, J.; Enattah, N.S.; Ylisaukko-oja, T.; Komu, H.; Varilo, T.; Peltonen, L.; Savilahti, E.; Jarvela, I. Mutations in the translated region of the lactase gene (LCT) underlie congenital lactase deficiency. Am. J. Hum. Genet. 2006, 78, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Sala, C.J.; Vinaixa, V.A.; Garcia, P.R. Congenital lactase deficiency: Identification of a new mutation. Anales de Pediatria 2015, 82, 365–366. [Google Scholar]
- Behrendt, M.; Keiser, M.; Hoch, M.; Naim, H.Y. Impaired trafficking and subcellular localization of a mutant lactase associated with congenital lactase deficiency. Gastroenterology 2009, 136, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Diekmann, L.; Pfeiffer, K.; Naim, H.Y. Congenital lactose intolerance is triggered by severe mutations on both alleles of the lactase gene. BMC Gastroenterol. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Schafer, C.; Reese, I.; Ballmer-Weber, B.K.; Beyer, K.; Erdmann, S.; Fuchs, T.; Henzgen, M.; Huttegger, I.; Jappe, U.; Kleine-Tebbe, J. Fruktosemalabsorption: stellungnahme der AG Nahrungsmittelallergie in der Deutschen Gesellschaft fur Allergologie und klinische Immunologie (DGAKI). Allergo J. 2010, 19, 66–69. [Google Scholar]
- Lomer, M.C.E.; Parkes, G.C.; Sanderson, J.D. Lactose intolerance in clinical practice-Myths and realities. Aliment. Pharmacol. Ther. 2008, 27, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Enattah, N.S.; Sahi, T.; Savilahti, E.; Terwilliger, J.D.; Peltonen, L.; Jarvela, I. Identification of a variant associated with adult-type hypolactasia. Nat. Genet. 2002, 30, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Fox, M.; Cong, Y.; Chu, H.; Shang, Y.; Fried, M.; Dai, N. Lactose intolerance in patients with chronic functional diarrhoea: The role of small intestinal bacterial overgrowth. Aliment. Pharmacol Ther. 2010, 31, 892–900. [Google Scholar] [PubMed]
- Tomba, C.; Baldassarri, A.; Coletta, M.; Cesana, B.M.; Basilisco, G. Is the subjective perception of lactose intolerance influenced by the psychological profile? Aliment. Pharmacol. Ther. 2012, 36, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Suchy, F.J.; Brannon, P.M.; Carpenter, T.O.; Fernandez, J.R.; Gilsanz, V.; Could, J.B.; Hall, K.; Hui, S.L.; Lupton, J.; Mennella, J.; et al. NIH consensus development conference statement: lactose intolerance and health. NIH Consens. State Sci. Statements 2010, 27, 1–27. [Google Scholar] [PubMed]
- Johnson, A.O.; Semenya, J.G.; Buchowski, M.S.; Enwonwu, C.O.; Scrimshaw, N.S. Adaptation of lactose maldigesters to continued milk intakes. Am. Soc. Clin. Nutr. 1993, 58, 879–881. [Google Scholar]
- Briet, F.; Pochart, P.; Marteau, P.; Flourie, B.; Arrigoni, E.; Rambaud, J.C. Improved clinical tolerance to chronic lactose ingestion in subjects with lactose intolerance: A placebo effect? Gut 1997, 41, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Di Rienzo, T.; D’angelo, G.; D’aversa, F.; Campanale, M.C.; Cesario, V.; Montalto, M.; Gasbarrini, A.; Ojetti, V. Lactose intolerance: from diagnosis to correct management. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 18–25. [Google Scholar] [PubMed]
- Marton, A.; Xue, X.; Szilagyi, A. Meta-analysis: The diagnostic accuracy of lactose breath hydrogen or lactose tolerance tests for predicting the North European lactase polymorphism C/T-13910. Aliment. Pharmacol. Ther. 2012, 35, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Aragòn, J.J.; Hermida, C.; Martínez-Costa, O.H.; Sánchez, V.; Martín, I.; Sánchez, J.J.; Codoceo, R.; Cano, J.; Cano, A.; Crespo, L. Noninvasive diagnosis of the hypolactasia with 4-Galactosylxylose (Gaxilose): A multicenter, open-label, phase IIB-III non randomized trial. J. Clin. Gastroenterol. 2014, 48, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Istituto Nazionale di Ricerca per gli Alimenti e la Nutrizione. Tabelle di composizione degli alimenti. Available online: http://www.clitt.it/contents/scienze-files/6160_rodato_quaderno-files/6160_TabelleComposAlim.pdf (accessed on 9 March 2016).
- Usai-Satta, P.; Scarpa, M.; Oppia, F.; Cabras, F. Lactose malabsorption and intolerance: what should be the best clinical management? World J. Gastrointest. Pharmacol. Ther. 2012, 3, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Ibba, I.; Gilli, A.; Boi, M.F.; Usai, P. Effects of exogenous lactase administration on hydrogen breath excretion and intestinal symptoms in patients presenting lactose malabsorption and intolerance. BioMed Res. Int. 2014, 2014, 680196. [Google Scholar] [CrossRef] [PubMed]
- Agostoni, M.; Berni Canani, R. EFSA Panel on dietetic products, nutrition and allergies (NDA). Scientific Opinion on Dietary Reference Values for calcium. EFSA J. 2015, 13. [Google Scholar] [CrossRef] [Green Version]
- Escobar, M.A.J.; Lustig, D.; Pflugeisen, B.M.; Amoroso, P.J.; Sherif, D.; Saeed, R.; Schamdeen, S.; Tuider, J.; Abdullah, B. Fructose intolerance/malabsorption and recurrent abdominal pain in children. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Sabrauskas, D.M.L.; Yalovsky, M.; Mishkin, S. Fructose and sorbitol malabsorption in ambulatory patients with functional dyspepsia: comparison with lactose maldigestion/malabsorption. Dig. Dis. Sci. 1997, 42, 2591–2598. [Google Scholar] [CrossRef]
- Montalto, M.; Gallo, A.; Ojetti, V.; Gasbarrini, A. Fructose, trehalose and sorbitol malabsorption. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 26–29. [Google Scholar] [PubMed]
- Wirth, S.; Klodt, C.; Wintermeyer, P.; Berrang, J.; Hensel, K.; Langer, T.; Heusch, A. Positive or negative fructose breath test results do not predict response to fructose restricted diet in children with recurrent abdominal pain: results from a prospective randomized trial. Klin. Padiatr. 2014, 226, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Imperato, M.R.; Leno, L.; Sorvillo, R.; Benigno, V.; Parenti, G.; Parini, R.; Vitagliano, L.; Zagari, A.; Salvatore, F. Hereditary fructose intolerance: functional study of two novel ALDOB natural variants and characterization of a partial gene deletion. Hum. Mutat. 2010, 31, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Duro, D.; Rising, R.; Cedillo, M.; Lifshitz, F. Association between infantile colic and carbohydrate malabsorption from fruit juice in infancy. Pediatrics 2002, 109, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.F.; Butler, R.N.; Moore, D.J.; Brooks, D.A. Developmental changes and fructose absorption in children: Effect on malabsorption testing and dietary management. Nutr. Rev. 2013, 71, 300–309. [Google Scholar] [CrossRef] [PubMed]
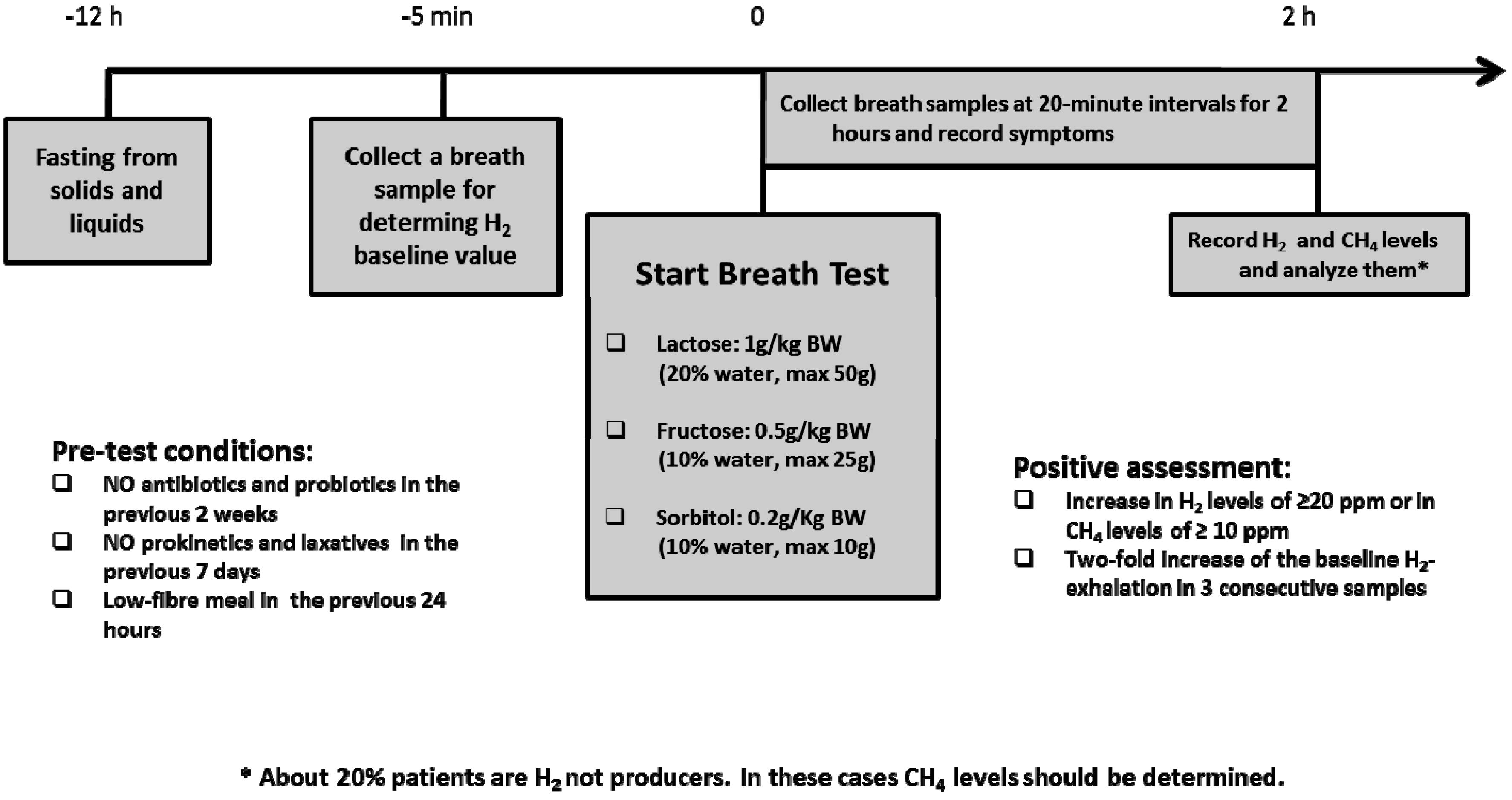
- Gasbarrini, A.; Corazza, G.R.; Gasbarrini, G.; Montalto, M.; Di Stefano, M.; Basilisco, G.; Usai-Satta, P.; Satta, P.U.; Vernia, P.; Anania, C.; Astegiano, M.; et al. Methodology and indications of H2-breath testing in gastrointestinal diseases: The Rome Consensus Conference. Aliment. Pharmacol. Ther. 2009, 29. [Google Scholar] [CrossRef]
- Lozinsky, A.C.; Boé, C.; Palmero, R.; Fagundes-Neto, N. Fructose malabsorption in children with functional digestive disorders. Arq. Gastroenterol. 2013, 50, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Kraft, N.; Zimmerman, B.; Jackson, M.; Rao, S.S.C. Fructose intolerance in IBS and utility of fructoserestricted diet. J. Clin. Gastroenterol. 2008, 42, 233–238. [Google Scholar] [PubMed]
- Jones, H.F.; Burt, E.; Dowling, K.; Davidson, G.; Brooks, D.A.; Butler, R.N. Effect of age on fructose malabsorption in children presenting with gastrointestinal symptoms. J. Pediatr. Gastroenterol. Nutr. 2011, 52, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Gibson, P.R.; Newnham, E.; Barrett, J.S.; Shepherd, S.J.; Muir, J.G. Review article: fructose malabsorption and the bigger picture. Aliment. Pharmacol. Ther. 2007, 25, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.C.S.; Attaluri, A.; Anderson, L.; Stumbo, P. Ability of the normal human small intestine to absorb fructose: evaluation by breath testing. Clin. Gastroenterol. Hepatol. 2007, 5, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, M.H.; Mayberry, J.F. Fructose malabsorption: true condition or a variance from normality. J. Clin. Gastroenterol. 2011, 45, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Doma, S.; Gaddipati, K.; Fernandez, A.; Bromer, M.; Parkman, H. Results of fructose breath test in healthy controls using different doses of fructose: which dose is best? Am. J. Gastroenterol. 2003, 98, S265–S266. [Google Scholar] [CrossRef]
- Eisenmann, A.; Amann, A.; Said, M.; Datta, B.; Ledochowski, M. Implementation and interpretation of hydrogen breath tests. J. Breath Res. 2008, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gijsbers, C.F.M.; Kneepkens, C.M.F.; Büller, H.A. Lactose and fructose malabsorption in children with recurrent abdominal pain: results of double-blinded testing. Acta Paediatr. 2012, 101, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Gomara, R.E.; Halata, M.S.; Newman, L.J.; Bostwick, H.E.; Berezin, S.H.; Cukaj, L.; See, M.C.; Medow, M.S. Fructose intolerance in children presenting with abdominal pain. J. Pediatr. Gastroenterol. Nutr. 2008, 47, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.F.; Butler, R.N. Intestinal fructose transport and malabsorption in humans. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G202–G206. [Google Scholar] [CrossRef] [PubMed]
- Ledochowsky, M.; Widner, B.; Bair, H.; Probst, T.; Fuchs, D. Fructose and sorbitol reduced diet improves mood and gastrointestinal disturbances in fructose malabsorbers. Scand. J. Gastroentrol. 2000, 35, 1048–1052. [Google Scholar]
- Komericki, P.; Akkilic-Materna, M.; Strimitzer, T.; Weyermair, K.; Hammer, H.F.; Aberer, W. Oral xylose isomerase decreases breath hydrogen excretion and improves gastrointestinal symptoms in fructose malabsorption—A double-blind, placebo-controlled study. Aliment. Pharmacol. Ther. 2012, 36, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Coupland, K.; Smith, J.A.; Ansell, I.D.; Long, R.G. Intestinal trehalase activity in a UK population: estabilishing a normal range and the effect of disease. Br. J. Nutr. 2000, 83, 241–245. [Google Scholar] [PubMed]
- Barrett, J.S.; Gibson, P.R. Fermentable oligosaccharides, disaccharides, monosaccharides and polyols (FODMAPs) and nonallergic food intolerance: FODMAPs or food chemicals? Ther. Adv. Gastroenterol. 2012, 5, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.S.; Gibson, P.R. Clinical ramifications of malabsorption of fructose and other short-chain carbohydrates. Pract. Gastroenterol. 2007, 31, 51–65. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Foods to avoid |
| Cereals with added sugars Apple, apricot, banana, cantaloupe, grapefruit, melon, mango, orange, peach, pineapple, tangerine Carrot, potato Beans, chickpeas, green peas, lentils, peas, soy Yogurt sweetened with sucrose, sweetened condensed milk, sweetened cream Sugar (sucrose), ice cream, all desserts made with sugar, marmalade, candies, jellies, chocolate, licorice, commercial cookies and cakes with added sugar, sweetened drinks |
| Foods allowed |
| Wheat, rice, corn, einkorn, oats, kamut, spelt, rye, bread, pasta, flour, cereals with no added sugar * Avocado, berries, cherries, fig, grapes, kiwi, lemon, lime, olives, papaya, pear, pomegranate, prune, strawberries All vegetables Milk, dairy product, butter, cream, cheeses, yogurt sweetened with dextrose or fructose All meat, fish, and eggs All fats Fructose, honey, cocoa, unsweetened juice, homemade low-sucrose cookies and cakes |
| Foods to avoid |
| All kinds of milk, butter, yogurt, cheeses and other dairy products Sugar (sucrose), ice cream, all desserts made with sugar, candies, gelatin desserts, chocolate, licorice, commercial cookies and cakes with added sugar, sweetened drinks Glucose, dextrose, dextrin, maltose, maltodextrin, corn syrup, glucose polymers, lactose, stevia |
| Foods allowed |
| Special formula without galactose and glucose Small amounts of: pasta, rice, potatoes, bread, unsweetened cereal, puffed wheat, puffed rice, oat, wholegrain cereals without a sugary coating, quinoa All legumes (beans, chickpeas, peas, lentils, soy) All vegetables All fruits All meat, fish, and eggs All vegetable fats Fructose, honey, cocoa, sugar-free marmalade, unsweetened juice, all fructose-sweetened desserts and snacks |
| Food | Lactose (g/100 g of Food) |
|---|---|
| Skimmed cow’s milk | 4.7 |
| Low-fat cow’s milk | 4.6 |
| Whole cow’s milk | 4.5 |
| Buttermilk | 4.1 |
| Free lactose milk | 0.5 |
| Whole powdered milk | 35.1 |
| Skimmed powdered milk | 50.5 |
| Goat’s milk | 4.2 |
| Buffalo milk | 4.9 |
| Yogurt | 3.2 |
| Butter | 4.0 |
| Cottage cheese | 2.6 |
| Mozzarella cheese | 1.5–2.0 |
| Goat cheese | 1.5–2.0 |
| Ricotta cheese | 4.0 |
| Parmigiano Reggiano | 0–0.9 |
| Cream cheese | 6.0 |
| Taleggio cheese | 0 |
| Fontina cheese | 0 |
| Provolone cheese | 0 |
| Gorgonzola cheese | 0 |
| Foods to limit |
| All kinds of milk: whole, low fat, nonfat, cream, powdered, condensed, evaporated, goat, acidophilus, and chocolate Butter, cottage cheese, ice cream, creamy/cheesy sauces, cream cheeses, soft cheeses (brie, ricotta), mozzarella, whipped cream, yogurt Fish and meat (breaded or creamed) Milk bread, crackers, creamed, scalloped, or au gratin potatoes Muffin, biscuit, waffle, pancake, and cake mixes; milk chocolate; bakery products and desserts that contain the ingredients listed above |
| Foods allowed |
| Lactose-free milk, soy milk Lactose-free dairy, hard cheeses (Parmigiano Reggiano, Pecorino, Grana Padano, fontina, taleggio, provolone, Swiss), gorgonzola All fruits All vegetables All legumes All cereals All meat, fish, and eggs All vegetable fats |
| Foods to Avoid |
| All fruits Broccoli, carrots, cauliflower, green beans, green peppers, sweet potatoes, tomatoes Beans, peas Corn Fructose, honey, high-fructose corn syrup, sorbitol, jams, gelatin desserts, candies, all desserts sweetened with fructose Condiments such as barbeque sauce, ketchup, sweet and sour sauce, pancake syrup, plum sauce, chutney, etc. |
| Foods Allowed |
| Asparagus, celery, chives, cucumber, kale, lettuce, parsnips, pumpkin, radish, scallions, spinach, spinach, white potatoes, shallots, zucchini All cereals All meat, fish, and eggs All dairy All fats Sugar (sucrose), molasses, saccharine |
| Foods to Avoid |
| Apple, apricot, blackberry, cherry, date, fig, nectarine, pear, peach, plum, raisin, and other dried fruits Sugar-free chewing gum and candies Diabetic foods and drinks Diet and light drinks Foods that contain the initials E420 in the list of ingredients |
| Foods Allowed |
| Banana, citrus fruit, kiwi, melon, pineapple, strawberry All legumes All cereals All vegetables All meat, fish, and eggs Milk and dairy products All fats Sugar, honey, fructose, cocoa, jams, gelatin desserts, molasses, all desserts made with sugar, marmalade, chocolate, commercial cookies and cakes with added sugar, sweetened drinks, artificial sweeteners: aspartame, saccharine, mannitol, isomalt, xylitol (cough drops, gums, mints) |
| Fructose | Lactose | Fructans | Galactans | Polyols |
|---|---|---|---|---|
| Fruits: watermelon, apple, mango, pear Jam Fruit juice Dried fruits Honey and molasses | Cow, sheep and goat milk Yogurt Cheese Mascarpone | Vegetables: asparagus, chicory, beets, broccoli, Brussels sprouts, cabbage, eggplant, fennel, onions, garlic, leeks Cereals: wheat, bread, biscuits, crackers, couscous, pasta Fruits: persimmon, watermelon | Legumes: beans, chickpeas, lentils | Fruits: apple, apricot, cherrys, lychee, peach, pear, plum, watermelon Vegetables: cauliflower, capsicum, mushrooms, corn Sweeteners: sorbitol, mannitol, maltitol, xylitol |
| Foods to limit |
| Milk (from cow, sheep, or goat), butter, cottage cheese, ice cream, creamy/cheesy sauces, sweetened condensed milk, soft cheeses (brie, ricotta), mozzarella, whipped cream, yogurt Legumes (beans, chickpeas, peas, lentils) Wheat, einkorn, emmer, kamut, spelt, rye Artichokes, asparagus, beets, leeks, broccoli, Brussels sprouts, cabbage, cauliflower, fennel, green beans, mushrooms, garlic, onions Avocado, apples, apricots, dates, canned fruit, cherries, dried fruits, figs, mango, pears, papaya, peaches, plums, prunes, persimmon, watermelon Honey, jams, jellies, molasses, artificial sweeteners: sorbitol, mannitol, isomalt, xylitol (cough drops, gum, mints) |
| Foods allowed |
| Lactose-free milk and lactose-free dairy, hard cheeses (cheddar, Parmesan, pecorino, Swiss) Wheat-free grains: rice, corn, quinoa, tapioca, buckwheat Banana, berry, cantaloupe, grape, grapefruit, kiwi, lemon, lime, mandarin, orange, pineapple Bell peppers, cucumbers, carrots, celery, eggplant, lettuce, leafy greens, olives, pumpkin, potatoes, tomatoes, zucchini, most spices and herbs Sugar All fats |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berni Canani, R.; Pezzella, V.; Amoroso, A.; Cozzolino, T.; Di Scala, C.; Passariello, A. Diagnosing and Treating Intolerance to Carbohydrates in Children. Nutrients 2016, 8, 157. https://doi.org/10.3390/nu8030157
Berni Canani R, Pezzella V, Amoroso A, Cozzolino T, Di Scala C, Passariello A. Diagnosing and Treating Intolerance to Carbohydrates in Children. Nutrients. 2016; 8(3):157. https://doi.org/10.3390/nu8030157
Chicago/Turabian StyleBerni Canani, Roberto, Vincenza Pezzella, Antonio Amoroso, Tommaso Cozzolino, Carmen Di Scala, and Annalisa Passariello. 2016. "Diagnosing and Treating Intolerance to Carbohydrates in Children" Nutrients 8, no. 3: 157. https://doi.org/10.3390/nu8030157
APA StyleBerni Canani, R., Pezzella, V., Amoroso, A., Cozzolino, T., Di Scala, C., & Passariello, A. (2016). Diagnosing and Treating Intolerance to Carbohydrates in Children. Nutrients, 8(3), 157. https://doi.org/10.3390/nu8030157