A Long-Read Genome Assembly of a Native Mite in China Pyemotes zhonghuajia Yu, Zhang & He (Prostigmata: Pyemotidae) Reveals Gene Expansion in Toxin-Related Gene Families

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Genome Assembly
2.2. Genome Annotation
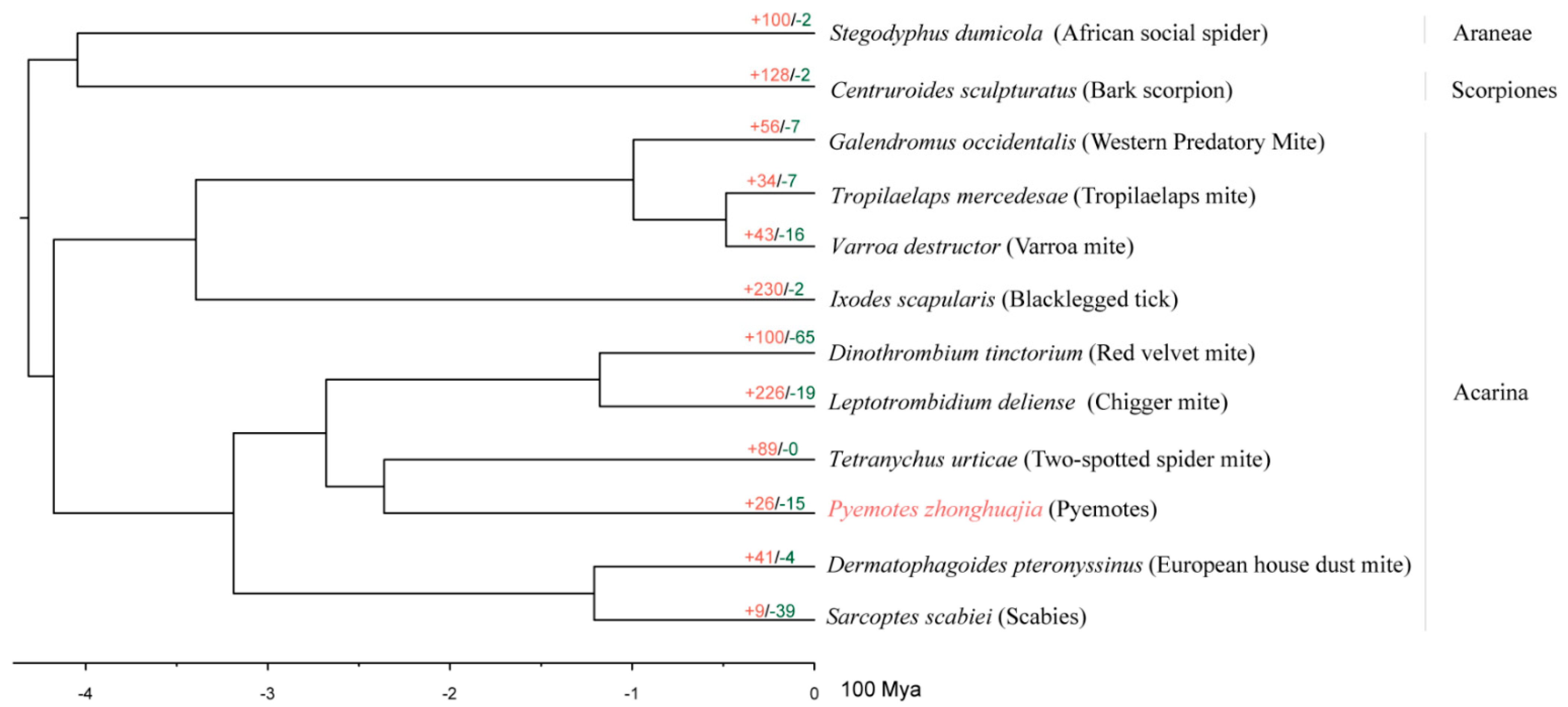
2.3. Species Phylogeny and Gene Family Evolution
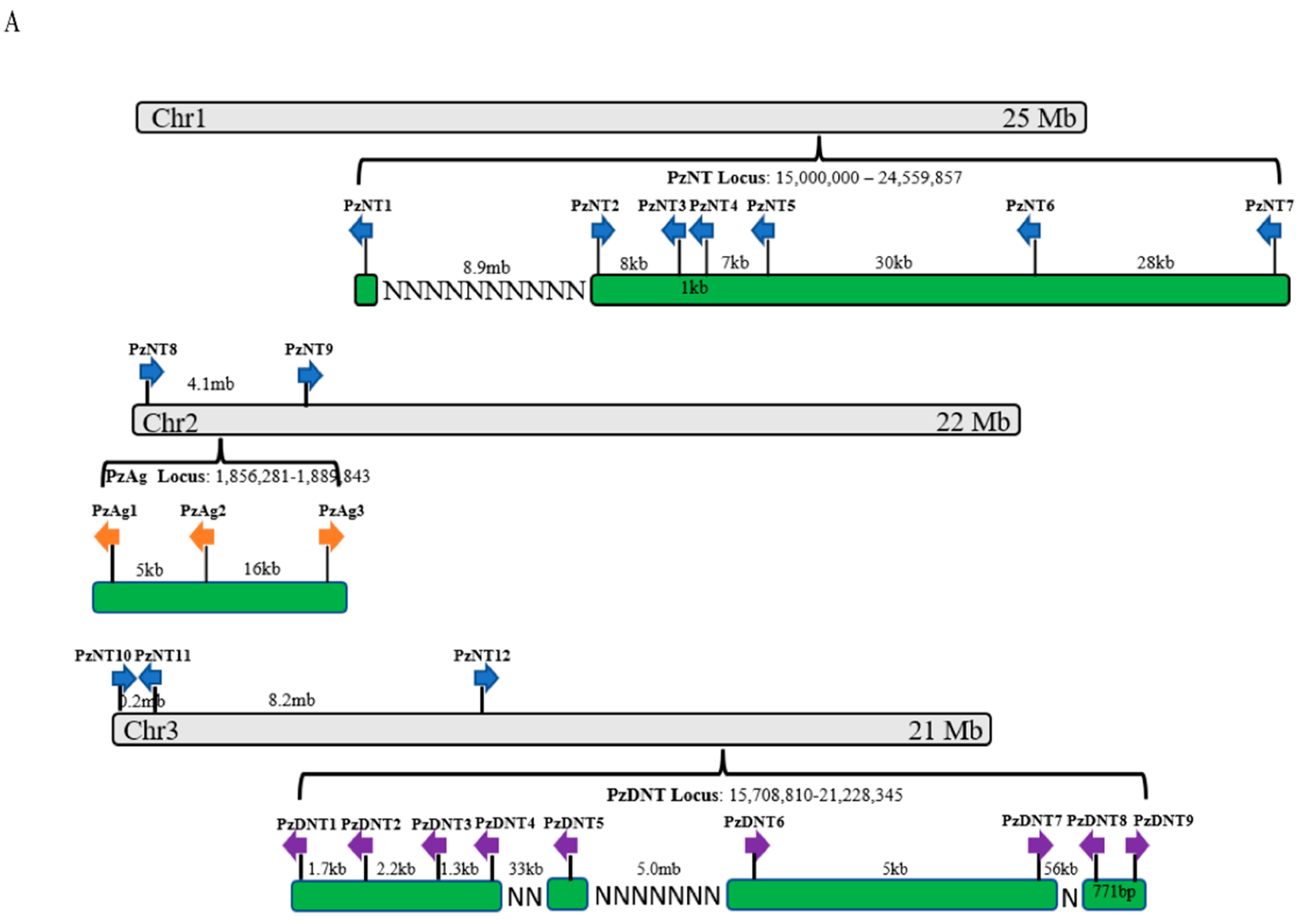
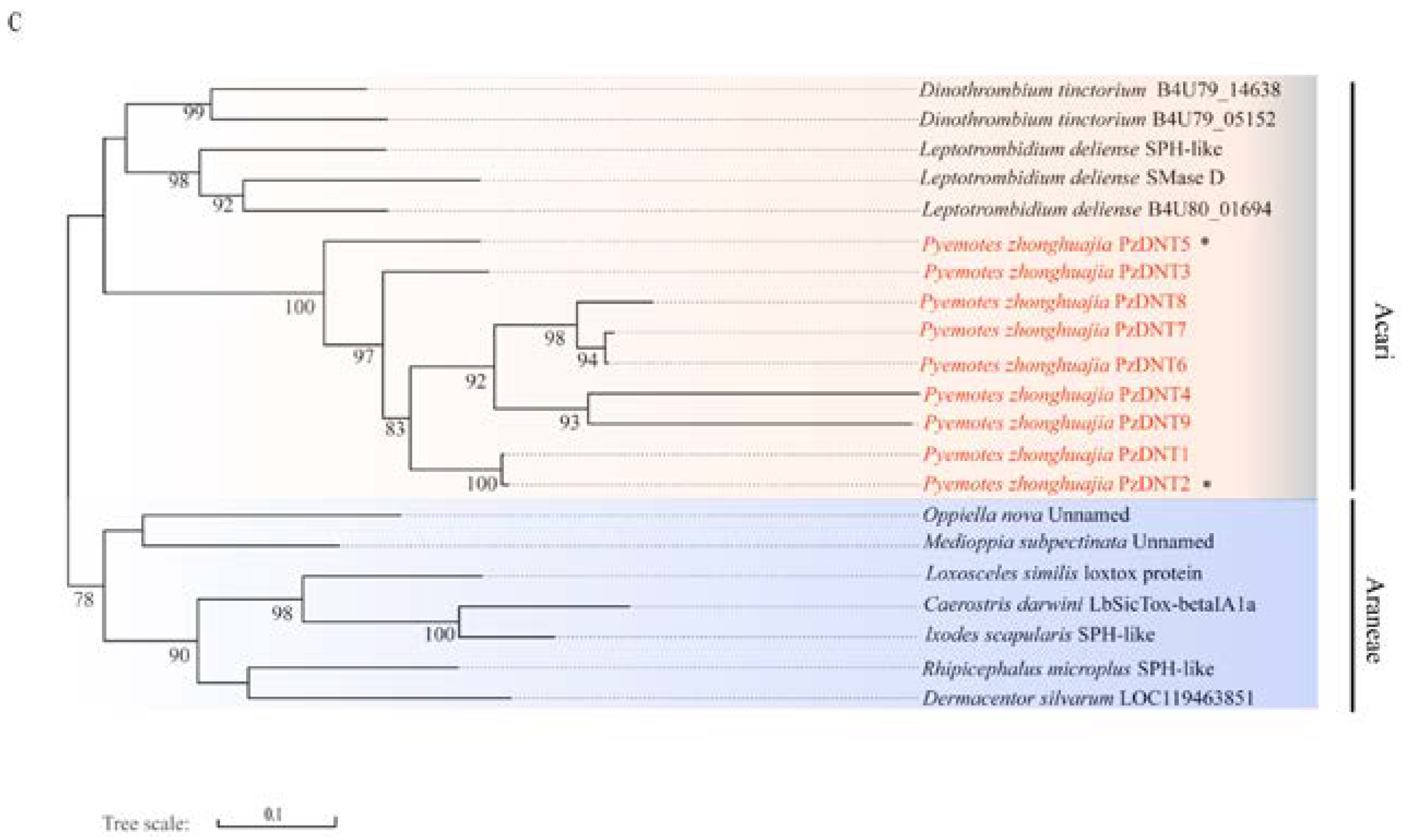
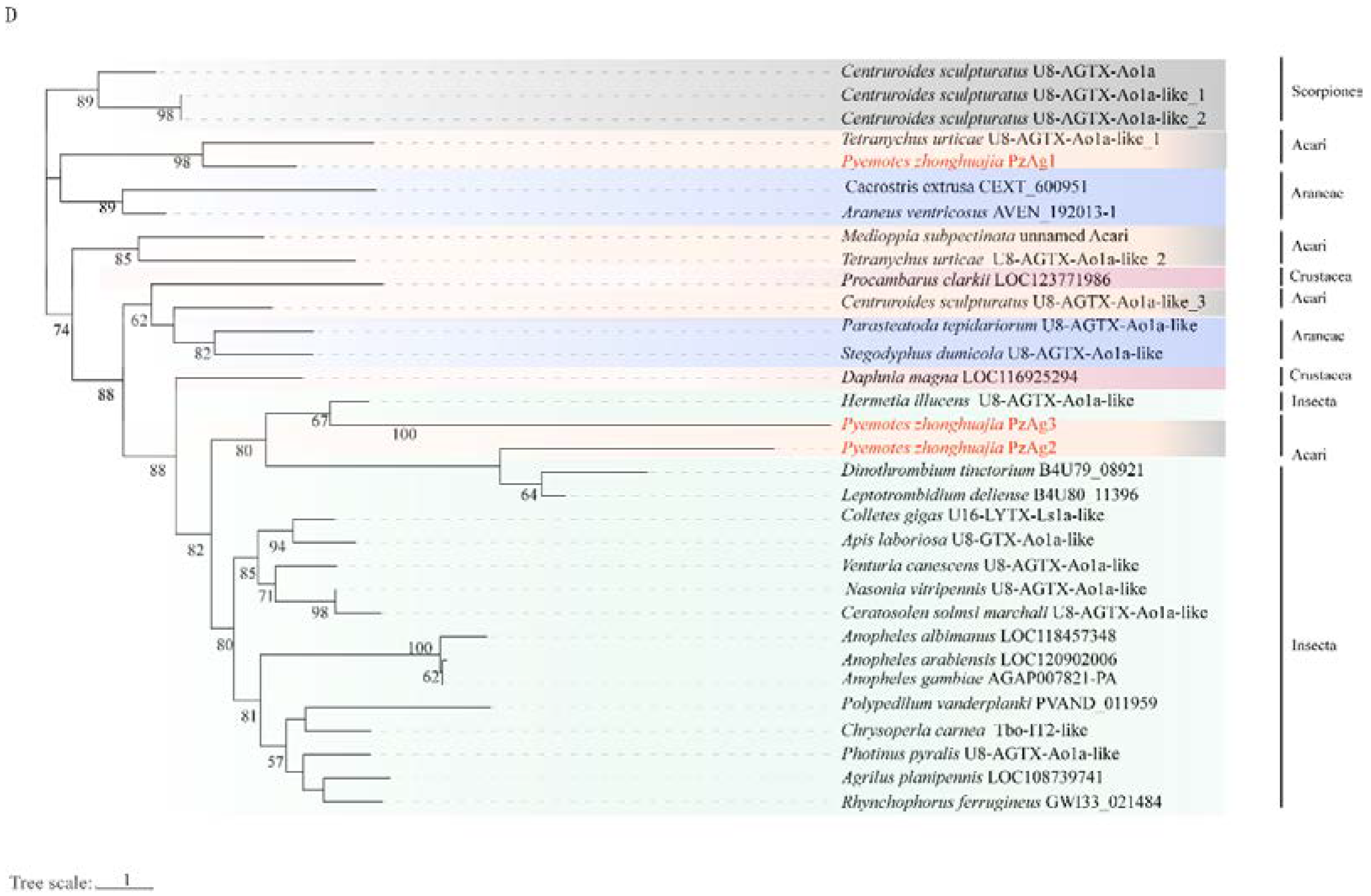
2.4. Neurotoxin, Dermonecrotic Toxin and Agatoxin Genes
3. Conclusions
4. Materials and Methods
4.1. Sample Collection and Sequencing
4.2. Genome Assembly
4.3. Genome Annotation
4.4. Gene Ontology Analysis and Species Evolution
4.5. Identification of Gene Family Expansion and Contraction
4.6. Phylogeny Construction and Gene Expression of Toxin-Related Gene Families
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, L.; Zhang, Z.Q.; He, L. Two New Species of Pyemotes Closely Related to P. Tritici (Acari: Pyemotidae). Zootaxa 2010, 2723, 1–40. [Google Scholar] [CrossRef] [Green Version]
- He, L.M.; Jiao, R.; Xu, C.X.; Hao, B.F.; Han, J.C.; Yu, L.C. Application of mtDNA COI Gene Sequence in Indentification of Pyemotes. J. Hebei Agric. Sci. 2010, 14, 46–50. [Google Scholar]
- Liu, J.F.; Tian, T.A.; Li, X.L.; Chen, Y.C.; Yu, X.F.; Tan, X.F.; Zhu, Y.; Yang, M.F. Is Pyemotes Zhonghuajia (Acari: Pyemotidae) a Suitable Biological Control Agent against the Fall Armyworm Spodoptera Frugiperda (Lepidoptera: Noctuidae)? Syst. Appl. Acarol. 2020, 25, 649–657. [Google Scholar] [CrossRef]
- Li, L.; He, L.; Yu, L.; He, X.Z.; Xu, C.; Jiao, R.; Zhang, L.; Liu, J. Preliminary Study on the Potential of Pyemotes Zhonghuajia (Acari: Pyemotidae) in Biological Control of Aphis Citricola (Hemiptera: Aphididae). Syst. Appl. Acarol. 2019, 24, 1116–1120. [Google Scholar] [CrossRef]
- De Sousa, A.H.; Mendonça, G.R.Q.; Lopes, L.M.; Faroni, L.R.D. Widespread Infestation of Pyemotes Tritici (Acari: Pyemotidae) in Colonies of Seven Species of Stored-Product Insects. Genet. Mol. Res. 2020, 19, 1–5. [Google Scholar] [CrossRef]
- Tian, T.A.; Yu, L.; Sun, G.J.; Yu, X.F.; Li, L.; Wu, C.X.; Chen, Y.C.; Yang, M.F.; Liu, J.F. Biological Control Efficiency of an Ectoparasitic Mite Pyemotes Zhonghuajia on Oriental Armyworm Mythimna Separata. Syst. Appl. Acarol. 2020, 25, 1683–1692. [Google Scholar]
- Chen, Y.C.; Tian, T.A.; Chen, Y.H.; Yu, L.C.; Hu, J.F.; Yu, X.F.; Liu, J.F.; Yang, M.F. The Biocontrol Agent Pyemotes zhonghuajia Has the Highest Lethal Weight Ratio Compared with Its Prey and the Most Dramatic Body Weight Change during Pregnancy. Insects 2021, 12, 490. [Google Scholar] [CrossRef]
- Tomalski, M.D.; Bruce, W.A.; Travis, J.; Blum, M.S. Preliminary Characterization of Toxins from the Straw Itch Mite, Pyemotes Tritici, Which Induce Paralysis in the Larvae of a Moth. Toxicon 1988, 26, 127–132. [Google Scholar] [CrossRef]
- Han, J.C.; He, L.M.; Jiao, R.; Hao, B.F.; Xu, Z.X.; Yu, L.C. Analysis of the toxin gene analogs cloned from Pyemotes phloeosinus sp. nov. J. Hebei Agric. Sci. 2008, 12, 72–74. [Google Scholar]
- Lu, H.L.; Li, L.T.; Yu, L.C.; He, L.M.; Ouyang, G.C.; Liang, G.W.; Lu, Y.Y. Ectoparasitic mite, Pyemotes zhonghuajia (Prostigmata: Pyemotidae), for biological control of Asian citrus psyllid, Diaphorina citri (Hemiptera: Liviidae). Syst. Appl. Acarol. 2019, 24, 520–524. [Google Scholar] [CrossRef]
- Guo, X.; Xu, Z.; Xiong, D.P. Study of utilizing Pyemotes zhonghuajia to control Semanotus bifasciatus beetles. Chin. Bull. Entomol. 2010, 47, 529–532. [Google Scholar]
- Li, Y.C.; Huang, C.Y.; Xia, Z.R.; Zhou, J.S.; Li, J.S.; Sun, Y.Z.; Xu, Y.; Wu, S.Q.; Zhang, F.P. Preliminary Study on Biocontrol Potential of Pyemotes Zhonghuajia on Monochamus Alternatus Hope. Genom. Appl. Biol. 2019, 38, 2516–2521. [Google Scholar]
- Zhang, Z.S.; Xiong, D.P.; Cheng, W. Control of Stem Borers by a Parasitoid, Pyemotes trittci Lagreze-Fossot & Montane. Chin. J. Biol. Control. 2008, 01, 1–6. [Google Scholar]
- Deng, S.Q.; Chen, J.T.; Li, W.W.; Chen, M.; Peng, H.J. Application of the scorpion neurotoxin AaIT against insect pests. Int. J. Mol. Sci. 2019, 20, 3467. [Google Scholar] [CrossRef] [Green Version]
- Harvey-Samuel, T.; Xu, X.; Lovett, E.; Dafa’alla, T.; Walker, A.; Norman, V.C.; Carter, R.; Teal, J.; Akilan, L.; Leftwich, P.T.; et al. Engineered expression of the invertebrate-specific scorpion toxin AaHIT reduces adult longevity and female fecundity in the diamondback moth Plutella xylostella. Pest Manag. Sci. 2021, 77, 3154–3164. [Google Scholar] [CrossRef]
- Li, H.; Xia, Y. Improving the secretory expression of active recombinant AaIT in Pichia pastoris by changing the expression strain and plasmid. World J. Microbiol. Biotechnol. 2018, 34, 104. [Google Scholar] [CrossRef]
- Tianpei, X.; Li, D.; Qiu, P.; Luo, J.; Zhu, Y.; Li, S. Scorpion peptide LqhIT2 activates phenylpropanoid pathways via jasmonate to increase rice resistance to rice leafrollers. Plant Sci. 2015, 230, 1–11. [Google Scholar] [CrossRef]
- Zlotkin, E.; Eitan, M.; Bindokas, V.P.; Adams, M.E.; Moyer, M.; Burkhart, W.; Fowler, E. Functional duality and structural uniqueness of the depressant insect-selective neurotoxins. Biochemistry 1991, 30, 4814–4821. [Google Scholar] [CrossRef]
- Kroemer, J.A.; Bonning, B.C.; Harrison, R.L. Expression, delivery and function of insecticidal proteins expressed by recombinant baculoviruses. Viruses 2015, 7, 422–455. [Google Scholar] [CrossRef] [Green Version]
- Windley, M.J.; Herzig, V.; Dziemborowicz, S.A.; Hardy, M.C.; King, G.F.; Nicholson, G.M. Spider-venom peptides as bioinsecticides. Toxins 2012, 4, 191–227. [Google Scholar] [CrossRef] [Green Version]
- Chaves-Moreira, D.; Senff-Ribeiro, A.; Wille, A.C.M.; Gremski, L.H.; Chaim, O.M.; Veiga, S.S. Highlights in the knowledge of brown spider toxins. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.E. Agatoxins: Ion channel specific toxins from the American funnel web spider, Agelenopsis aperta. Toxicon 2004, 43, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Nentwig, W.; Kuhn-Nentwig, L. Main components of spider venoms. In Spider Ecophysiology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 191–202. [Google Scholar]
- Kuhn-Nentwig, L.; Stöcklin, R.; Nentwig, W. Venom composition and strategies in spiders: Is everything possible? Adv. Insect Phys. 2011, 40, 1–86. [Google Scholar]
- Tomalski, M.D.; Hutchinson, K.; Todd, J.; Miller, L.K. Identification and Characterization of Tox21A: A Mite CDNA Encoding a Paralytic Neurotoxin Related to TxP-I. Toxicon 1993, 31, 319–326. [Google Scholar] [CrossRef]
- Burden, J.P.; Hails, R.S.; Windass, J.D.; Suner, M.M.; Cory, J.S. Infectivity, Speed of Kill, and Productivity of a Baculovirus Expressing the Itch Mite Toxin Txp-1 in Second and Fourth Instar Larvae of Trichoplusia Ni. J. Invertebr. Pathol. 2000, 75, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, N.; Liu, C.; Wu, X.; Liu, K.; Yin, Z.; Zhou, X.; Xie, L. A Highly Contiguous Genome Assembly of a Polyphagous Predatory Mite Stratiolaelaps scimitus (Womersley) (Acari: Laelapidae). Genome Biol. Evol. 2021, 13, evab011. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, K.; Dawson, R.J.P.; Locher, K.P. Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 2007, 17, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Acosta, F.; Planas, L.; Penneys, N. Demodex mites contain immunoreactive lipase. Arch. Dermatol. 1989, 125, 1436–1437. [Google Scholar] [CrossRef]
- Ando, T.; Homma, R.; Ino, Y.; Ito, G.; Miyahara, A.; Yanagihara, T.; Kimura, H.; Ikeda, S.; Yamakawa, H.; Iwaki, M.; et al. Trypsin-like protease of mites: Purification and characterization of trypsin-like protease from mite faecal extract Dermatophagoides farinae. Relationship between trypsin-like protease and Der f III. Clin. Exp. Allergy 1993, 23, 777–784. [Google Scholar] [CrossRef]
- Chaim, O.M.; Sade, Y.B.; Da Silveira, R.B.; Toma, L.; Kalapothakis, E.; Chávez-Olórtegui, C.; Mangili, O.C.; Gremski, W.; Von Dietrich, C.P.; Nader, H.B.; et al. Brown Spider Dermonecrotic Toxin Directly Induces Nephrotoxicity. Toxicol. Appl. Pharmacol. 2006, 211, 64–77. [Google Scholar] [CrossRef]
- Krantz, G.W. Dolichocybe Keiferi, A New Genus and New Species of Pyemotid Mite, with a Description of a New Species of Siteroptes (Acarina: Pyemotidae). Ann. Entomol. Soc. Am. 1957, 50, 259–264. [Google Scholar] [CrossRef]
- Shen, Z.; Jacobs-Lorena, M. Evolution of Chitin-Binding Proteins in Invertebrates. J. Mol. Evol. 1999, 48, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Peigneur, S.; Tytgat, J. Toxins in Drug Discovery and Pharmacology. Toxins 2018, 10, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn-Nentwig, L.; Langenegger, N.; Heller, M.; Koua, D.; Nentwig, W. The Dual Prey-Inactivation Strategy of Spiders-In-Depth Venomic Analysis of Cupiennius salei. Toxins 2019, 11, 167. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B. BBtools. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 4 April 2020).
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [Green Version]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long errorprone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Guan, D.; McCarthy, S.A.; Wood, J.; Howe, K.; Wang, Y.; Durbin, R. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 2020, 36, 2896–2898. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A fast and efficient genome polishing tool for long read assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST +: Architecture and applications. BMC Bioinform. 2009, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, R.M.; Seppey, M.; Simao, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for Automated Genomic Discovery of Transposable Element Families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Hubley, R.; Finn, R.D.; Clements, J.; Eddy, S.R.; Jones, T.A.; Bao, W.; Smit, A.F.A.; Wheeler, T.J. The Dfam Database of Repetitive DNA Families. Nucleic Acids Res. 2016, 44, D81–D89. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.D.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Repeat Masker Open-4.0. 2013–2015. Available online: http://www.repeatmasker.org (accessed on 8 January 2020).
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome- database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [Green Version]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Tanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. [Google Scholar]
- Lomsadze, A.; Ter-Hovhannisyan, V.; Chernoff, Y.O.; Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005, 33, 6494–6506. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Kriventseva, E.V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; Simão, F.A.; Zdobnov, E.M. OrthoDB V10: Sampling the Diversity of Animal, Plant, Fungal, Protist, Bacterial and Viral Genomes for Evolutionary and Functional Annotations of Orthologs. Nucleic Acids Res. 2019, 47, D807–D811. [Google Scholar] [CrossRef] [Green Version]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome Assembly from Long-Read RNA-Seq Alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods. 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.Y.; Dosztányi, Z.; El-Gebali, S.; et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam Protein Families Database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Letunic, L.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.E.; Sillitoe, I.; Dawson, N.; Lam, S.D.; Clarke, T.; Lee, D.; Orengo, C.; Lees, J. Gene3D: Extensive Prediction of Globular Domains in Proteins. Nucleic Acids Res. 2018, 46, D435–D439. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.; Pethica, R.; Zhou, Y.; Talbot, C.; Vogel, C.; Madera, M.; Chothia, C.; Gough, J. SUPERFAMILY—Sophisticated Comparative Genomics, Data Mining, Visualization and Phylogeny. Nucleic Acids Res. 2009, 37, 380–386. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional Classification of Proteins via Subfamily Domain Architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. EggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-Fold Faster RNA Homology Searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2019; Volume 1962, pp. 1–14. [Google Scholar]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol. 2018, 20, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): A new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010, 10, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kück, P.; Longo, G.C. FASconCAT-G: Extensive Functions for Multiple Sequence Alignment Preparations Concerning Phylogenetic Studies. Front. Zool. 2014, 11, 81. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Michael, D.; Haeseler, A.; Von Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Han, M.V.; Thomas, G.W.C.; Lugo-Martinez, J.; Hahn, M.W. Estimating Gene Gain and Loss Rates in the Presence of Error in Genome Assembly and Annotation Using CAFE 3. Mol. Biol. Evol. 2013, 30, 1987–1997. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Vizueta, J.; Sánchez-Gracia, A.; Rozas, J. BITACORA: A comprehensive tool for the identification and annotation of gene families in genome assemblies. Mol. Ecol. Resour. 2020, 20, 1445–1452. [Google Scholar] [CrossRef]
- Potter, S.C.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D.; Hmmer, T. HMMER Web Server: 2018 Update. Nucleic Acids Res. 2018, 46, 200–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Gaujoux, R.; Seoighe, C. A Flexible R Package for Nonnegative Matrix Factorization. BMC Bioinform. 2010, 11, 367. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Elements | Pyemotes zhonghuajia | Stratiolaelaps scimitus | Tetranychus urticae |
|---|---|---|---|
| Genome assembly | |||
| Assembly size (Mb) | 71.943 | 426.50 | 89.6 |
| Number of scaffolds/contigs | 19/20 | 158 | -/2035 |
| Longest scaffold/contig (Mb) | 25.136/22.128 | 31.29 | 7/0.929 |
| N50 scaffold/contig length (Mb) | 22.128/21.248 | 7.66 | 10/120 |
| GC (%) | 25.02 | 45.85 | - |
| Gaps (%) | 0.00 | 0.00 | - |
| BUSCO completeness (%) | 90.6 | 93.1 | - |
| Annotation | |||
| Protein-coding genes | 11,183 | 13,305 | 18,414 |
| Mean protein length (aa) | 480.97 | 500.59 | - |
| Mean gene length (bp) | 3243.42 | 7870.13 | 2652 |
| Exons/introns per gene | 3.59/2.45 | 6.24/- | 3.82/- |
| Exon (%) | 26.77 | 7.25 | - |
| Mean exon length | 475.53 | 372.35 | 178 |
| Intron (%) | 24.07 | 5.02 | - |
| Mean intron length | 626.12 | 1105.66 | 400 |
| BUSCO completeness (%) | 91.2 | 95.8 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.-F.; Yu, L.-C.; Yang, M.-F.; Ye, S.; Yan, B.; Li, L.-T.; Wu, C.; Liu, J.-F. A Long-Read Genome Assembly of a Native Mite in China Pyemotes zhonghuajia Yu, Zhang & He (Prostigmata: Pyemotidae) Reveals Gene Expansion in Toxin-Related Gene Families. Toxins 2022, 14, 571. https://doi.org/10.3390/toxins14080571
Song Y-F, Yu L-C, Yang M-F, Ye S, Yan B, Li L-T, Wu C, Liu J-F. A Long-Read Genome Assembly of a Native Mite in China Pyemotes zhonghuajia Yu, Zhang & He (Prostigmata: Pyemotidae) Reveals Gene Expansion in Toxin-Related Gene Families. Toxins. 2022; 14(8):571. https://doi.org/10.3390/toxins14080571
Chicago/Turabian StyleSong, Yan-Fei, Li-Chen Yu, Mao-Fa Yang, Shuai Ye, Bin Yan, Li-Tao Li, Chen Wu, and Jian-Feng Liu. 2022. "A Long-Read Genome Assembly of a Native Mite in China Pyemotes zhonghuajia Yu, Zhang & He (Prostigmata: Pyemotidae) Reveals Gene Expansion in Toxin-Related Gene Families" Toxins 14, no. 8: 571. https://doi.org/10.3390/toxins14080571