
Lipid-Induced Insulin Resistance in Skeletal Muscle: The Chase for the Culprit Goes from Total Intramuscular Fat to Lipid Intermediates, and Finally to Species of Lipid Intermediates

Abstract
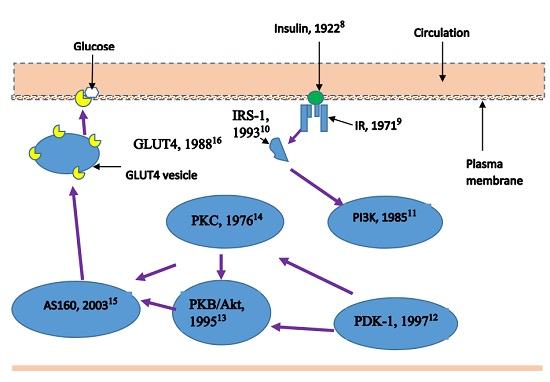
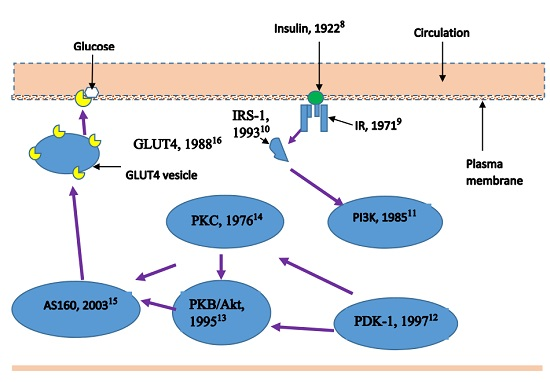
:
1. Diabetes Epidemic and Insulin Resistance
2. Lipid Transport into Muscle Cells
3. Total Intramuscular Fat
4. DAG in Skeletal Muscle IR
5. Ceramides in Skeletal Muscle IR
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Pournaras, D.J.; le Roux, C.W. Type 2 diabetes: Multimodal treatment of a complex disease. Lancet 2015, 386, 936–937. [Google Scholar] [CrossRef]
- Centre for Disease Control (CDC). Available online: http://www.cdc.gov/diabetes/basics/prediabetes.html (accessed on 11 August 2015).
- Reaven, G.M. Banting Lecture 1988. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Integrating mechanisms of insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Petersen, K.-F.; Shulman, G.I. Lipid-Induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar] [CrossRef]
- Warram, J.H.; Martin, B.C.; Krolewski, A.S.; Soeldner, J.S.; Khan, C.R. Slow glucose removal rate and hyperinsulinemia precede the development of Type II diabetes in the offspring of diabetic patients. Ann. Intern. Med. 1990, 113, 909–915. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is primary defect in Type 2 Diabetes. Diabetes Car. 2009, 32 (Suppl. S2), S157–S163. [Google Scholar] [CrossRef] [PubMed]
- Banting, F.G.; Best, C.H. The internal secretion of the pancreas. J. Lab. Clin. Med. 1922, 7, 251–266. [Google Scholar]
- Freychet, P.; Roth, J.; Neville, D.M., Jr. Insulin receptors in the liver: specific binding of (125 I) insulin to the plasma membrane and its relation to insulin bioactivity. Proc. Natl. Acad. Sci. USA 1971, 68, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G.; White, M.F. Insulin receptor substrate-1 and proteins with SH2 domains. Diabetes 1993, 42, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Whitman, M.; Kaplan, D.R.; Schaffhausen, B.; Cantley, L.; Roberts, T.M. Association of phosphatidylinositol kinase activity with with polyoma middle-T component for transformation. Nature 1985, 315, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Allesi, D.R.; Deak, M.; Casamayor, A.; Caudwell, F.B.; Morrice, N.; Norman, D.G.; Gaffney, P.; Reese, C.B.; MacDougal, C.N.; Harbison, D.; et al. 3-phosphoinositide dependent protein kinase-1 (PDK1): Structural and functional homology with the Drosophila DSTPK61 kinase. Curr. Biol. 1997, 7, 776–789. [Google Scholar] [CrossRef]
- Franke, T.F.; Yang, S.I.; Chan, T.O.; Datta, K.; Kazlauskas, A.; Morrison, D.K.; Kaplan, D.R.; Tsichlis, P.N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 1995, 81, 727–736. [Google Scholar] [CrossRef]
- Inoue, M.; Kishimoto, A.; Takai, Y.; Nishizuka, Y. Guanosine 3′:5′-Monophosphate-dependent protein kinase from silkworm, properties of a catalytic fragment obtained by limited proteolysis. J. Biol. Chem. 1976, 251, 4476–4478. [Google Scholar] [PubMed]
- Sano, H.; Kane, S.; Sano, E.; Minea, C.P.; Asara, J.M.; Lane, W.S.; Garner, C.W.; Lienhard, G.E. Insulin-Stimulated phosphorylation of a Rab GTPase activating protein regulates GLUT4 translocation. J. Biol. Chem. 2003, 278, 14599–14602. [Google Scholar] [CrossRef] [PubMed]
- James, D.E.; Brown, R.; Navarro, J.; Pilch, P.F. Insulin regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature 1988, 333, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J. Fuel selection in animals. Biochem. Soc. Trans. 1985, 14, 799–806. [Google Scholar] [CrossRef]
- McFarlan, J.T.; Yoshida, Y.; Jain, S.S.; Han, X.X.; Snook, L.A.; Lally, J.; Smith, B.K.; Glatz, J.F.; Luiken, J.J.; Sayer, R.A.; et al. In vivo, fatty acid translocase (CD36) critically regulates skeletal muscle fuel selection, exercise performance, and training-induced adaptation of fatty acid oxidation. J. Biol. Chem. 2012, 287, 23502–23516. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.T.; Steele, R.; Altszuker, N.; Dunn, A.; Bishop, J.S.; DeBodo, R.C. Regulation of plasma free fatty acid turnover. Am. J. Physiol. 1961, 201, 9–15. [Google Scholar] [PubMed]
- Issekutz, B., Jr.; Bortz, W.M.; Miller, H.I.; Paul, P. Turnover rate of plasma FFA in humans and dogs. Metabolism 1967, 16, 1001–1009. [Google Scholar] [CrossRef]
- Turcotte, L.P.; Kiens, B.; Richter, E.A. Saturation kinetics of palmitate uptake in perfused skeletal muscle. FEBS Lett. 1991, 279, 327–329. [Google Scholar] [CrossRef]
- Kiens, B.; Kristiansen, S.; Jensen, P.; Richter, E.A.; Turcotte, L.P. Membrane associated fatty acid binding protein (FABPpm) in human skeletal muscle is increased by endurance training. Biochem. Biophys. Res. Commun. 1997, 231, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.C.; Luiken, J.J.F.P.; Bonen, A. Membrance fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef] [PubMed]
- Kazantzis, M.; Stahl, A. Fatty acid transport proteins, implications in physiology and disease. Biochim. Biophys. Acta 2012, 1821, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Aitman, T.J.; Glazier, A.M.; Wallace, C.A.; Cooper, L.D.; Nortsworthy, J.K.; Wahid, F.N.; Al-Majali, K.M.; Trembling, P.M.; Mann, C.J.; Shoulders, C.C.; et al. Identification of Cd36 (Fat) as an insulin-resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat. Genet. 1999, 21, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Luiken, J.J.; Arumugam, Y.; Bell, R.C.; Calles-Escandon, J.; Tandon, N.N.; Glatz, J.F.; Bonen, A. Changes in fatty acid transport and transporters are related to the severity of insulin deficiency. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E612–E621. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Cooney, G.J.; Kraegen, E.W.; Bruce, C.R. Fatty acid metabolism, energy expenditure and insulin resistance in muscle. J. Endocrinol. 2014, 220, T61–T79. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, U.; Christensen, B.; Nielsen, T.S.; Pedersen, S.B.; Ørskov, L.; Lund, S.; Møller, N.; Jessen, N. GLUT4 and UBC9 protein expression is reduced in muscle from type 2 diabetic patients with severe insulin resistance. PLoS ONE 2011, 6, e27854. [Google Scholar] [CrossRef] [PubMed]
- Funai, K.; Semenkovich, C.F. Skeletal muscle lipid flux: Running water carries no poison. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E245–E251. [Google Scholar] [CrossRef] [PubMed]
- Zamora, M.; Josep, A.; Villena, J.A. Targeting mitochondrial biogenesis to treat insulin resistance. Curr. Pharm. Des. 2014, 20, 5527–5557. [Google Scholar] [CrossRef] [PubMed]
- Valero, T. Mitochondrial biogenesis: Pharmacological approaches. Curr. Pharm. Des. 2014, 20, 5507–5509. [Google Scholar] [CrossRef] [PubMed]
- Besseiche, A.; Riveline, J.-P.; Gautier, J.-F.; Breant, B.; Blondeau, B. Metabolic roles of PGC-α and its implications for type 2 diabetes. Diabetes Metab. 2015, 41, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Ward, W.F. PGC-1α: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Hoeks, J.; Schrauwen, P. Muscle mitochondria and insulin resistance: A human perspective. Trends Endocrinol. Metab. 2012, 23, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Gavrilova, O.; Pack, S.; Jou, W.; Mullen, S.; Summer, A.E.; Cushman, S.W.; Periwal, V. Hypertrophy and/or hyperplasia: Dynamics of adipose tissue growth. PLoS Comput. Biol. 2009, 5, e1000324. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; He, J.; Watkins, S.; Kelley, D.E. Skeletal muscle lipid content and insulin resistance: Evidence for a paradox in endurance-trained athletes. J. Clin. Endocrinol. Metab. 2001, 86, 5755–5761. [Google Scholar] [CrossRef] [PubMed]
- Dubé, J.J.; Amati, F.; Stefanovic-Racic, M.; Toledo, F.G.S.; Sauers, S.E.; Goodpaster, B.H. Exercise-Induced alterations in intramyocellular lipids and insulin resistance: The athlete’s paradox revisited. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E882–E888. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM: Genetic and clinical implications. Diabetes 1995, 44, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Amti, F. Revisiting the diacylglycerol-induced insulin resistance hypothesis. Obes. Rev. 2012, 13 (Suppl. S2), 40–50. [Google Scholar] [CrossRef] [PubMed]
- Erion, D.M.; Shulman, G.I. Diacylglycerol-Mediated insulin resistance. Nat. Med. 2010, 16, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zhong, H.; Wang, Y.; Bergeron, R.; Kim, Y.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.E.; Marcucci, M.J.; Cline, G.W.; Bell, K.; Barucci, N.; Lee, D.; Goodyear, L.J.; Kraegen, E.W.; White, M.F.; Shulman, G.I. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C θ and alterations in insulin signalling cascade. Diabetes 1999, 48, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkB-α. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Veinberg, S.G.; Bezy, O.; O’Neill, B.T.; Kahn, C.R. Role of PKCδ in insulin sensitivity and skeletal muscle metabolism. Diabetes 2015, 64, 4023–4032. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.Y.; Hatzinikolas, G.; Weir, J.M.; Janovska, A.; McAinch, A.J.; Game, P.; Meikle, P.J.; Wittert, G.A. Insulin-Stimulated glucose uptake and pathways regulating energy metabolism in skeletal muscle cells: The effects of subcutaneous and visceral fat, and long-chain saturated, n-3 and n-6 polyunsaturated fatty acids. Biochim. Biophy. Acta 2011, 1811, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Selathurai, A.; Kowalski, G.M.; Burch, M.L.; Sepulveda, P.; Risis, S.; Lee-Young, R.S.; Lamon, S.; Meikle, P.J.; Genders, A.J.; McGee, S.L.; et al. The CDP-Ethanolamine pathway regulates skeletal muscle diacylglycerol content and mitochondrial biogenesis without altering insulin sensitivity. Cell Metab. 2015, 21, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Szendroedi, J.; Yoshimura, T.; Phielix, E.; Koliaki, C.; Marucci, M.; Zhang, D.; Jelenik, T.; Muller, J.; Herder, C.; Nowotny, P.; et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 9597–9602. [Google Scholar] [CrossRef] [PubMed]
- Ritter, O.; Jelenik, T.; Roden, M. Lipid-Induced muscle insulin resistance: different fat, different pathways. J. Mol. Med. 2015, 93, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Takai, Y.; Kishimoto, A.; Kikkawa, U.; Mori, T.; Nishizuka, Y. Unsaturated diacylglycerol as a possible messenger for the activation of calcium-activated, phospholipid-dependent protein kinase system. Biochem. Biophys. Res. Commun. 1979, 91, 1218–1224. [Google Scholar] [CrossRef]
- Nishizuka, Y. Discovery and prospect of protein kinase C research: Epilogue. J. Biochem. 2003, 133, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Kowalski, G.M.; Leslie, S.J.; Risis, S.; Yang, C.; Lee-Young, R.S.; Babb, J.R.; Meikle, P.J.; Lancaster, G.I.; Hentridge, D.C.; et al. Distinct patterns of tissue specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 2013, 56, 1638–1648. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, A.H.; Lina, M.O. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar]
- Lee, J.S.; Pinnamaneni, S.K.; Eo, S.J.; Cho, I.H.; Pyo, J.H.; Kim, C.K.; Sinclair, A.J.; Febrraio, M.A.; Watt, M.J. Saturated, but not n-6 polyunsaturated, fatty acids induce insulin resistance: Role of intramuscular accumulation of lipid metabolites. J. Appl. Physiol. 2006, 100, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Newson, S.A.; Everett, A.C.; Park, S.; Van Pelt, D.W.; Hinko, A.; Horowitz, J.F. Lipid mixtures containing a very high proportion of saturated fatty acids only modestly impair insulin signalling in cultured muscle cells. PLoS ONE 2015, 10, e0120871. [Google Scholar] [CrossRef]
- Merrill, A.H., Jr. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem. Rev. 2011, 111, 6387–6422. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.Q.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Siddique, M.M.; Wang, S.T.; Ching, J.H.; Shayman, J.A.; Summers, S.A. Ceramides and glucosylceramides are independent antagonists of insulin signaling. J. Biol. Chem. 2014, 289, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.H.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-Induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Holand, W.L.; Summers, S.A. Sphingolipids, insulin resistance, and metabolic disease: New insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev. 2008, 29, 381–402. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A. Sphingolipids and insulin resistance: The five Ws. Curr. Opin. Lipidol. 2010, 21, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. A ceramide-centric view of insulin resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, B.; Summers, S.A. Ceramides-Lipotoxic inducers of metabolic disorders. Trends Endocrinol. Metab. 2015, 26, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Siddique, M.M.; Li, Y.; Chaurasia, B.; Kaddai, V.A.; Summers, S.A. Dihydroceramides: From bit players to lead actors. J. Biol. Chem. 2015, 290, 15371–15379. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro, M.; Higa, M.; Zhou, Y.T.; Wang, M.Y.; Newgard, C.B.; Unger, R.H. Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J. Biol. Chem. 1998, 273, 32487–32490. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.L.; Coghlan, M.; Hundal, H.S. Modulating serine palmitoyl transferase (SPT) expression and activity unveils a crucial role in lipid-induced insulin resistance in rat skeletal muscle cells. Biochem. J. 2009, 417, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Skovbro, M.; Baranowski, M.; Skov-Jensen, C.; Flint, A.; Dela, F.; Gorski, J.; Helge, J.W. Human skeletal muscle ceramide content is not a major factor in muscle insulin sensitivity. Diabetologia 2008, 51, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Helge, J.W.; Stalknectt, B.; Drachmann, T.; Hellgren, L.I.; Jimenez-Jimenez, R.; Andersen, J.L.; Richelsen, B.; Bruun, J.M. Improved glucose tolerance after intensive life style intervention occurs without change in muscle ceramide or triacylglycerol in morbidly obese subjects. Acta Physiol. (Oxf.) 2011, 201, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Dubé, J.J.; Amati, F.; Stefanovic-Racic, M.; Rossi, A.; Coen, P.; Goodpaster, B.H. Effects of weight loss and exercise on insulin resistance, and on intramyocellular triacylglycerol, diacylglycerol and ceramide. Diabetologia 2011, 54, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Raicur, S.; Wang, T.; Chan, P.W.; Li, Y.; Ching, J.; Chaurasia, B.; Dogra, S.; Ohman, M.K.; Takeda, K.; Sugii, S.; et al. CerS2 haploinsufficiency inhibits β-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab. 2014, 20, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Maur, J.; Xu, E.; Hammerschmidt, P.; Bronneke, H.S.; et al. Obesity-induced Cers6-dependent c16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Bergman, B.C.; Brozinick, J.T.; Straus, A.; Bacon, S.; Kerege, A.; Bui, H.H.; Sanders, P.; Siddall, P.; Wei, T.; Thomas, M.K.; et al. Muscle sphingolipids during rest and exercise: C18:0 signature for insulin resistance. Diabetologia 2016, 59, 785–798. [Google Scholar] [CrossRef] [PubMed]
- De La Maza, M.P.; Rodriguez, J.M.; Hirsch, S.; Leiva, L.; Barrera, G.; Bunout, D. Skeletal muscle ceramide species in men with abdominal obesity. J. Nutr. Health Aging 2015, 19, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Kasumov, T.; Solomon, T.P.J.; Hwang, K.; Hunag, H.; Haus, J.M.; Zhang, R.; Kirwan, J.P. Improved insulin sensitivity after exercise training is linked to reduced plasma C14:0 ceramide in obesity and Type 2 Diabetes. Obesity 2015, 23, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Caricilli, A.M.; Saad, M.J.A. The role of gut microbiota on insulin resistance. Nutrients 2013, 5, 829–851. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Transporter | Major Site/Organ |
|---|---|
| FATP1 | White adipose tissue (WAT), brown adipose tissue (BAT), skeletal muscle, heart (lesser extent: pancreas, lung, kidney and brain) |
| FATP2 | Kidney and liver |
| FATP3 | Lung, liver, pancreas and endothelial cells of capillaries in many organs |
| FATP4 | Broadly distributed; heart, liver, kidney, skeletal muscle, brain, skin, and endothelial cells. The predominant FATP in small intestines |
| FATP5 | A liver-specific protein |
| FATP6 | Exclusive to heart |
| Model | Fat Intervention/s | Muscle Lipid Change | Glucose Uptake/Insulin Resistance | Reference |
|---|---|---|---|---|
| L6 myotubes | PA, LA, DHA at 0.4 mmol/L | Muscle DAG: | Muscle glucose uptake: | [45] |
| ||||
|
| |||
|
| |||
|
| |||
| Muscle Ceramide (in presence of PA): | Myriocin inhibits ceramide synthesis | |||
| DHA increases hydrolysis of sphingomyelin | |||
| ||||
| L6 myotubes | Palmitic acid (Palmitate), Linoleic acid (Linoleate) | Muscle DAG: | Muscle glucose uptake: | [53] |
| ||||
| ||||
| 0.5 mmol/L | Muscle Ceramide: |
| ||
| Unaffected by LA | |||
| ||||
| C12C12 myotubes | Normal FA mixture (40% SFA), high SFA FA mixture (60% SFA) & 100% Palmitic acid | Muscle DAG: | Akt phosphorylation impaired (p < 0.05) by 100% PA. | [54] |
| Doses: 0.1, 0.2, 0.4 or 0.8 mmol/L |
| Modest impact of the two mixtures. No difference between 40% & 60% SFA. | ||
| ||||
| Vastus lateralis biopsies (Obese non-diabetic men & women) | Normal FA mixture (40% SFA), high SFA FA mixture (60% SFA) & 100% Palmitic acid | Muscle DAG: | Akt phosphorylation impaired (p > 0.05) by 100% PA. | [54] |
| Dose: 0.4 mmol/L |
| Modest impact of the two mixtures. No difference between 40% & 60% SFA. |
| Experimental Background | Dietary Fat Intervention/s | Muscle Lipid Change | Glucose Uptake/Insulin Resistance | Reference |
|---|---|---|---|---|
| Muscle specific ECT Knock out mice, 4 weeks duration | 5% Cal from fat | DAG: 200%↑ | No change detected | [46] |
| Control versus ECT KO | ||||
| 18 week old male mice | ||||
| 42% Cal from fat | DAG: 200%↑ | No change detected | [46] | |
| Control versus ECT KO | ||||
| 6-week old male mice | ||||
| Sprague–Dawley rats, male, 95–110 g, 8 weeks duration | Control: 15.7% fat | Muscle DAG: (SFA = PUFA) > Control | HOMA-IR: SFA > Control PUFA < Control | [53] |
| High SFA: 52.8% fat from lard and coconut oil. | (SFA = PUFA) > Control | |||
| High PUFA: 52.8% fat from safflower oil | ||||
| C57Bl/6 mice, 8–12 weeks old | Std chow, 5% energy from fat versus HFD (45% en from fat), endpoints at 1, 3, 6 and 16 weeks | At 3 weeks, muscle DAG increased over control. | At 3 weeks, muscle IR detected in HFD group | [51] |
| At 3 weeks, muscle ceramide 18:0 increased over control. |
| Phase | Hypothesis | Size of Bioactive in Muscle | Main Suggested Mechanism |
|---|---|---|---|
| Initial association studies | Total IMCL | 1–5 g/100 g muscle | General interference with glucose metabolism |
| Detailed lipid metabolism studies | Total DAG and/or Ceramide | pg/g muscle | Interference with insulin signaling (PKC pathway) |
| Lipidomics | Specific DAG and/or ceramide species | A fraction of pg/g muscle | Interference with insulin signaling (PKC pathway) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitessa, S.M.; Abeywardena, M.Y. Lipid-Induced Insulin Resistance in Skeletal Muscle: The Chase for the Culprit Goes from Total Intramuscular Fat to Lipid Intermediates, and Finally to Species of Lipid Intermediates. Nutrients 2016, 8, 466. https://doi.org/10.3390/nu8080466
Kitessa SM, Abeywardena MY. Lipid-Induced Insulin Resistance in Skeletal Muscle: The Chase for the Culprit Goes from Total Intramuscular Fat to Lipid Intermediates, and Finally to Species of Lipid Intermediates. Nutrients. 2016; 8(8):466. https://doi.org/10.3390/nu8080466
Chicago/Turabian StyleKitessa, Soressa M., and Mahinda Y. Abeywardena. 2016. "Lipid-Induced Insulin Resistance in Skeletal Muscle: The Chase for the Culprit Goes from Total Intramuscular Fat to Lipid Intermediates, and Finally to Species of Lipid Intermediates" Nutrients 8, no. 8: 466. https://doi.org/10.3390/nu8080466
APA StyleKitessa, S. M., & Abeywardena, M. Y. (2016). Lipid-Induced Insulin Resistance in Skeletal Muscle: The Chase for the Culprit Goes from Total Intramuscular Fat to Lipid Intermediates, and Finally to Species of Lipid Intermediates. Nutrients, 8(8), 466. https://doi.org/10.3390/nu8080466