Hederagenin Supplementation Alleviates the Pro-Inflammatory and Apoptotic Response to Alcohol in Rats

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
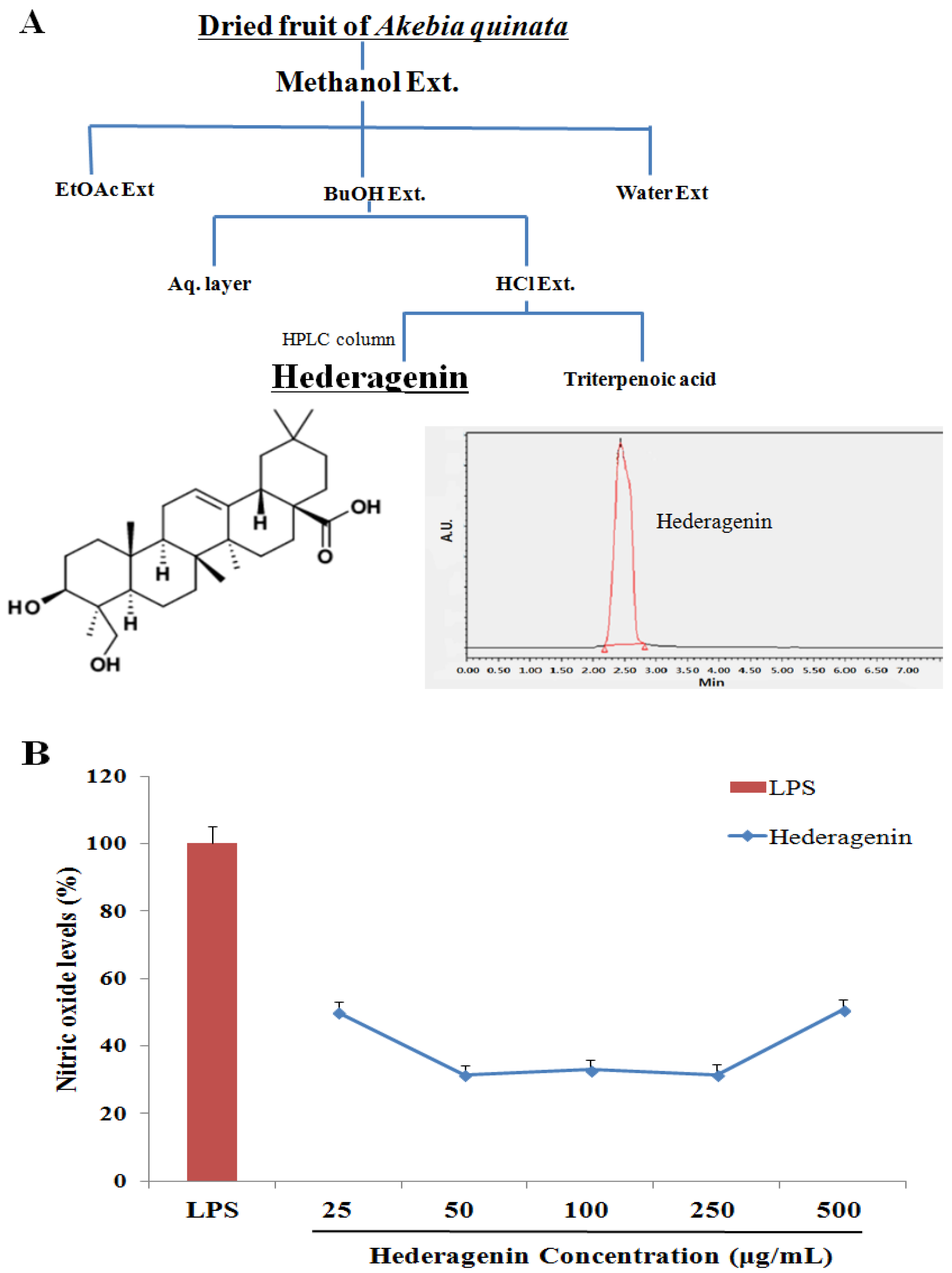
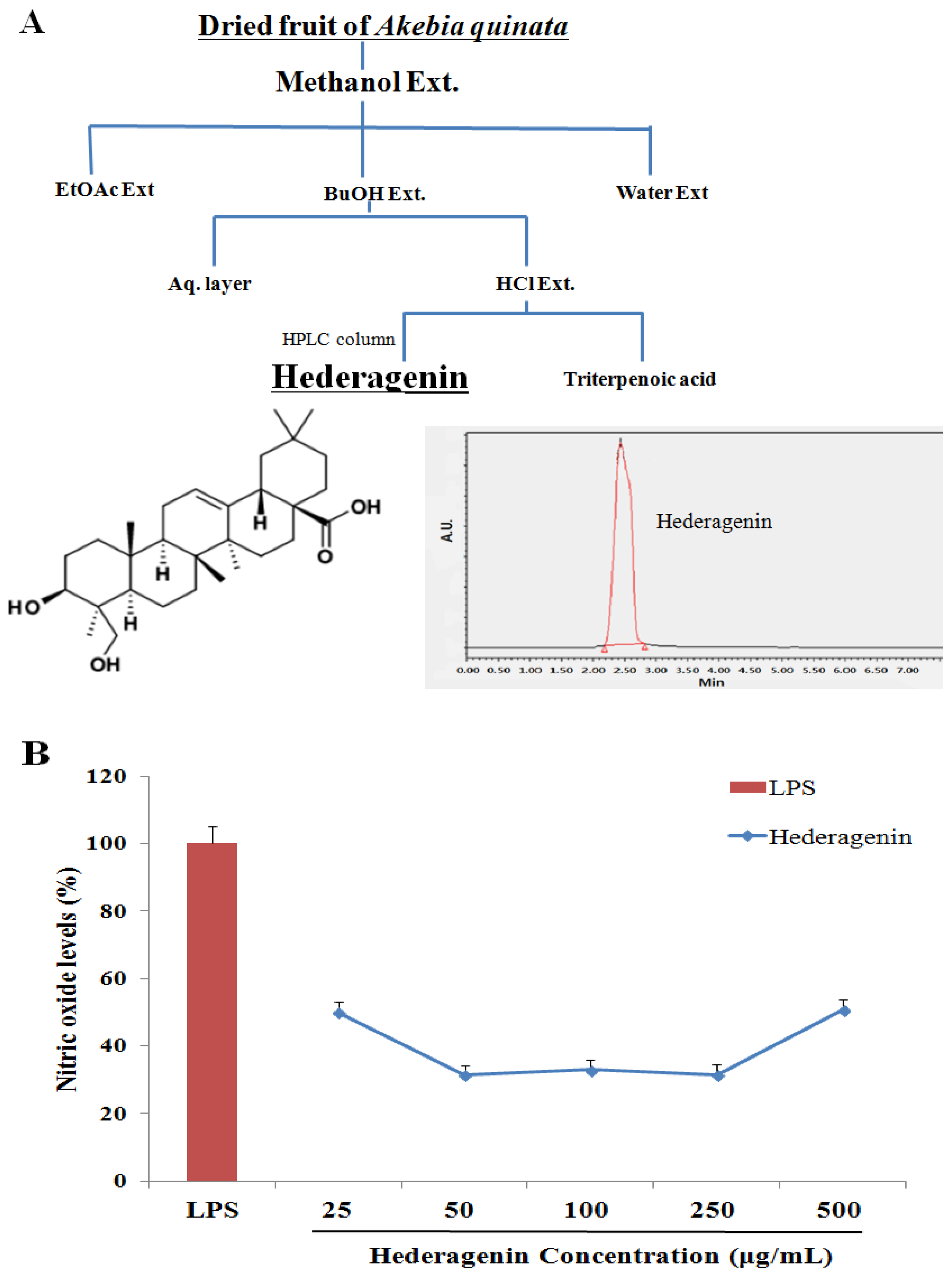
2.2. Akebia Quinata (AQ) Extraction and Isolation of Hederagenin
2.3. Nitric Oxide (NO) Assay
2.4. Animal Experiments
2.5. Biochemical Assays
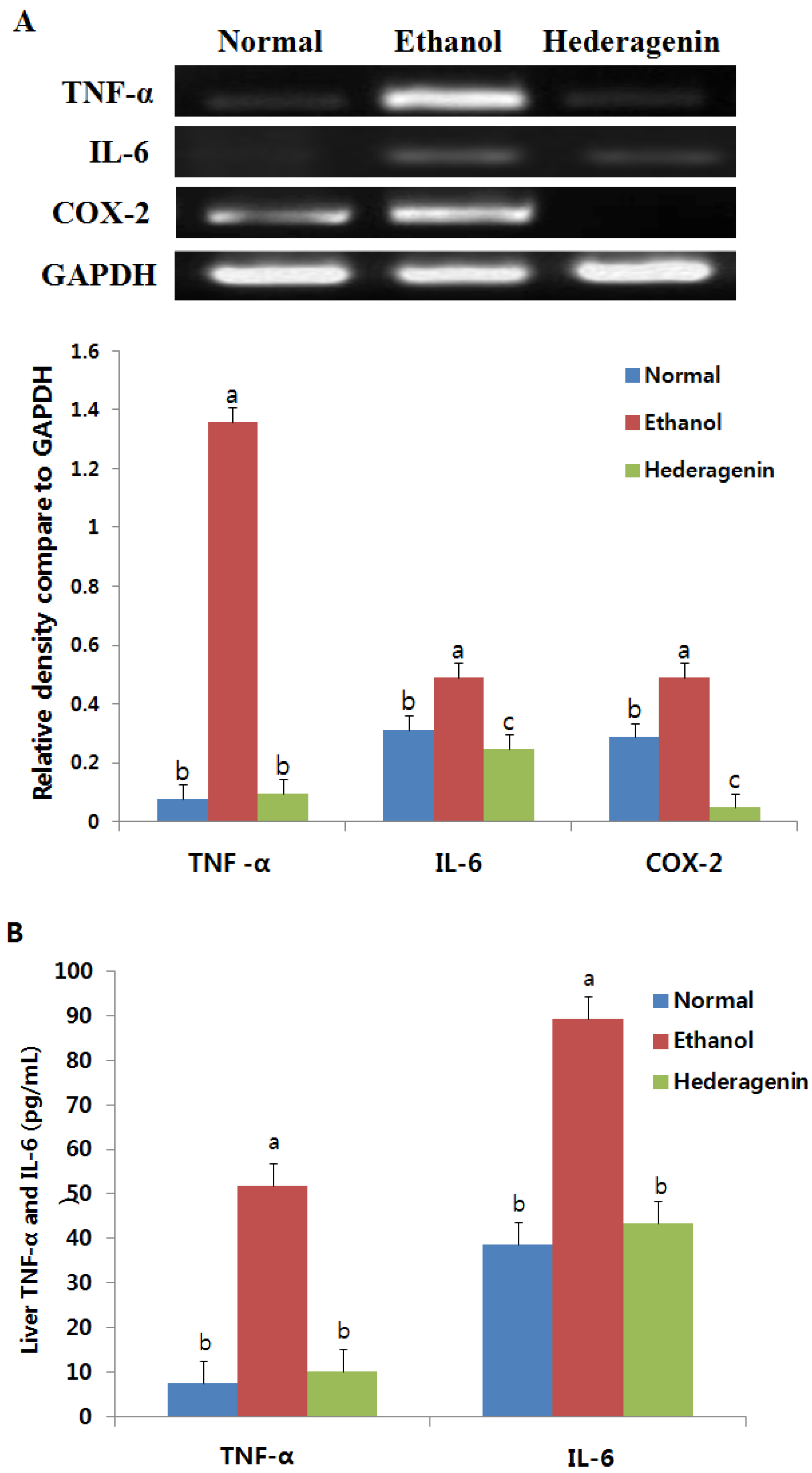
2.6. Enzyme-Linked Immunosorbent Assay (ELISA) of TNFα and IL6
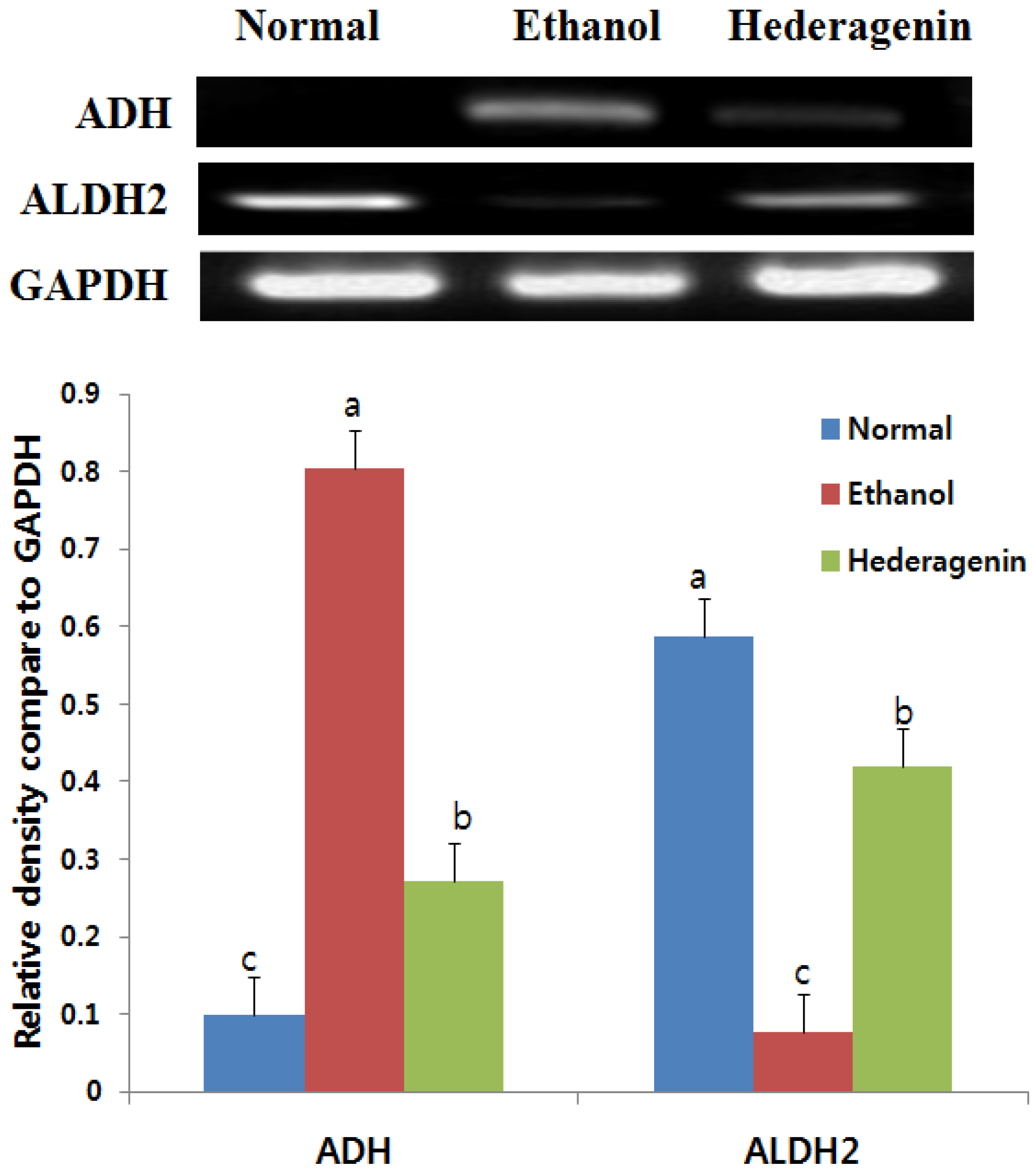
2.7. Reverse Transcription-PCR
2.8. Western Blotting Analysis
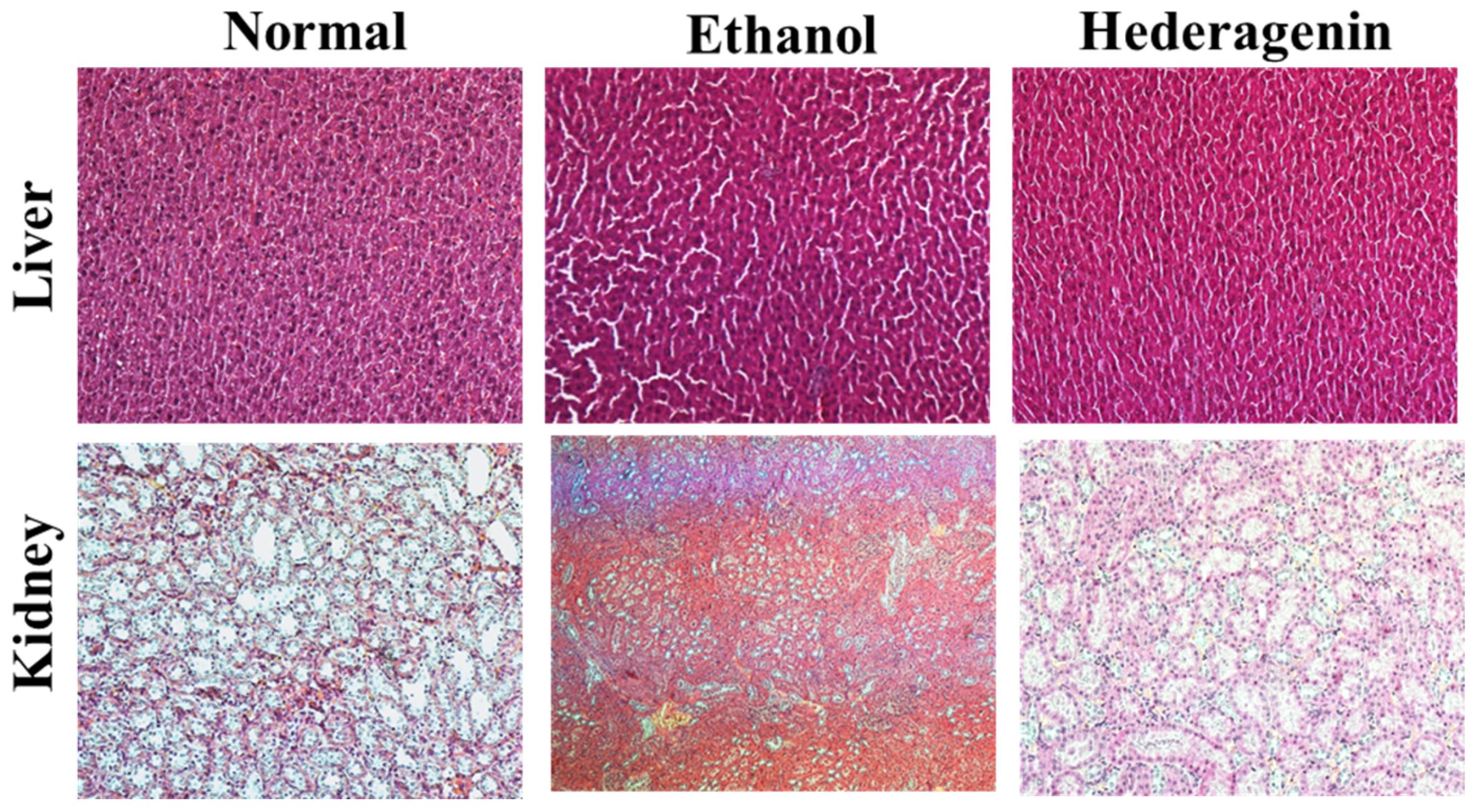
2.9. Histopathological Observation
2.10. Statistical Analysis
3. Results
3.1. Inhibition of NO of Hederagenin from AQ
3.2. Weight Gain and Liver Weight
3.3. Effects of Hederagenin on Biomarkers of Liver Injury
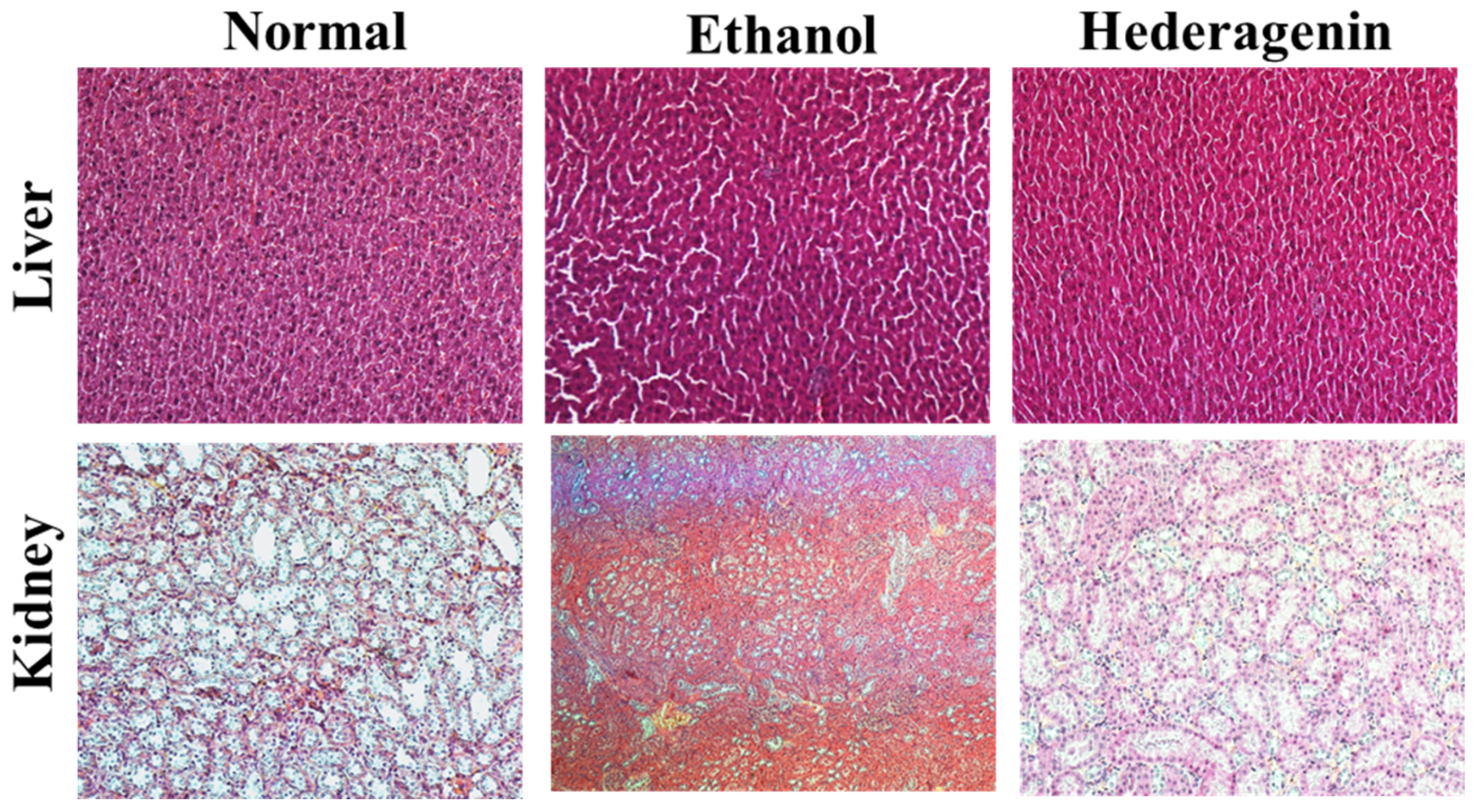
3.4. Histological Analysis
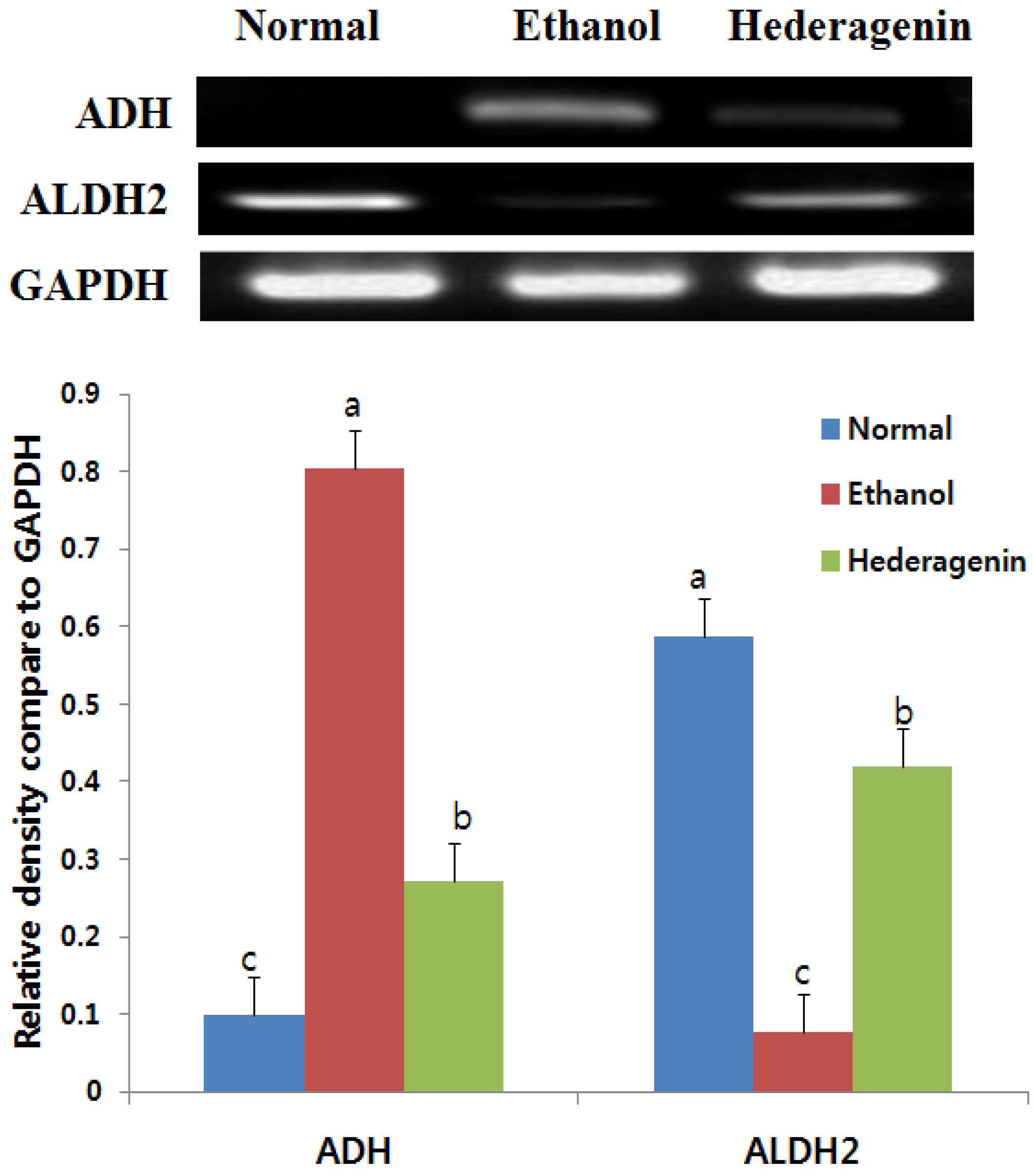
3.5. Effects of Hederagenin on Hepatic ADH and ALDH2 mRNA Expression
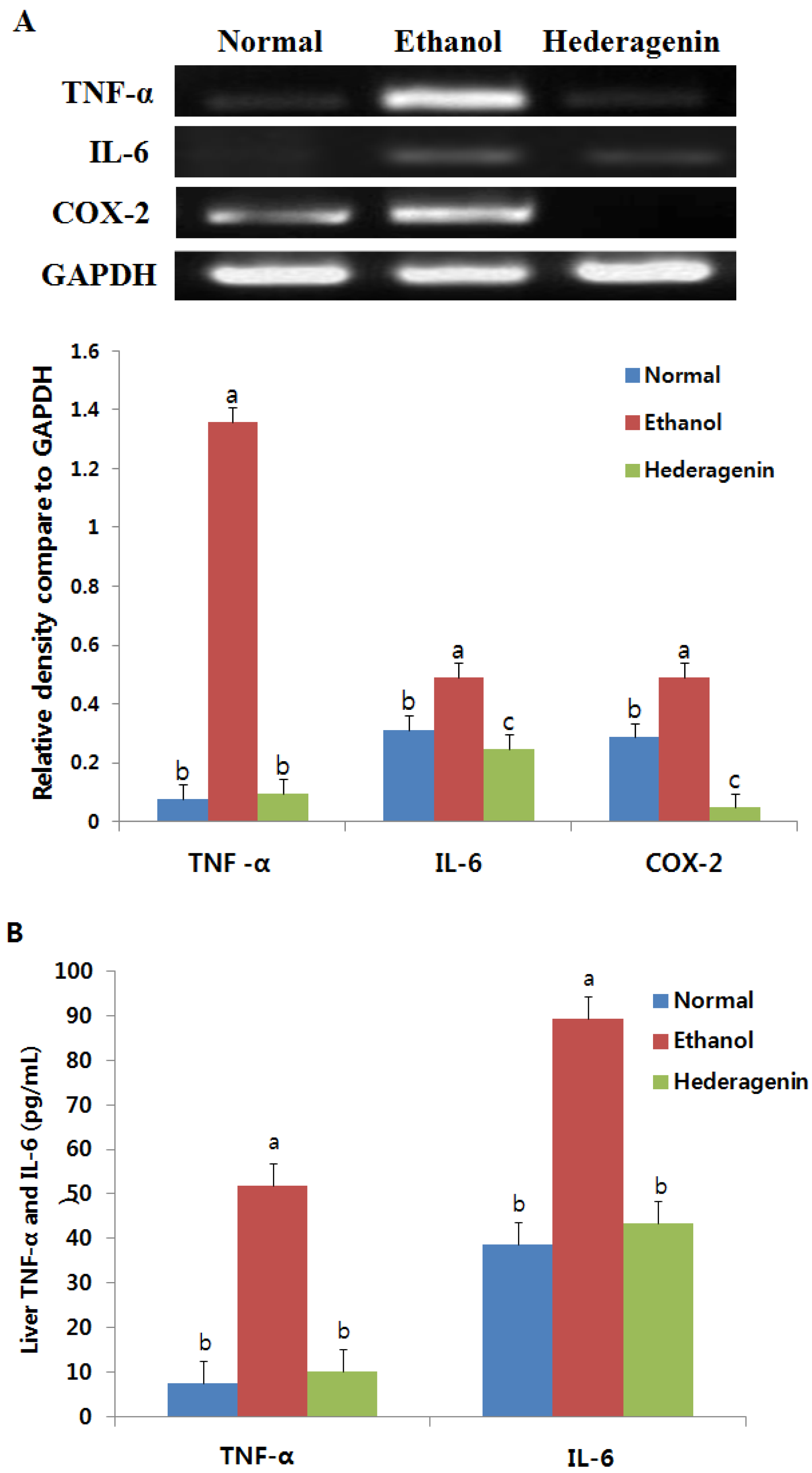
3.6. Effect of Hederagenin on Inflammation
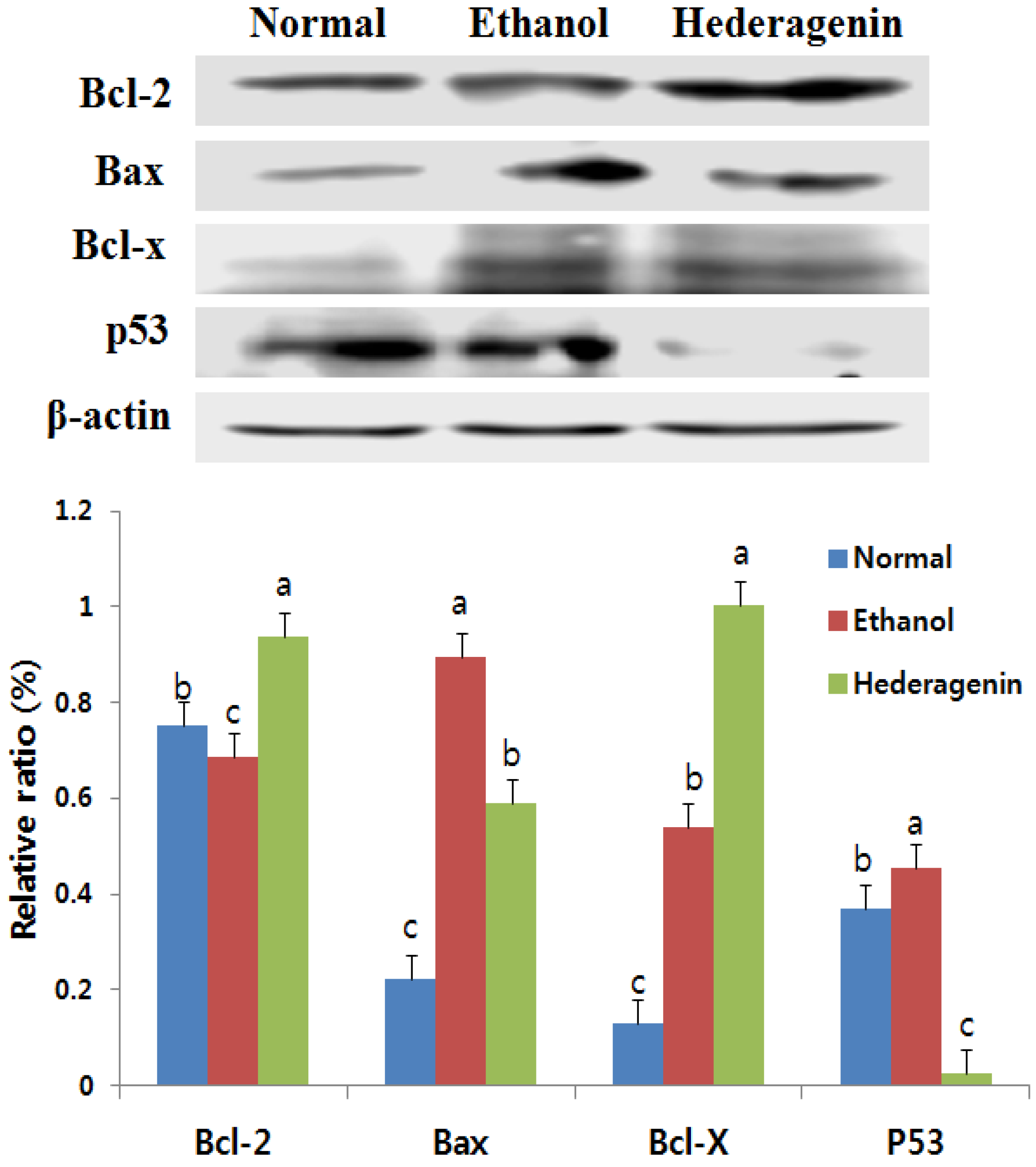
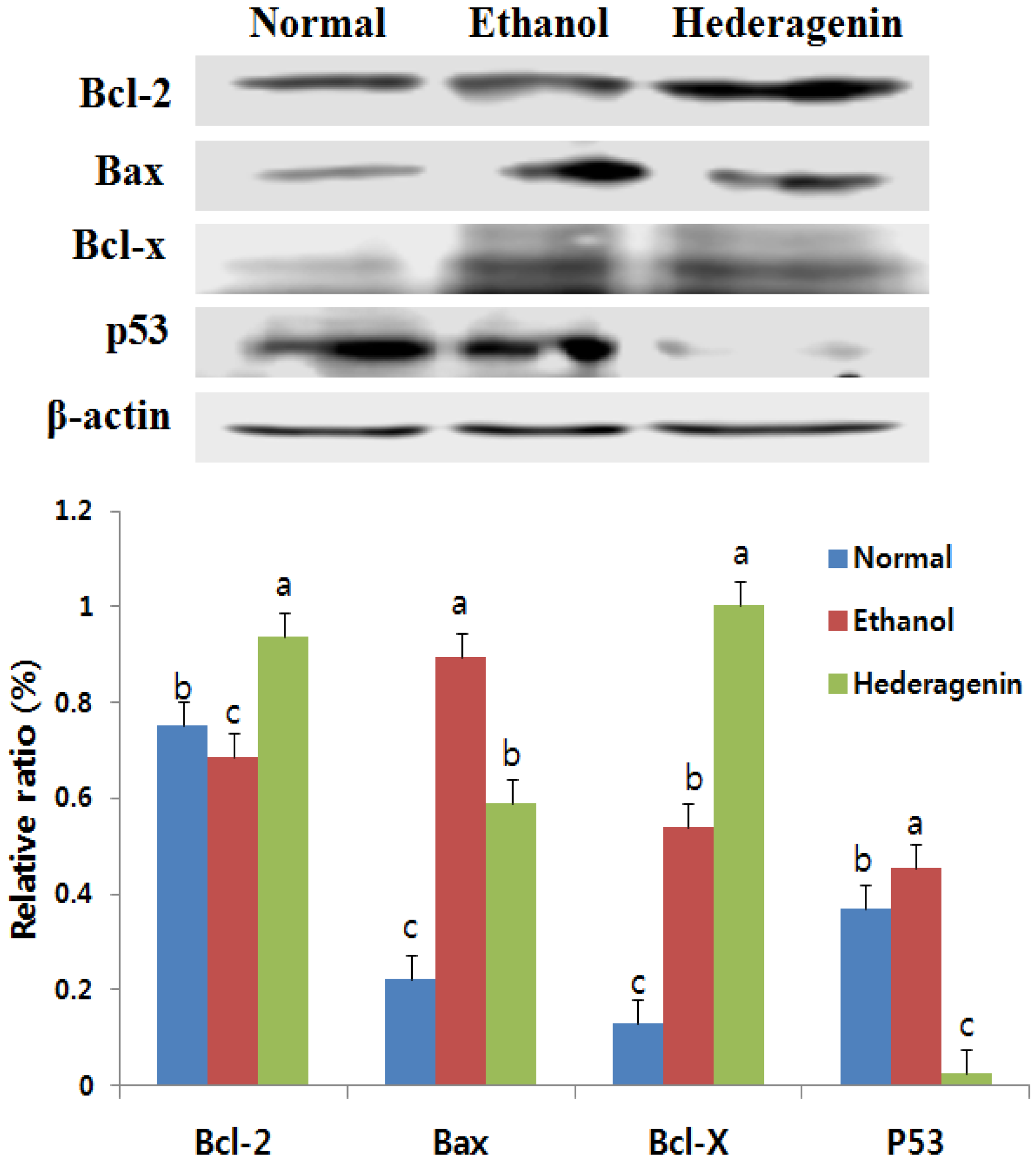
3.7. Effects of Hederagenin on the Expression of Bcl-2, Bax, Bcl-x, and p53
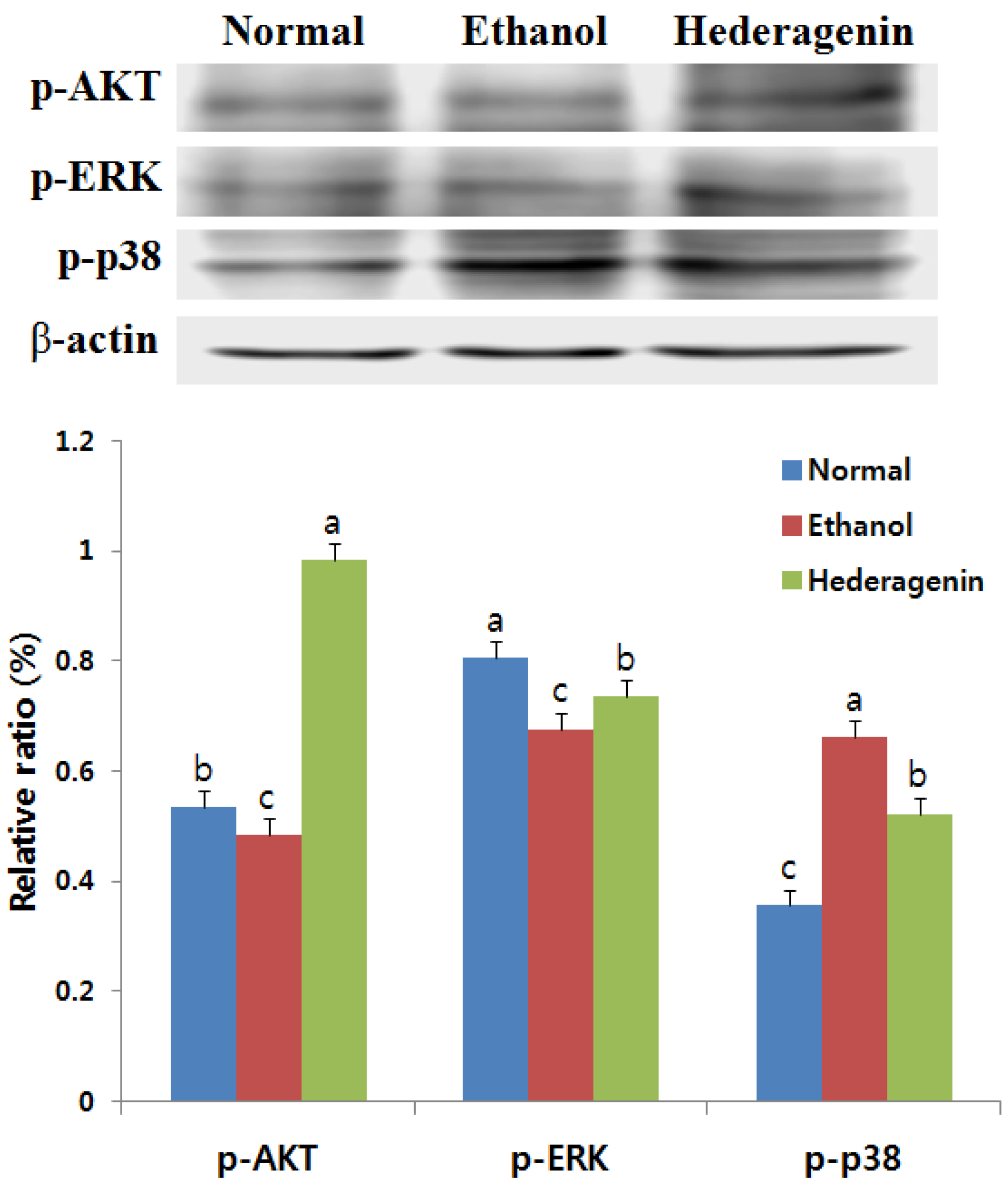
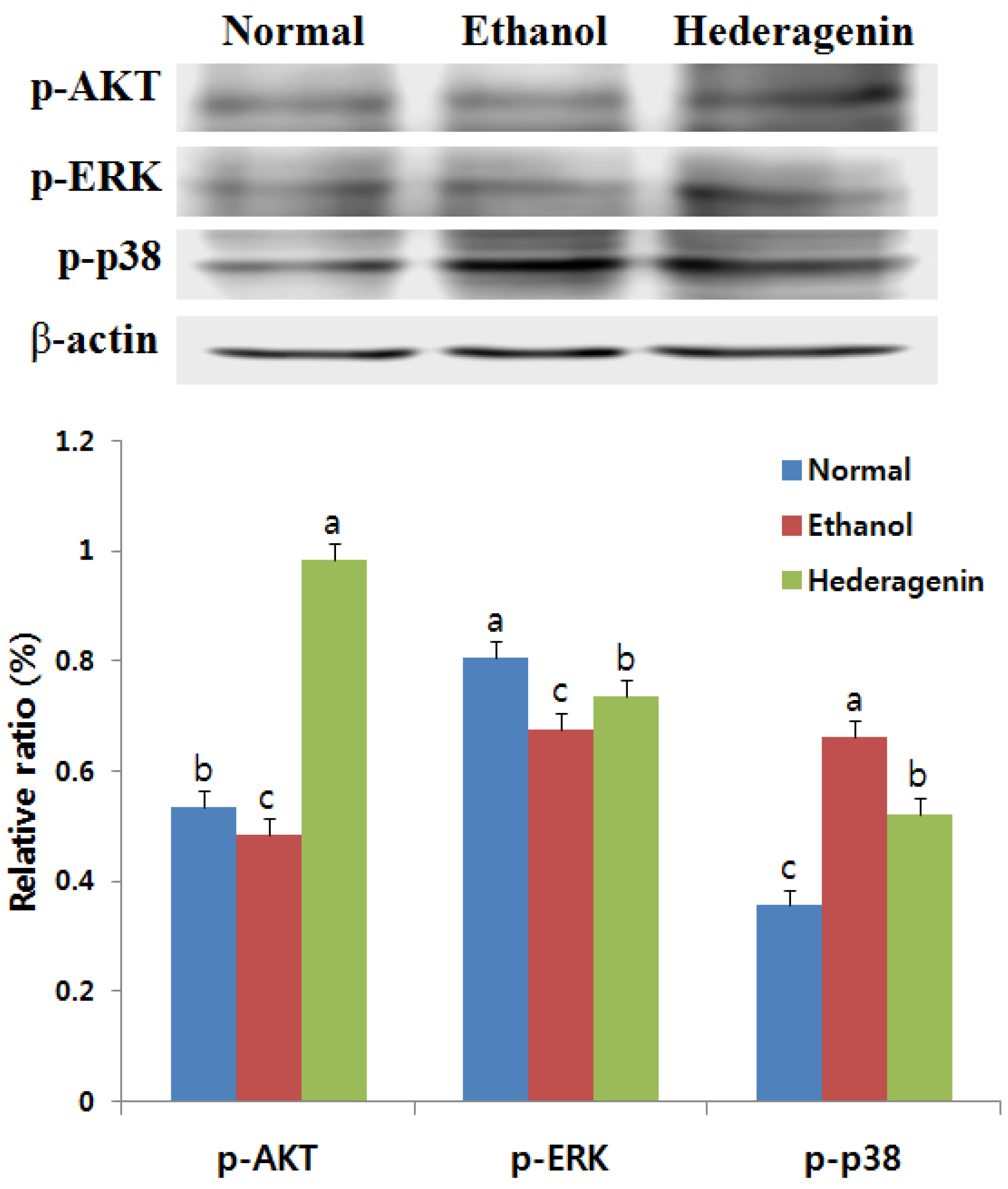
3.8. Effects of Hederagenin on Phosphorylation of AKT, ERK, and p38 MAPK
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Asante, L.S.; Chun, S.; Yun, M.; Newell, M. Social supply of alcohol to Korean high school students: A cross-sectional International Alcohol Control Study. BMJ Open 2013, 4, e003462. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Zheng, L.; Xu, L.; Yin, L.; Qi, Y.; Xu, Y.; Han, X.; Peng, J. Protective effects of dioscin against alcohol-induced liver injury. Arch. Toxicol. 2014, 88, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.J.; Koh, E.J.; Kim, C.S.; Zee, O.P.; Kwak, J.H.; Jeong, W.J.; Kim, J.H.; Lee, S.M. Agrimonia eupatoria protects against chronic ethanol-induced liver injury in rats. Food Chem. Toxicol. 2012, 50, 2335–2341. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.; Peng, Z.; Zhang, T.; Wu, H.; Yu, W.; Kong, D.; Liu, Y.; Bai, H.; Liu, R.; et al. The effects of Insulin Pre-Administration in Mice Exposed to Ethanol: Alleviating Hepatic Oxidative Injury through Anti-Oxidative, Anti-Apoptotic Activities and Deteriorating Hepatic Steatosis through SRBEP-1c Activation. Int. J. Biol. Sci. 2015, 11, 569–586. [Google Scholar] [CrossRef] [PubMed]
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxid. Med. Cell. Longev. 2010, 3, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Barry, R.E. Role of acetaldehyde in the pathogenesis of alcoholic liver disease. Br. J. Addict. 1988, 83, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Zhu, K.X.; Meng, F.L. Relationship between ALDH gene polymorphism and alcoholic liver diseases. Zhonghua Gan Zang Bing Za Zhi 2003, 11, 654–656. [Google Scholar] [PubMed]
- Huang, X.; Li, X.; Ma, Q.; Xu, Q.; Duan, W.; Lei, J.; Zhang, L.; Wu, Z. Chronic alcohol exposure exacerbates inflammatory and triggers pancreatic acinar-to-ductal metaplasia through PI3K/Akt/IKK. Int. J. Mol. Med. 2014, 35, 653–663. [Google Scholar] [PubMed]
- Boe, D.M.; Richens, T.R.; Horstmann, S.A. Acute and chronic alcohol exposure impair the phagocytosis of apoptotic cells and enhance the pulmonary inflammatory response. Alcohol. Clin. Exp. Res. 2010, 34, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Park, S.M.; Zhao, R.; Lee, C.; Chun, W.; Son, Y.; Kim, S.H.; Jung, J.Y.; Jegal, K.H.; Cho, I.J.; et al. Hederagenin, a major component of Clematis mandshurica Ruprecht root, attenuates inflammatory responses in RAW 264.7 cells and in mice. Int. Immunopharmacol. 2015, 29, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J.; Gautam, P.; Kushwaha, P. Lead and Ethanol Co-Exposure Lead to Blood Oxidative Stress and Subsequent Neuronal Apoptosis in Rats. Alcohol Alcohol. 2012, 47, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Jana, K.; Jana, N.; De, D.K.; Guha, S.K. Ethanol induces mouse spermatogenic cell apoptosis in vivo through over-expression of Fas/Fas-L, p53, and Caspase-3 along with cytochrome C translocation and glutathione depletion. Mol. Reprod. Dev. 2010, 77, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Ok, D.W.; Kwon, D.N.; Chung, J.I.; Kim, H.C.; Yeo, S.M.; Kim, T.; Seo, H.G.; Kim, J.H. Murine male germ cell apoptosis inducedby busulfan treatment correlates with loss of c-kit-expression in a Fas/Fas-L-and p53-independent manner. FEBS Lett. 2004, 575, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J.; Saxena, G.; Mehta, A. Reversal of lead-induced neuronal apoptosis by chelation treatment in rats: Role of ROS and intracellular Ca2+. J. Pharmacol. Exp. Ther. 2007, 322, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.H.; Guan, J.H.; Huang, Y.L.; Pan, Y.W.; Yang, W.; Lan, H.; Huang, S.; Hu, J.; Zhao, G.P. Experimental study of antiatherosclerosis effects with hederagenin in rats. Evid.-Based Complement. Altern. Med. 2015, 2015, 456354. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Han, Y.N.; Lee, K.T. Anti-lipid peroxidative principles from the stem bark of Kalopanax pictus Nakai. Arch. Pharm. Res. 2001, 24, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Takagi, K.; Park, E.H.; Kato, H. Anti-inflammatory activities of hederagenin and crude saponin isolated from Sapindus mukorossi Gaertn. Chem. Pharm. Bull. 1980, 28, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.G.; Zeng, W.; Wong, V.; Kam, W.; Zhu, Y.Z.; Lo, A.C.Y.; Liu, L.; Law, B.Y.K. Hederagenin and α-hederin promote degradation of proteins in neurodegenerative diseases and improve motor deficits in MPTP-mice. Pharmacol. Res. 2017, 115, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Lee, C.O.; Lee, K.T.; Choi, J.; Park, H.J. Structure-activity relationship of oleanane disaccharides isolated from akebia quinata versus cytotoxicity against cancer cells and NO inhibition. Biol. Pharm. Bull. 2004, 27, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Green, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S. Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Anal. Biochem. 1982, 126, 131–138. [Google Scholar] [CrossRef]
- Reitman, S.; Frankel, S. A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. Am. J. Clin. Path. 1957, 28, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, H.O.; Chaker, H.; Leaming, R.; Johnson, A.; Brechtel, G. Obesity/insulin resistance is associated with endothelial dysfunction implications for the syndrome of insulin resistance. J. Clin. Investig. 1996, 97, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.L.; Liang, B.; Wang, X.W.; Fan, F.G.; Jin, J.; Lan, R.; Yang, J.H.; Wang, X.C.; Jin, L.; Cao, Q. Glycyrrhizic acid attenuates CCl4-induced hepatocyte apoptosis in rats via a p53-mediated pathway. World J. Gastroenterol. 2013, 19, 3781–3791. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.X.Z.; Zhou, J.Y.; Li, Y.; Zou, X.; Wu, J.; Gu, J.F.; Yuan, J.R.; Zhao, B.J.; Feng, L.; Jia, X.B.; et al. Hederagenin from the leaves of ivy (Hedera helix L.) induces apoptosis in human LoVo colon cells through the mitochondrial pathway. BMC Complement. Altern. Med. 2014, 14, 412. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.H.; Lan, R.; Zhen, X.D.; Zhang, W.; Xiang, J. An-Gong-Niu-Huang Wan protects against cerebral ischemia induced apoptosis in rats: Up-regulation of Bcl-2 and down-regulation of Bax and caspase-3. J. Ethnophapmacol. 2014, 154, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Zhao, Y.; Zhang, X.; Lin, X. Astaxantin inhibits acetaldehyde-induced cytotoxicity in SH-SY5Y cells by modulating Akt/CREB and p38MAPK/ERK signailing pathways. Mar. Drugs 2016, 14, 56. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Normal | Ethanol | Hederagenin |
|---|---|---|---|
| Initial body weight (g) | 128.5 ± 3.39 a | 128.71 ± 5.31 a | 130.17 ± 3.19 a |
| Final body weight (g) | 245.13 ± 41.49 a | 270.01 ± 31.01 a | 260.86 ± 9.68 a |
| Body weight gain/day (g) | 5.55 ± 1.81 a | 6.72 ± 1.22 a | 6.22 ± 0.31 a |
| Liver weight (g) | 10.90 ± 1.43 a | 9.61 ± 0.92 a | 9.39 ± 1.07 a |
| Kidney weight (g) | 2.61 ± 0.11 a | 2.44 ± 0.17 a | 2.34 ± 0.24 a |
| LW/BW | 0.045 ± 0.034 a | 0.036 ± 0.030 a | 0.036 ± 0.110 a |
| Treatment | AST (IU/L) | ALT (IU/L) | TC (mg/dL) | TG (mg/dL) |
|---|---|---|---|---|
| Normal (n = 6) | 113.03 ± 28.20 c | 35.14 ± 2.41 a | 85.55 ± 4.06 b | 30.28 ± 10.55 c |
| Ethanol (n = 7) | 235.93 ± 45.38 a | 42.17 ± 20.48 a | 95.47 ± 8.65 a | 55.32 ± 9.80 a |
| Hederagenin (n = 6) | 208.65 ± 32.94 b | 27.53 ± 7.38 a | 79.75 ± 5.24 b | 41 ± 9.79 b |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, G.-J.; Song, D.H.; Yoo, H.S.; Chung, K.-H.; Lee, K.J.; An, J.H. Hederagenin Supplementation Alleviates the Pro-Inflammatory and Apoptotic Response to Alcohol in Rats. Nutrients 2017, 9, 41. https://doi.org/10.3390/nu9010041
Kim G-J, Song DH, Yoo HS, Chung K-H, Lee KJ, An JH. Hederagenin Supplementation Alleviates the Pro-Inflammatory and Apoptotic Response to Alcohol in Rats. Nutrients. 2017; 9(1):41. https://doi.org/10.3390/nu9010041
Chicago/Turabian StyleKim, Gyeong-Ji, Da Hye Song, Han Seok Yoo, Kang-Hyun Chung, Kwon Jai Lee, and Jeung Hee An. 2017. "Hederagenin Supplementation Alleviates the Pro-Inflammatory and Apoptotic Response to Alcohol in Rats" Nutrients 9, no. 1: 41. https://doi.org/10.3390/nu9010041