Interaction of Ochratoxin A and Its Thermal Degradation Product 2′R-Ochratoxin A with Human Serum Albumin


 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
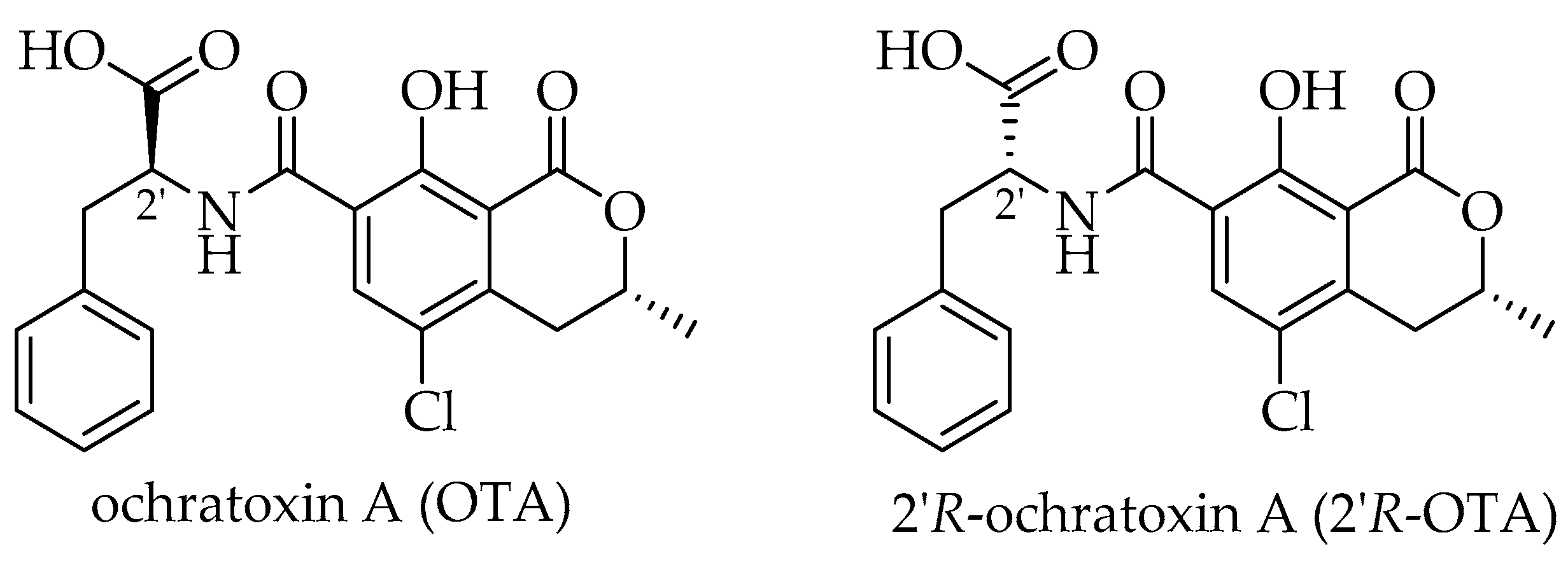
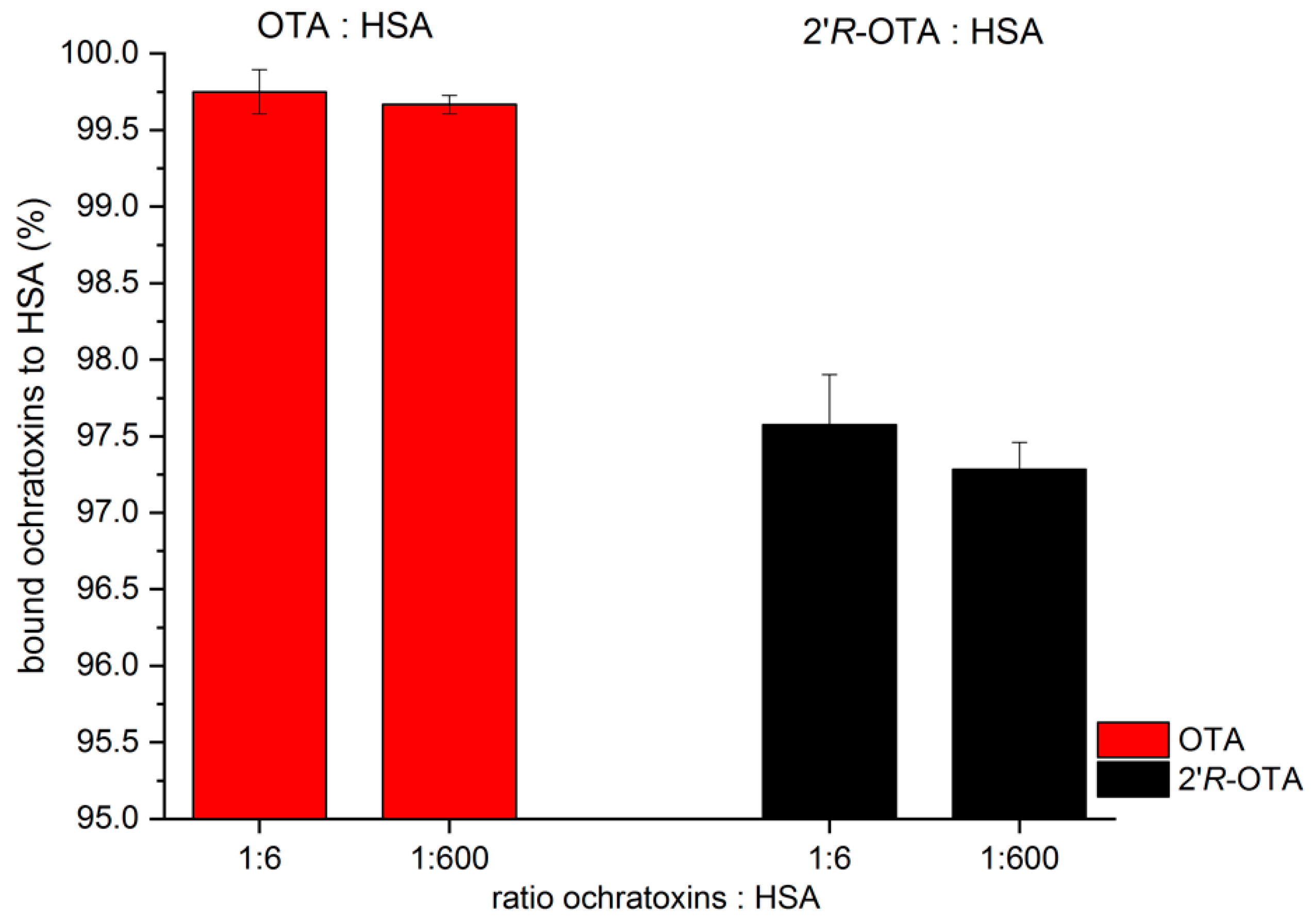
2.1. Dialysis of OTA and 2′R-OTA with HSA
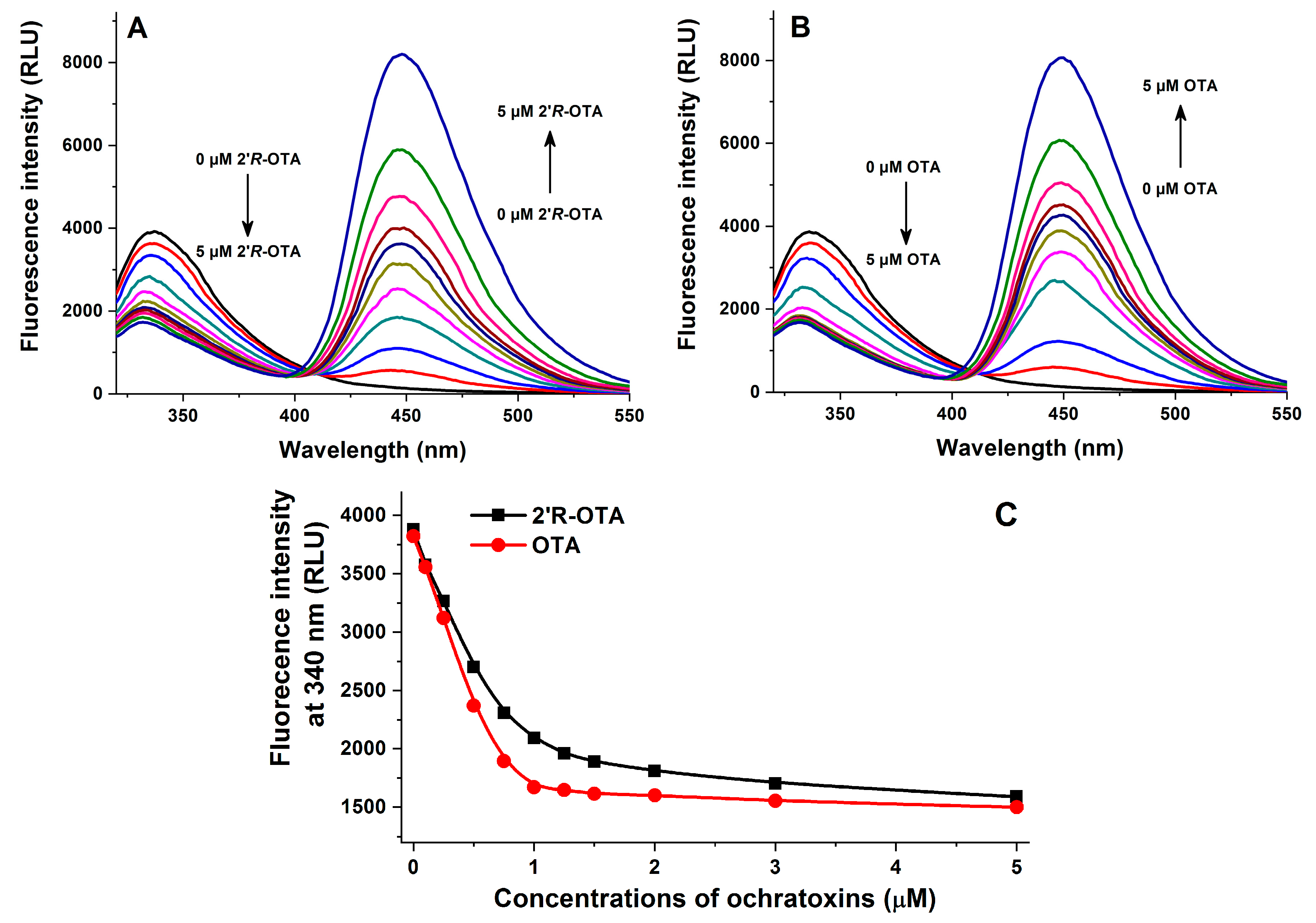
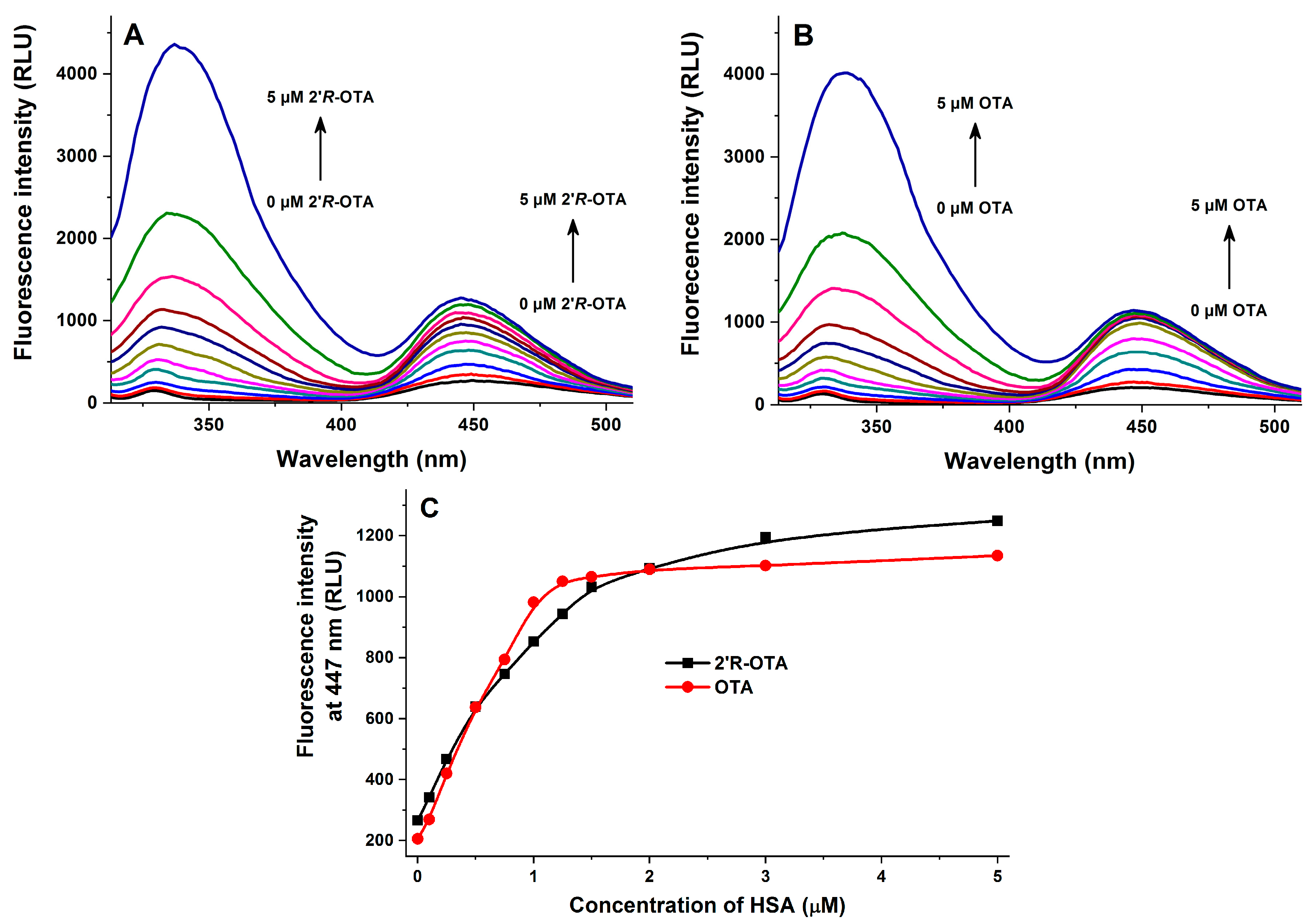
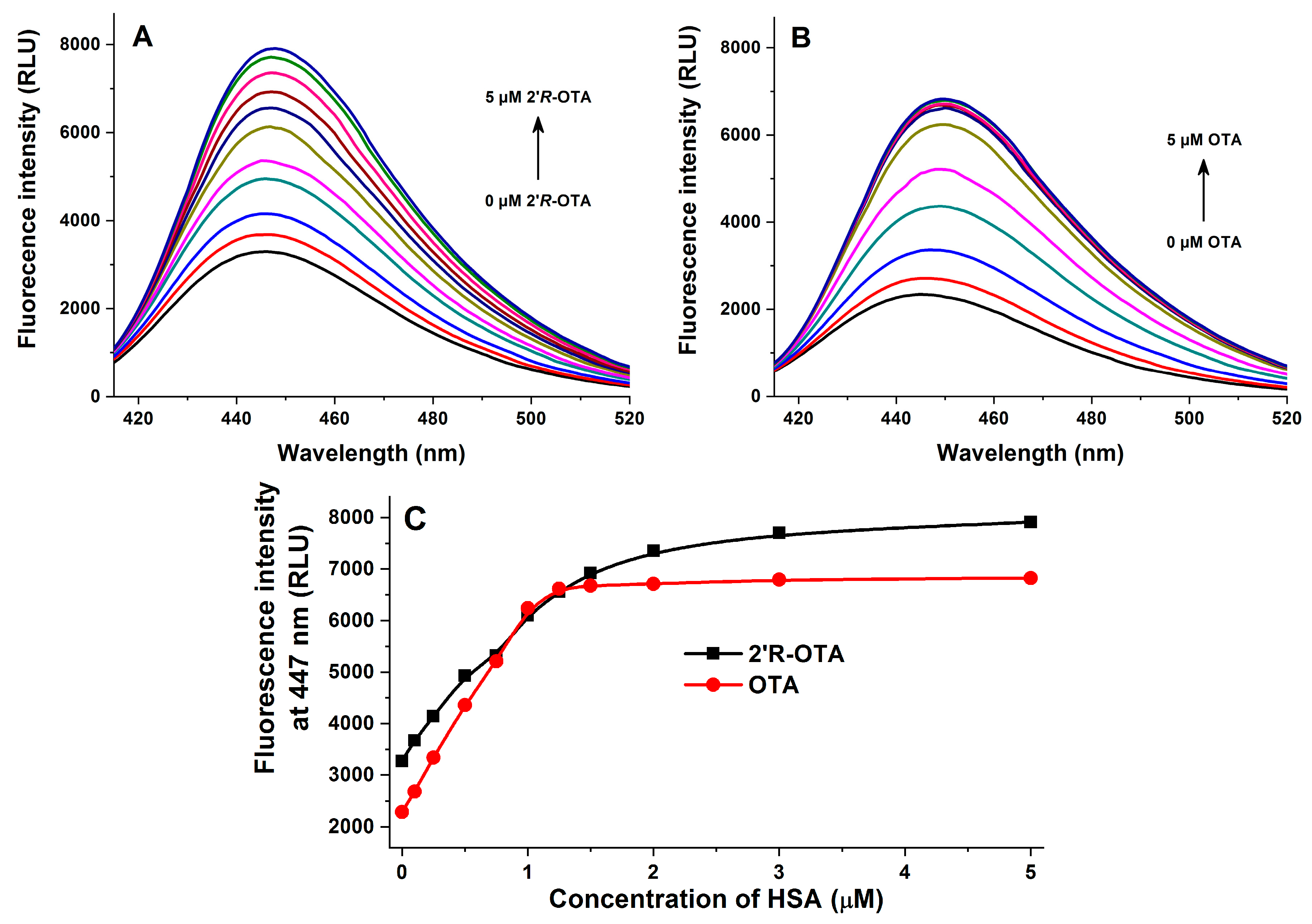
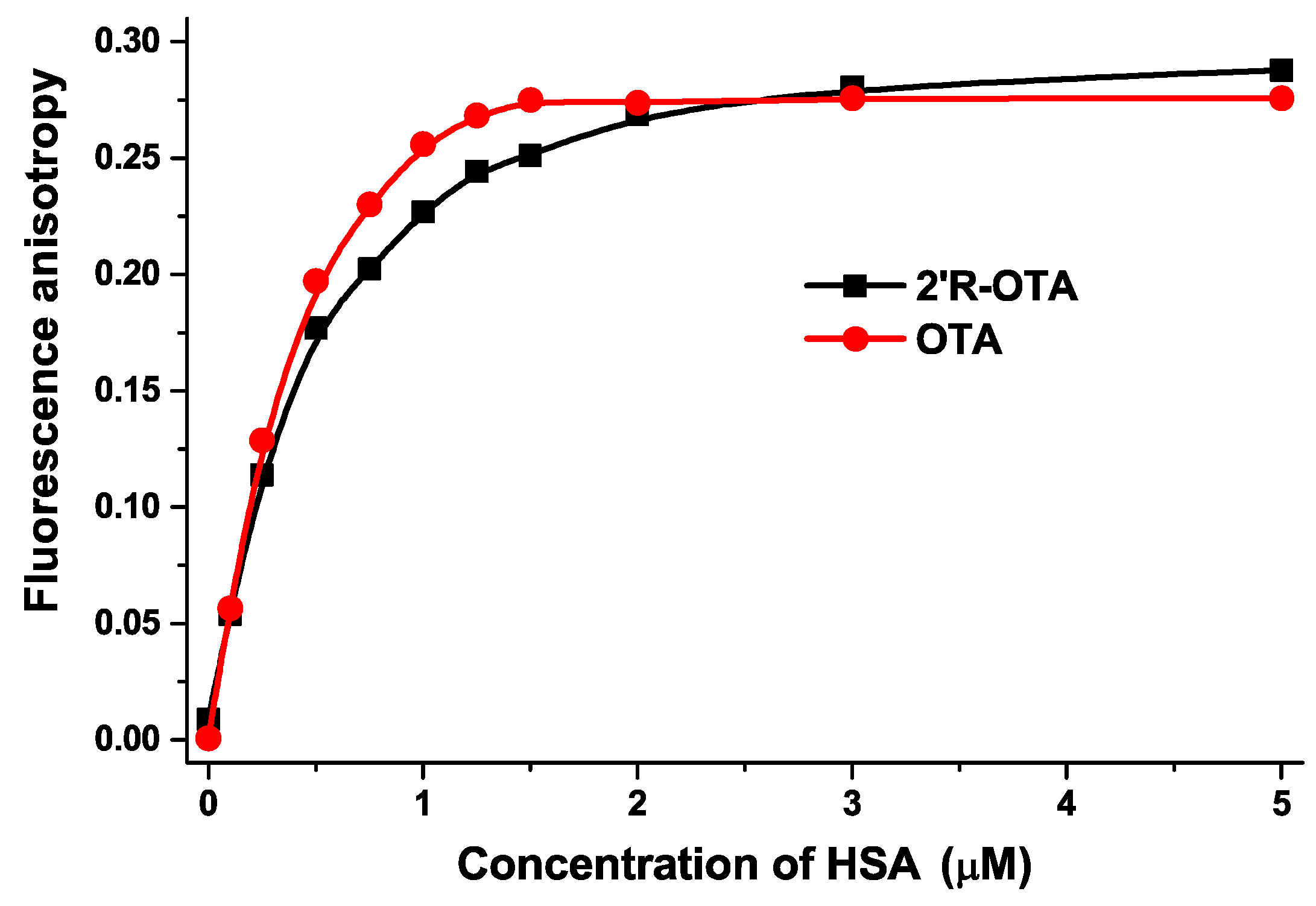
2.2. Fluorescence Spectroscopic Investigation of OTA and 2′R-OTA with HSA
2.3. High-Performance Affinity Chromatography of HSA with OTA and 2′R-OTA
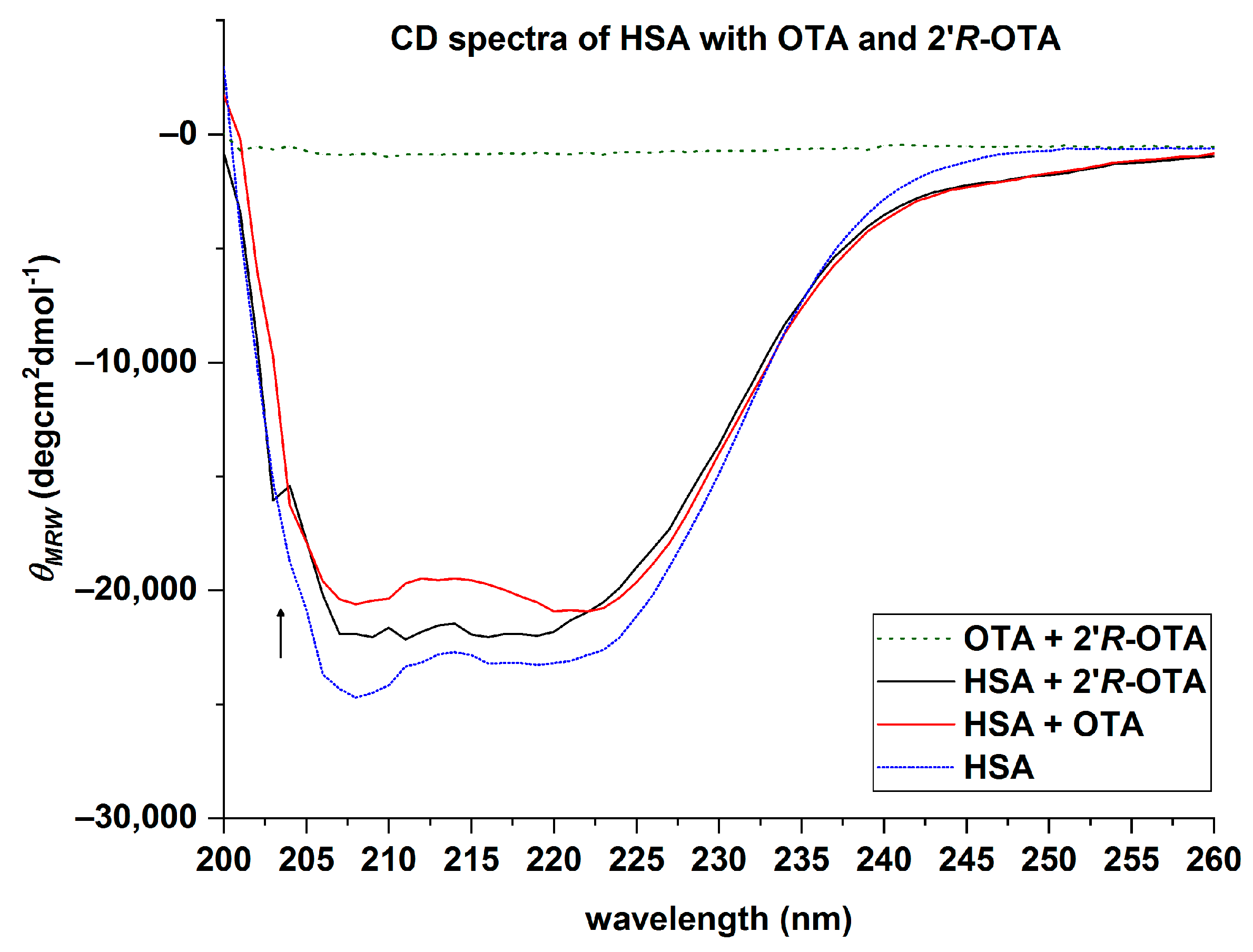
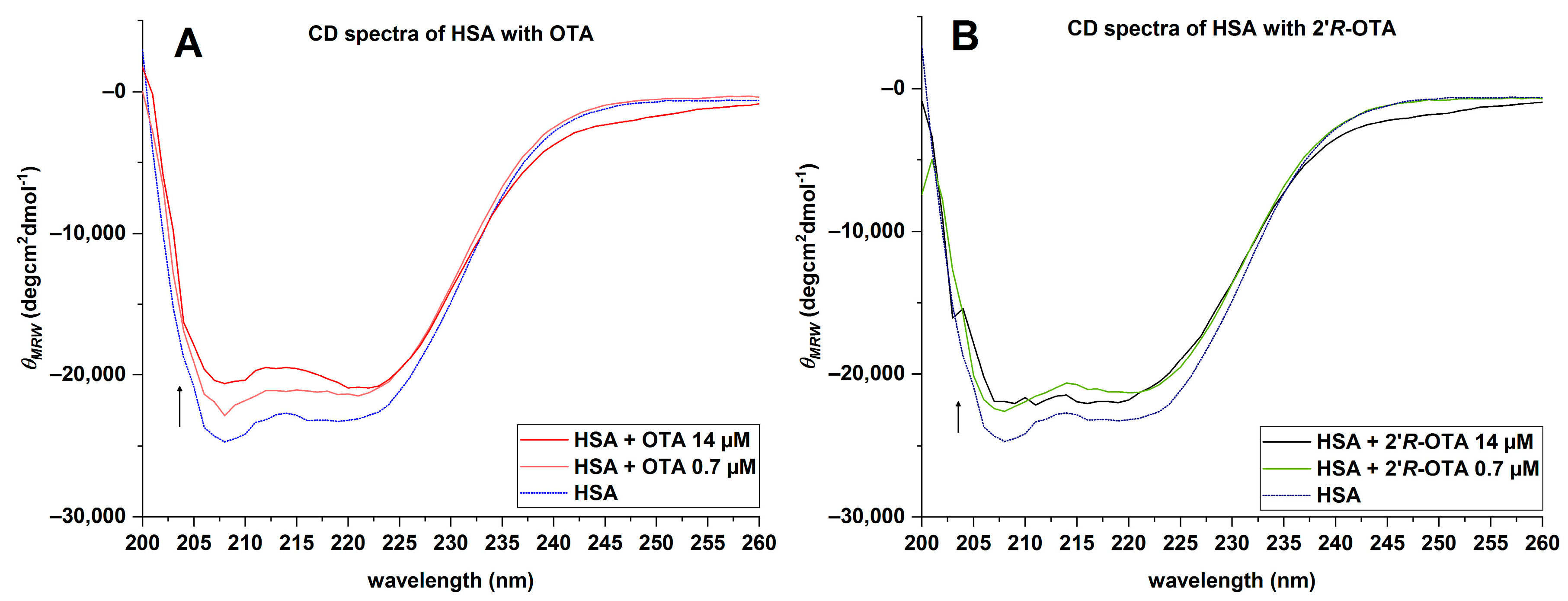
2.4. Circular Dichroism (CD) of HSA with OTA and 2′R-OTA
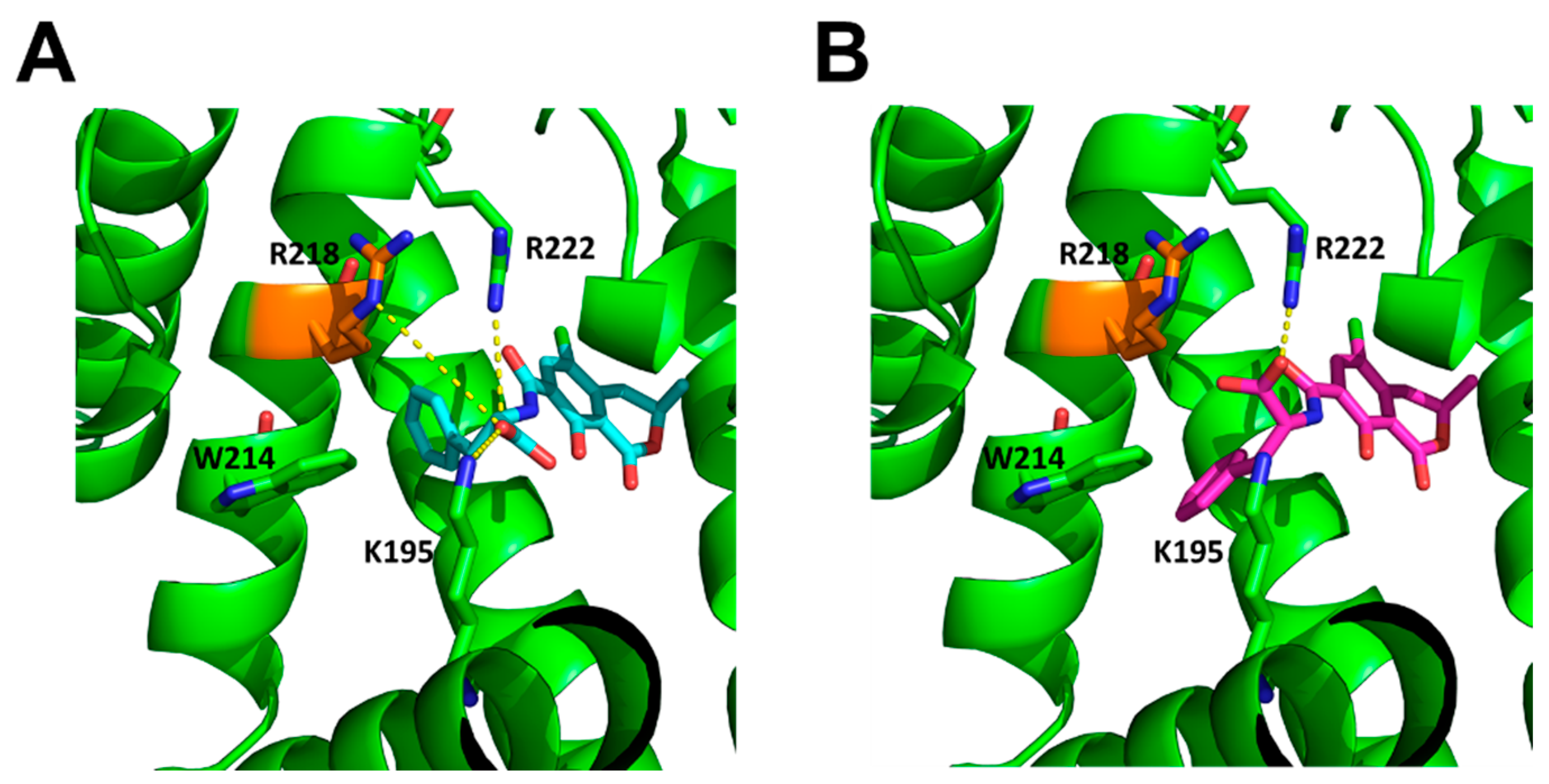
2.5. Molecular Modelling Studies of OTA and 2′R-OTA with HSA
3. Conclusions
4. Materials and Methods
4.1. Reagents
4.2. Biosynthesis of Standards
4.3. Dialysis Experiments
4.4. HPLC-MS/MS Parameters for the Dialysis Experiments
4.5. Fluorescence Spectroscopic Measurments
4.6. HPAC-MS Measurments
4.7. Circular Dichroism Measurements
4.8. Molecular Modeling Studies
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- EU. SCOOP Task 3.2.7. Reports on Tasks for Scientific Cooperation: Assessment of Dietary Intake of Ochratoxin A by the Population of EU Member States. 2002. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/cs_contaminants_catalogue_ochratoxin_task_3-2-7_en.pdf (accessed on 21 June 2018).
- Cramer, B.; Königs, M.; Humpf, H.-U. Identification and in vitro cytotoxicity of ochratoxin A degradation products formed during coffee roasting. J. Agric. Food Chem. 2008, 56, 5673–5681. [Google Scholar] [CrossRef] [PubMed]
- Studer-Rohr, I.; Dietrich, D.R.; Schlatter, J.; Schlatter, C. The Occurrence of Ochratoxin A in Coffee. Food Chem. Toxicol. 1995, 33, 341–355. [Google Scholar] [CrossRef]
- Cramer, B.; Osteresch, B.; Muñoz, K.A.; Hillmann, H.; Sibrowski, W.; Humpf, H.-U. Biomonitoring using dried blood spots: Detection of ochratoxin A and its degradation product 2′R-ochratoxin A in blood from coffee drinkers. Mol. Nutr. Food Res. 2015, 59, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Viegas, S.; Osteresch, B.; Almeida, A.; Cramer, B.; Humpf, H.-U.; Viegas, C. Enniatin B and ochratoxin A in the blood serum of workers from the waste management setting. Mycotoxin Res. 2017, 34, 85–90. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer (IARC). Monographs on the Evaluation of Carcinogenic Risks to Humans: Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins; IARC: Lyon, France, 1993; pp. 607–618. [Google Scholar]
- Cramer, B.; Harrer, H.; Nakamura, K.; Uemura, D.; Humpf, H.-U. Total synthesis and cytotoxicity evaluation of all ochratoxin A stereoisomers. Bioorg. Med. Chem. 2010, 18, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Malir, F.; Ostry, V.; Pfohl-Leszkowicz, A.; Malir, J.; Toman, J. Ochratoxin A: 50 Years of Research. Toxins 2016, 8, 191. [Google Scholar] [CrossRef] [PubMed]
- Hagelberg, S.; Hult, K.; Fuchs, R. Toxicokinetics of ochratoxin A in several species and its plasma-binding properties. J. Appl. Toxicol. 1989, 9, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Galtier, P.; Charpenteau, J.-L.; Alvinerie, M.; Labouche, C. The pharmacokinetic profile of ochratoxin A in the rat after oral and intravenous administration. J. Pharmacol. Exp. Ther. 1979, 7, 429–434. [Google Scholar]
- Studer-Rohr, I.; Schlatter, J.; Dietrich, D.R. Kinetic parameters and intraindividual fluctuations of ochratoxin A plasma levels in humans. Arch. Toxicol. 2000, 74, 499–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, R.; Hult, K. Ochratoxin A in blood and its pharmacokinetic properties. Food Chem. Toxicol. 1992, 30, 201–204. [Google Scholar] [CrossRef]
- Fanali, G.; Di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.L.; Christensen, T.; Goldsmith, M.R.; Toone, E.J.; Beratan, D.N.; Simon, J.D. Binding of Ochratoxin A to Human Serum Albumin Stabilized by a Protein−Ligand Ion Pair. J. Phys. Chem. B 2003, 107, 7884–7888. [Google Scholar] [CrossRef]
- Il'ichev, Y.V.; Perry, J.L.; Rüker, F.; Dockal, M.; Simon, J.D. Interaction of ochratoxin A with human serum albumin. Binding sites localized by competitive interactions with the native protein and its recombinant fragments. Chem. Biol. Interact. 2002, 141, 275–293. [Google Scholar] [CrossRef]
- Poór, M.; Kunsági-Máté, S.; Bencsik, T.; Petrik, J.; Vladimir-Knežević, S.; Kőszegi, T. Flavonoid aglycones can compete with Ochratoxin A for human serum albumin: A new possible mode of action. Int. J. Biol. Macromol. 2012, 51, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Poór, M.; Li, Y.; Matisz, G.; Kiss, L.; Kunsági-Máté, S.; Kőszegi, T. Quantitation of species differences in albumin–ligand interactions for bovine, human and rat serum albumins using fluorescence spectroscopy: A test case with some Sudlow’s site I ligands. J. Lumin. 2014, 145, 767–773. [Google Scholar] [CrossRef]
- Li, Y.; Czibulya, Z.; Poór, M.; Lecomte, S.; Kiss, L.; Harte, E.; Kőszegi, T.; Kunsági-Máté, S. Thermodynamic study of the effects of ethanol on the interaction of ochratoxin A with human serum albumin. J. Lumin. 2014, 148, 18–25. [Google Scholar] [CrossRef]
- Singh, S.S.; Mehta, J. Measurement of drug-protein binding by immobilized human serum albumin-HPLC and comparison with ultrafiltration. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 834, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yan, J.; He, J.; Bai, K.; Li, H. Characterization of the interaction between 3-Oxotabersonine and two serum albumins by using spectroscopic techniques. J. Lumin. 2013, 138, 1–7. [Google Scholar] [CrossRef]
- Poór, M.; Bálint, M.; Hetényi, C.; Gődér, B.; Kunsági-Máté, S.; Kőszegi, T.; Lemli, B. Investigation of Non-Covalent Interactions of Aflatoxins (B1, B2, G1, G2, and M1) with Serum Albumin. Toxins 2017, 9, 339. [Google Scholar] [CrossRef] [PubMed]
- Poór, M.; Li, Y.; Kunsági-Máté, S.; Varga, Z.; Hunyadi, A.; Dankó, B.; Chang, F.-R.; Wu, Y.-C.; Kőszegi, T. Protoapigenone derivatives: Albumin binding properties and effects on HepG2 cells. J. Photochem. Photobiol. B Biol. 2013, 124, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Ajmal, M.R.; Nusrat, S.; Alam, P.; Zaidi, N.; Khan, M.V.; Zaman, M.; Shahein, Y.E.; Mahmoud, M.H.; Badr, G.; Khan, R.H. Interaction of anticancer drug clofarabine with human serum albumin and human α-1 acid glycoprotein. Spectroscopic and molecular docking approach. J. Pharm. Biomed. Anal. 2017, 135, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Louis-Jeune, C.; Andrade-Navarro, M.A.; Perez-Iratxeta, C. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins 2012, 80, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.; Amman, H.; Doran, D.; Gerber, P.; Gubernator, K.; Schrepfer, G. MOLOC: A molecular modeling program. Bull. Soc. Chim. Belg. 1988, 97, 655–667. [Google Scholar]
- Bas, D.C.; Rogers, D.M.; Jensen, J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins 2008, 73, 765–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Case, D.A.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Greene, D.; Homeyer, N.; et al. Amber 16, AmberTools17. San Francisco, CA, USA, 2017. Available online: http://ambermd.org/ (accessed on 21 June 2018).
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. FEW: A workflow tool for free energy calculations of ligand binding. J. Comput. Chem. 2013, 34, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Case, D.A. Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J. Comput. Chem. 2004, 25, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Weis, A.; Katebzadeh, K.; Söderhjelm, P.; Nilsson, I.; Ryde, U. Ligand affinities predicted with the MM/PBSA method: Dependence on the simulation method and the force field. J. Chem. Inf. Model. 2006, 49, 6596–6606. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | logKSV (SV-Plot, Figure 3) | logK (Hyperquad, Figure 3) | logK (Hyperquad, Figure 5) | logK (Hyperquad, Figure 6) | logK (Anisotropy, Figure 7) |
|---|---|---|---|---|---|
| 2′R-OTA-HSA | 5.94 ± 0.02 | 6.23 ± 0.01 | 6.36 ± 0.06 | 6.40 ± 0.24 | 6.18 ± 0.01 |
| Compound | Retention Time ± SD (min) 1 |
|---|---|
| OTA | 20.1 ± 0.9 |
| 2′R-OTA | 16.9 ± 0.2 |
| HSA + Ochratoxin (Ratio) | θMRE (×102) (deg × cm2 × dmol−1) | α-Helix * (%) | α-Helix ** (%) | Rel. Differences to HSA | ||
|---|---|---|---|---|---|---|
| 208 nm | 222 nm | 208 nm | 222 nm | |||
| HSA | −247 | −228 | 71.4 | 65.0 | 66.9 | - |
| HSA + 2′R-OTA (1:20) | −220 | −210 | 61.8 | 58.5 | 65.3 | 2–13% |
| HSA + OTA (1:20) | −206 | −209 | 57.3 | 58.3 | 61.5 | 8–20% |
| Q1 Mass (m/z) | Q3 Mass (m/z) | Transition Time (ms) | DP (V) | CE (V) | EP (V) |
|---|---|---|---|---|---|
| 404.1 | 239.0 (quantifier) | 100 | +70 | +31 | +10 |
| 404.1 | 221.1 (qualifier) | 100 | +70 | +47 | +10 |
| 404.1 | 102.0 (qualifier) | 100 | +77 | +88 | +10 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sueck, F.; Poór, M.; Faisal, Z.; Gertzen, C.G.W.; Cramer, B.; Lemli, B.; Kunsági-Máté, S.; Gohlke, H.; Humpf, H.-U. Interaction of Ochratoxin A and Its Thermal Degradation Product 2′R-Ochratoxin A with Human Serum Albumin. Toxins 2018, 10, 256. https://doi.org/10.3390/toxins10070256
Sueck F, Poór M, Faisal Z, Gertzen CGW, Cramer B, Lemli B, Kunsági-Máté S, Gohlke H, Humpf H-U. Interaction of Ochratoxin A and Its Thermal Degradation Product 2′R-Ochratoxin A with Human Serum Albumin. Toxins. 2018; 10(7):256. https://doi.org/10.3390/toxins10070256
Chicago/Turabian StyleSueck, Franziska, Miklós Poór, Zelma Faisal, Christoph G. W. Gertzen, Benedikt Cramer, Beáta Lemli, Sándor Kunsági-Máté, Holger Gohlke, and Hans-Ulrich Humpf. 2018. "Interaction of Ochratoxin A and Its Thermal Degradation Product 2′R-Ochratoxin A with Human Serum Albumin" Toxins 10, no. 7: 256. https://doi.org/10.3390/toxins10070256
APA StyleSueck, F., Poór, M., Faisal, Z., Gertzen, C. G. W., Cramer, B., Lemli, B., Kunsági-Máté, S., Gohlke, H., & Humpf, H.-U. (2018). Interaction of Ochratoxin A and Its Thermal Degradation Product 2′R-Ochratoxin A with Human Serum Albumin. Toxins, 10(7), 256. https://doi.org/10.3390/toxins10070256