Exposure to the Harmful Algal Bloom (HAB) Toxin Microcystin-LR (MC-LR) Prolongs and Increases Severity of Dextran Sulfate Sodium (DSS)-Induced Colitis

 , ,
, , 
 , , and
, , and
Abstract
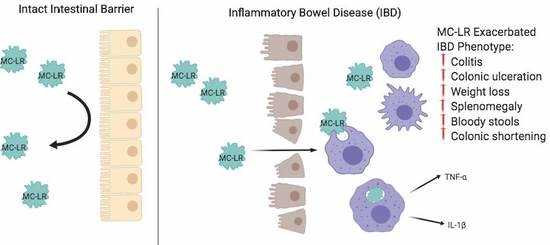
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
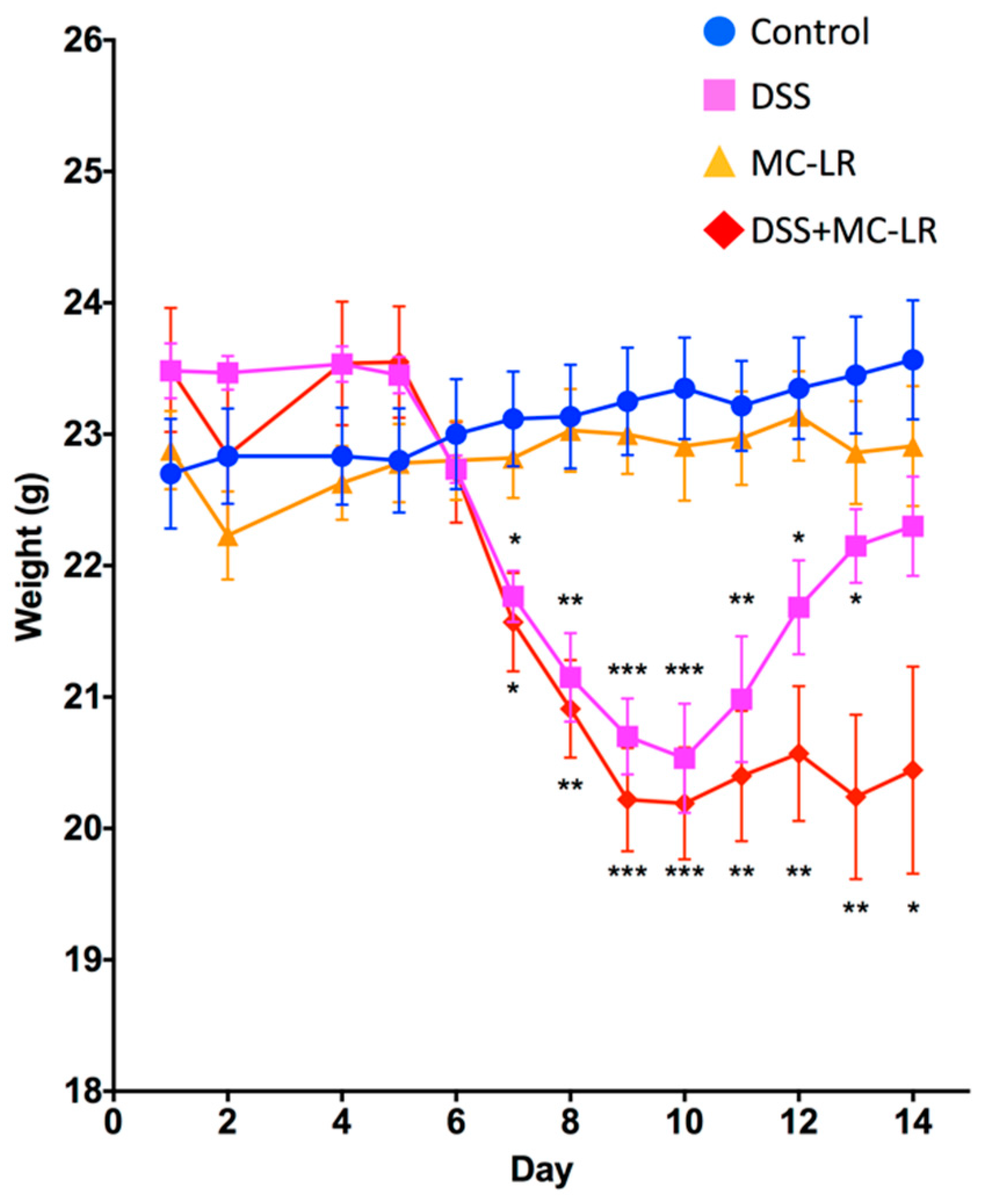
2.1. Body Weight and Survival
2.2. Stool Grading
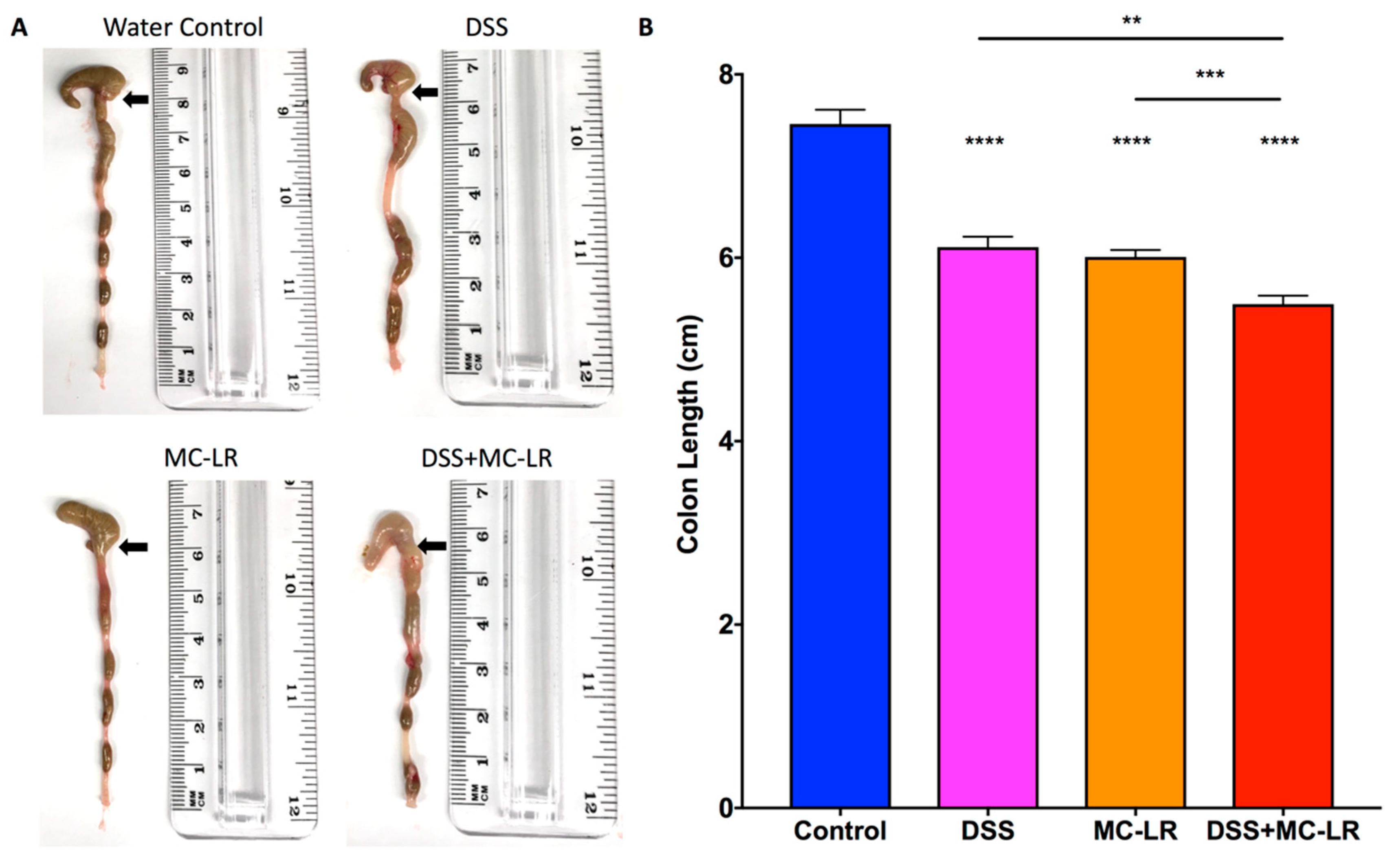
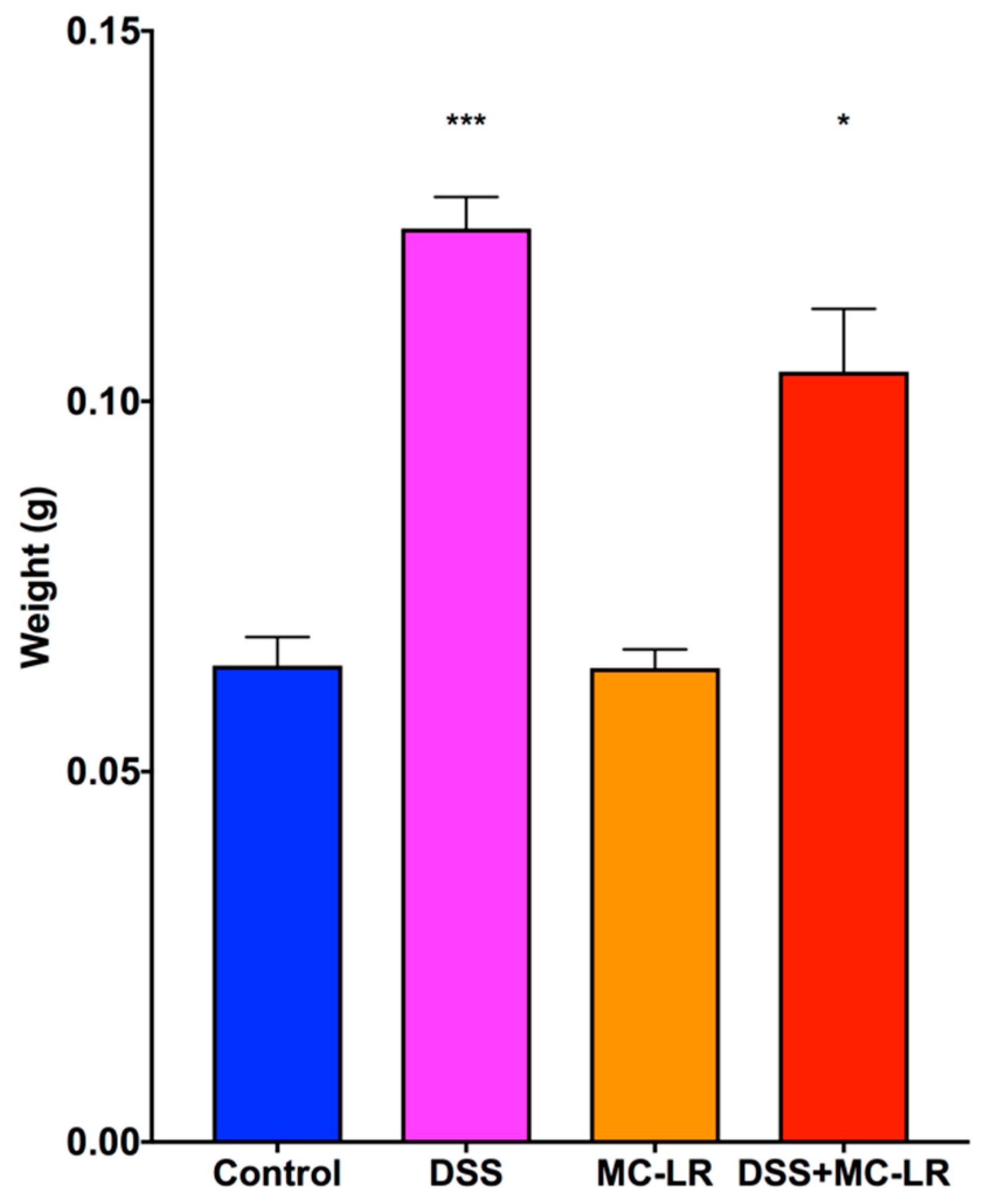
2.3. Colon Length and Spleen Weight
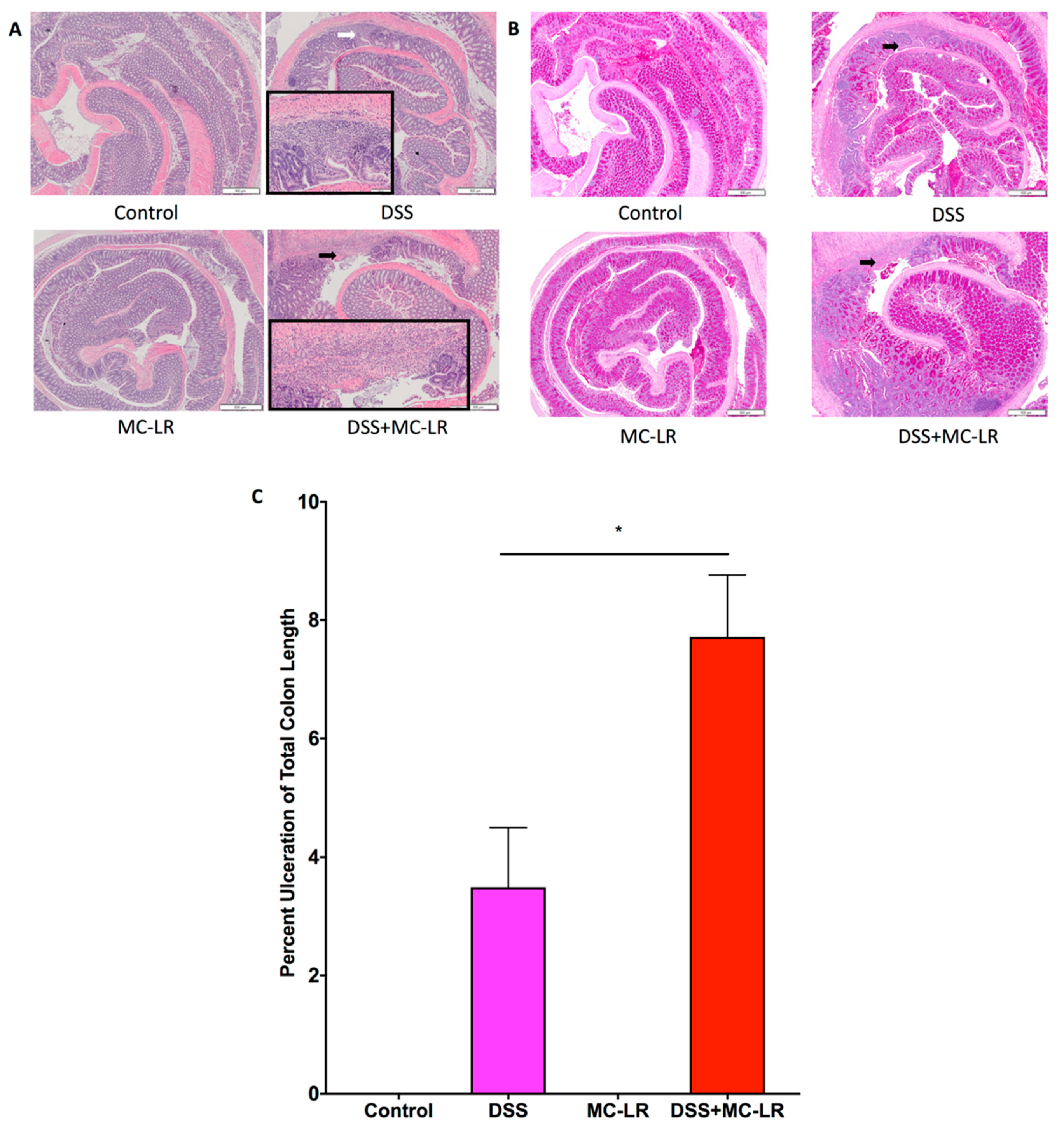
2.4. Histopathology
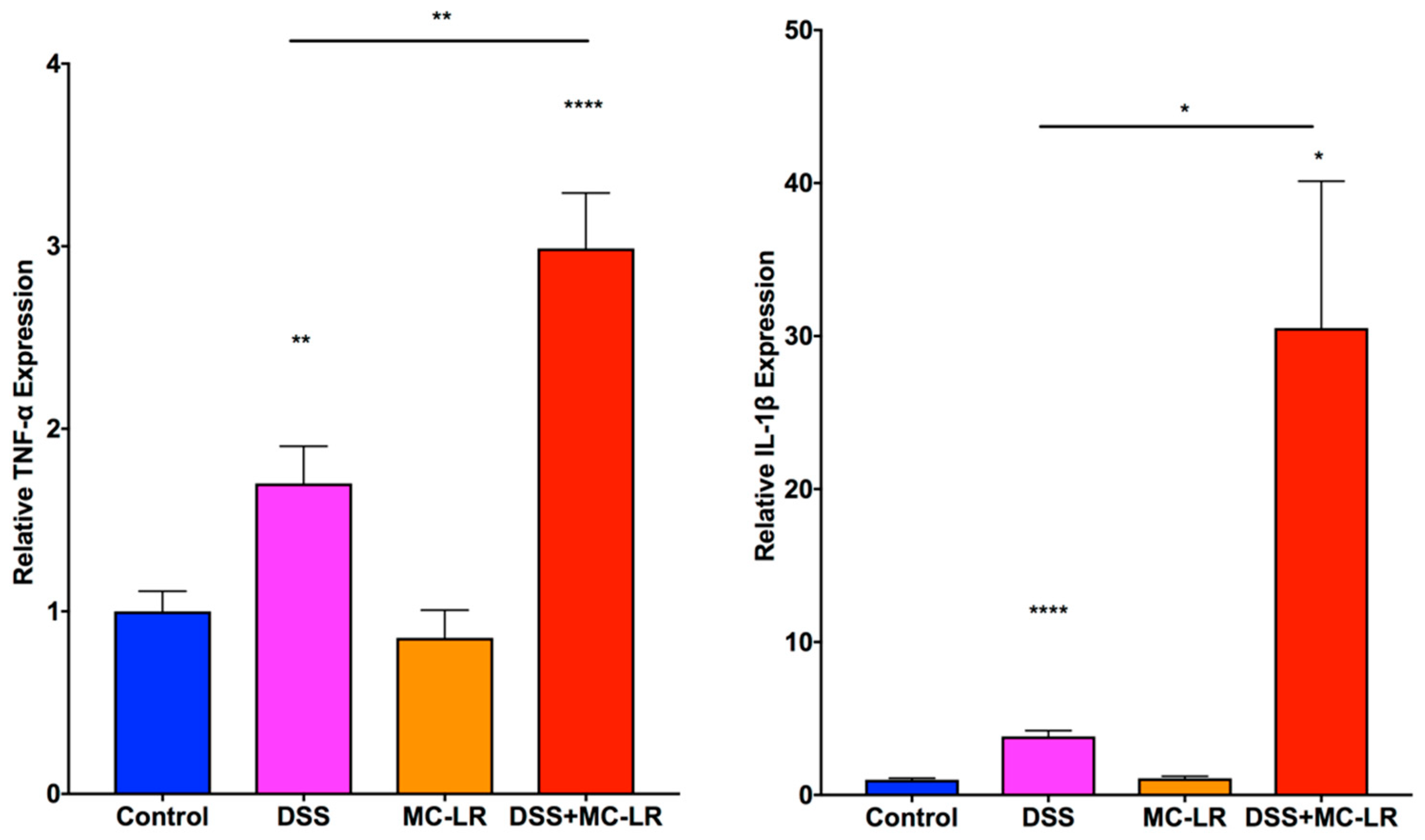
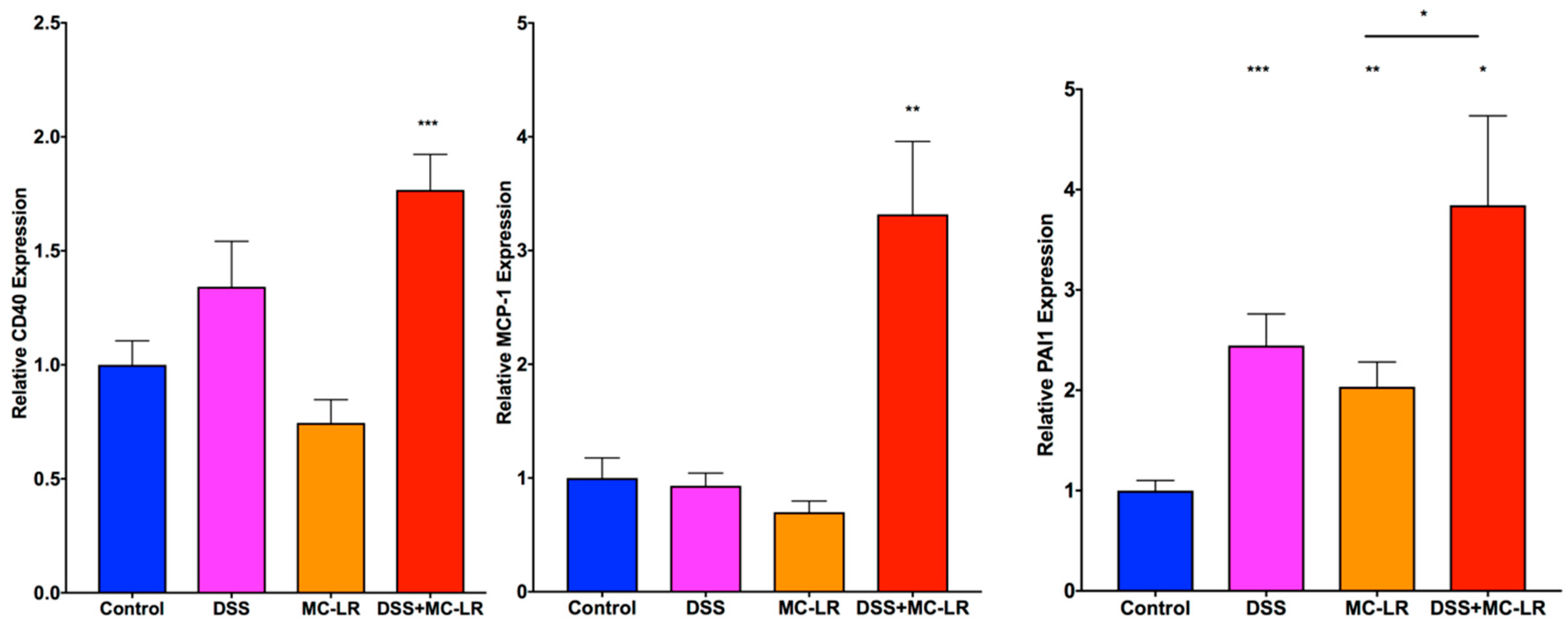
2.5. Gene Expression in the Colon
3. Discussion
4. Materials and Methods
4.1. Mice
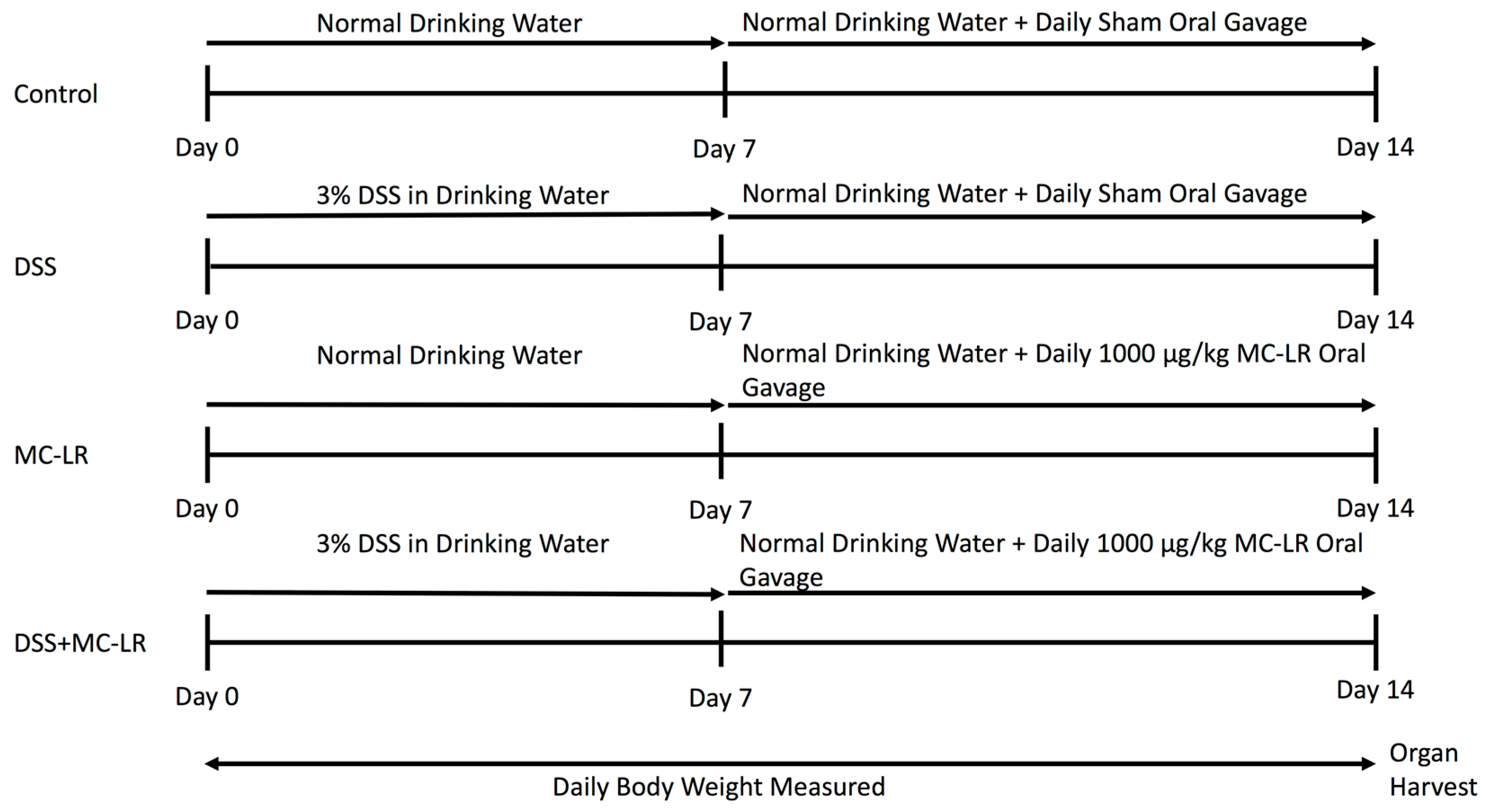
4.2. Colitis Induction and MC-LR Exposure Protocol
4.3. Histology
4.4. RNA Extraction and RT-qPCR Method
4.5. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G. The global burden of IBD: from 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720. [Google Scholar] [CrossRef] [PubMed]
- Mehta, F. Report: Economic implications of inflammatory bowel disease and its management. Am. J. Manag. Care 2016, 22, s51–s60. [Google Scholar] [PubMed]
- Park, K.T.; Ehrlich, O.G.; Allen, J.I.; Meadows, P.; Szigethy, E.M.; Henrichsen, K.; Kim, S.C.; Lawton, R.C.; Murphy, S.M.; Regueiro, M.; et al. The Cost of Inflammatory Bowel Disease: An Initiative From the Crohn’s & Colitis Foundation. Inflamm. Bowel. Dis. 2019. [Google Scholar] [CrossRef]
- Jeengar, M.K.; Thummuri, D.; Magnusson, M.; Naidu, V.G.M.; Uppugunduri, S. Uridine Ameliorates Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Sci. Rep. 2017, 7, 3924. [Google Scholar] [CrossRef] [PubMed]
- Molodecky, N.A.; Kaplan, G.G. Environmental risk factors for inflammatory bowel disease. Gastroenterol. Hepatol. 2010, 6, 339–346. [Google Scholar]
- Campos, A.; Vasconcelos, V. Molecular Mechanisms of Microcystin Toxicity in Animal Cells. Int. J. Mol. Sci. 2010, 11, 268–287. [Google Scholar] [CrossRef] [Green Version]
- Lone, Y.; Koiri, R.K.; Bhide, M. An overview of the toxic effect of potential human carcinogen Microcystin-LR on testis. Toxicol. Rep. 2015, 2, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Chorus, I. Introduction: Cyanotoxins—Research for Environmental Safety and Human Health. In Cyanotoxins: Occurrence, Causes, Consequences; Chorus, I., Ed.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2001; pp. 1–4. [Google Scholar] [CrossRef]
- Greer, B.; Meneely, J.P.; Elliott, C.T. Uptake and accumulation of Microcystin-LR based on exposure through drinking water: An animal model assessing the human health risk. Sci. Rep. 2018, 8, 4913. [Google Scholar] [CrossRef]
- Svircev, Z.; Drobac, D.; Tokodi, N.; Mijovic, B.; Codd, G.A.; Meriluoto, J. Toxicology of microcystins with reference to cases of human intoxications and epidemiological investigations of exposures to cyanobacteria and cyanotoxins. Arch. Toxicol. 2017, 91, 621–650. [Google Scholar] [CrossRef]
- Jochimsen, E.M.; Carmichael, W.W.; An, J.S.; Cardo, D.M.; Cookson, S.T.; Holmes, C.E.; Antunes, M.B.; de Melo Filho, D.A.; Lyra, T.M.; Barreto, V.S.; et al. Liver failure and death after exposure to microcystins at a hemodialysis center in Brazil. N. Engl. J. Med. 1998, 338, 873–878. [Google Scholar] [CrossRef]
- Carmichael, W.W.; Azevedo, S.M.; An, J.S.; Molica, R.J.; Jochimsen, E.M.; Lau, S.; Rinehart, K.L.; Shaw, G.R.; Eaglesham, G.K. Human fatalities from cyanobacteria: chemical and biological evidence for cyanotoxins. Environ. Health Perspect. 2001, 109, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, J.; Huet, S.; Jarry, G.; Fessard, V. In vivo DNA damage induced by the cyanotoxin microcystin-LR: comparison of intra-peritoneal and oral administrations by use of the comet assay. Mutat. Res. 2008, 652, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Sedan, D.; Laguens, M.; Copparoni, G.; Aranda, J.O.; Giannuzzi, L.; Marra, C.A.; Andrinolo, D. Hepatic and intestine alterations in mice after prolonged exposure to low oral doses of Microcystin-LR. Toxicon 2015, 104, 26–33. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Zhang, J.-Y.; Hong, Y.; Chen, Y.-X. Evaluation of organ distribution of microcystins in the freshwater phytoplanktivorous fish Hypophthalmichthys molitrix. J. Zhejiang Univ. Sci. B 2007, 8, 116–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15–25. [Google Scholar] [CrossRef]
- Melgar, S.; Karlsson, L.; Rehnström, E.; Karlsson, A.; Utkovic, H.; Jansson, L.; Michaëlsson, E. Validation of murine dextran sulfate sodium-induced colitis using four therapeutic agents for human inflammatory bowel disease. Int. Immunopharmacol. 2008, 8, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Melgar, S.; Karlsson, A.; Michaëlsson, E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: correlation between symptoms and inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1328–G1338. [Google Scholar] [CrossRef]
- Fischer, A.; Hoeger, S.J.; Stemmer, K.; Feurstein, D.J.; Knobeloch, D.; Nussler, A.; Dietrich, D.R. The role of organic anion transporting polypeptides (OATPs/SLCOs) in the toxicity of different microcystin congeners in vitro: a comparison of primary human hepatocytes and OATP-transfected HEK293 cells. Toxicol. Appl. Pharmacol. 2010, 245, 9–20. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Y.; Wang, J.; Wang, L.; Xiang, Z.; Li, D.; Han, X. Microcystin-Leucine Arginine Causes Cytotoxic Effects in Sertoli Cells Resulting in Reproductive Dysfunction in Male Mice. Sci. Rep. 2016, 6, 39238. [Google Scholar] [CrossRef]
- Adegoke, E.O.; Wang, C.; Machebe, N.S.; Wang, X.; Wang, H.; Adeniran, S.O.; Zhang, H.; Zheng, P.; Zhang, G. Microcystin-leucine arginine (MC-LR) induced inflammatory response in bovine sertoli cell via TLR4/NF-kB signaling pathway. Environ. Toxicol. Pharmacol. 2018, 63, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Adegoke, E.O.; Adeniran, S.O.; Zeng, Y.; Wang, X.; Wang, H.; Wang, C.; Zhang, H.; Zheng, P.; Zhang, G. Pharmacological inhibition of TLR4/NF-kappaB with TLR4-IN-C34 attenuated microcystin-leucine arginine toxicity in bovine Sertoli cells. J. Appl. Toxicol. 2019, 39, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Hou, J.; Guo, H.; Qiu, Y.; Li, L.; Li, D.; Tang, R. Dualistic immunomodulation of sub-chronic microcystin-LR exposure on the innate-immune defense system in male zebrafish. Chemosphere 2017, 183, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Guzman, R.E.; Solter, P.F. Hepatic oxidative stress following prolonged sublethal microcystin LR exposure. Toxicol. Pathol. 1999, 27, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, Y.; Duan, H.; Sivakumar, R.; Li, X. Chronic exposure of nanomolar MC-LR caused oxidative stress and inflammatory responses in HepG2 cells. Chemosphere 2018, 192, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Vindigni, S.M.; Zisman, T.L.; Suskind, D.L.; Damman, C.J. The intestinal microbiome, barrier function, and immune system in inflammatory bowel disease: a tripartite pathophysiological circuit with implications for new therapeutic directions. Ther. Adv. Gastroenterol. 2016, 9, 606–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borcherding, F.; Nitschke, M.; Hundorfean, G.; Rupp, J.; von Smolinski, D.; Bieber, K.; van Kooten, C.; Lehnert, H.; Fellermann, K.; Buning, J. The CD40-CD40L pathway contributes to the proinflammatory function of intestinal epithelial cells in inflammatory bowel disease. Am. J. Pathol. 2010, 176, 1816–1827. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Sans, M.; Fiocchi, C. The CD40/CD40L costimulatory pathway in inflammatory bowel disease. Gut 2004, 53, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polese, L.; Angriman, I.; Cecchetto, A.; Norberto, L.; Scarpa, M.; Ruffolo, C.; Barollo, M.; Sommariva, A.; D’Amico, D.F. The role of CD40 in ulcerative colitis: histochemical analysis and clinical correlation. Eur. J. Gastroenterol. Hepatol. 2002, 14, 237–241. [Google Scholar] [CrossRef]
- Gelbmann, C.M.; Leeb, S.N.; Vogl, D.; Maendel, M.; Herfarth, H.; Schölmerich, J.; Falk, W.; Rogler, G. Inducible CD40 expression mediates NFkappaB activation and cytokine secretion in human colonic fibroblasts. Gut 2003, 52, 1448–1456. [Google Scholar] [CrossRef]
- Miller, M.A.; Kudela, R.M.; Mekebri, A.; Crane, D.; Oates, S.C.; Tinker, M.T.; Staedler, M.; Miller, W.A.; Toy-Choutka, S.; Dominik, C.; et al. Evidence for a novel marine harmful algal bloom: cyanotoxin (microcystin) transfer from land to sea otters. PLoS ONE 2010, 5, e12576. [Google Scholar] [CrossRef] [PubMed]
- Organization, W.H. Cyanobacterial toxins: Microcystin-LR in Drinking Water. Guidel. Drink. Water Qual. 1998, 2, 83–127. [Google Scholar]
- Fawell, J.K.; Mitchell, R.E.; Everett, D.J.; Hill, R.E. The toxicity of cyanobacterial toxins in the mouse: I microcystin-LR. Hum. Exp. Toxicol. 1999, 18, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Viennois, E.; Tahsin, A.; Merlin, D. Purification of Total RNA from DSS-treated Murine Tissue via Lithium Chloride Precipitation. Bio. Protoc. 2018, 8, e2829. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Khalaf, F.K.; Sheehy, B.; Weber, M.E.; Agatisa-Boyle, B.; Conic, J.; Hauser, K.; Medert, C.M.; Westfall, K.; Bucur, P.; et al. Telocinobufagin, a Novel Cardiotonic Steroid, Promotes Renal Fibrosis via Na(+)/K(+)-ATPase Profibrotic Signaling Pathways. Int. J. Mol. Sci. 2018, 19, 2566. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, R.C.; Blomquist, T.M.; Kleinhenz, A.L.; Khalaf, F.K.; Dube, P.; Lad, A.; Breidenbach, J.D.; Mohammed, C.J.; Zhang, S.; Baum, C.E.; et al. Exposure to the Harmful Algal Bloom (HAB) Toxin Microcystin-LR (MC-LR) Prolongs and Increases Severity of Dextran Sulfate Sodium (DSS)-Induced Colitis. Toxins 2019, 11, 371. https://doi.org/10.3390/toxins11060371
Su RC, Blomquist TM, Kleinhenz AL, Khalaf FK, Dube P, Lad A, Breidenbach JD, Mohammed CJ, Zhang S, Baum CE, et al. Exposure to the Harmful Algal Bloom (HAB) Toxin Microcystin-LR (MC-LR) Prolongs and Increases Severity of Dextran Sulfate Sodium (DSS)-Induced Colitis. Toxins. 2019; 11(6):371. https://doi.org/10.3390/toxins11060371
Chicago/Turabian StyleSu, Robin C., Thomas M. Blomquist, Andrew L. Kleinhenz, Fatimah K. Khalaf, Prabhatchandra Dube, Apurva Lad, Joshua D. Breidenbach, Chrysan J. Mohammed, Shungang Zhang, Caitlin E. Baum, and et al. 2019. "Exposure to the Harmful Algal Bloom (HAB) Toxin Microcystin-LR (MC-LR) Prolongs and Increases Severity of Dextran Sulfate Sodium (DSS)-Induced Colitis" Toxins 11, no. 6: 371. https://doi.org/10.3390/toxins11060371
APA StyleSu, R. C., Blomquist, T. M., Kleinhenz, A. L., Khalaf, F. K., Dube, P., Lad, A., Breidenbach, J. D., Mohammed, C. J., Zhang, S., Baum, C. E., Malhotra, D., Kennedy, D. J., & Haller, S. T. (2019). Exposure to the Harmful Algal Bloom (HAB) Toxin Microcystin-LR (MC-LR) Prolongs and Increases Severity of Dextran Sulfate Sodium (DSS)-Induced Colitis. Toxins, 11(6), 371. https://doi.org/10.3390/toxins11060371