Isolation and Optimization of Aflatoxin B1 Degradation by Uniform Design and Complete Genome Sequencing of Novel Deep-Sea Kocuria rosea Strain 13

 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Screening and Identification of Degrading Strain 13
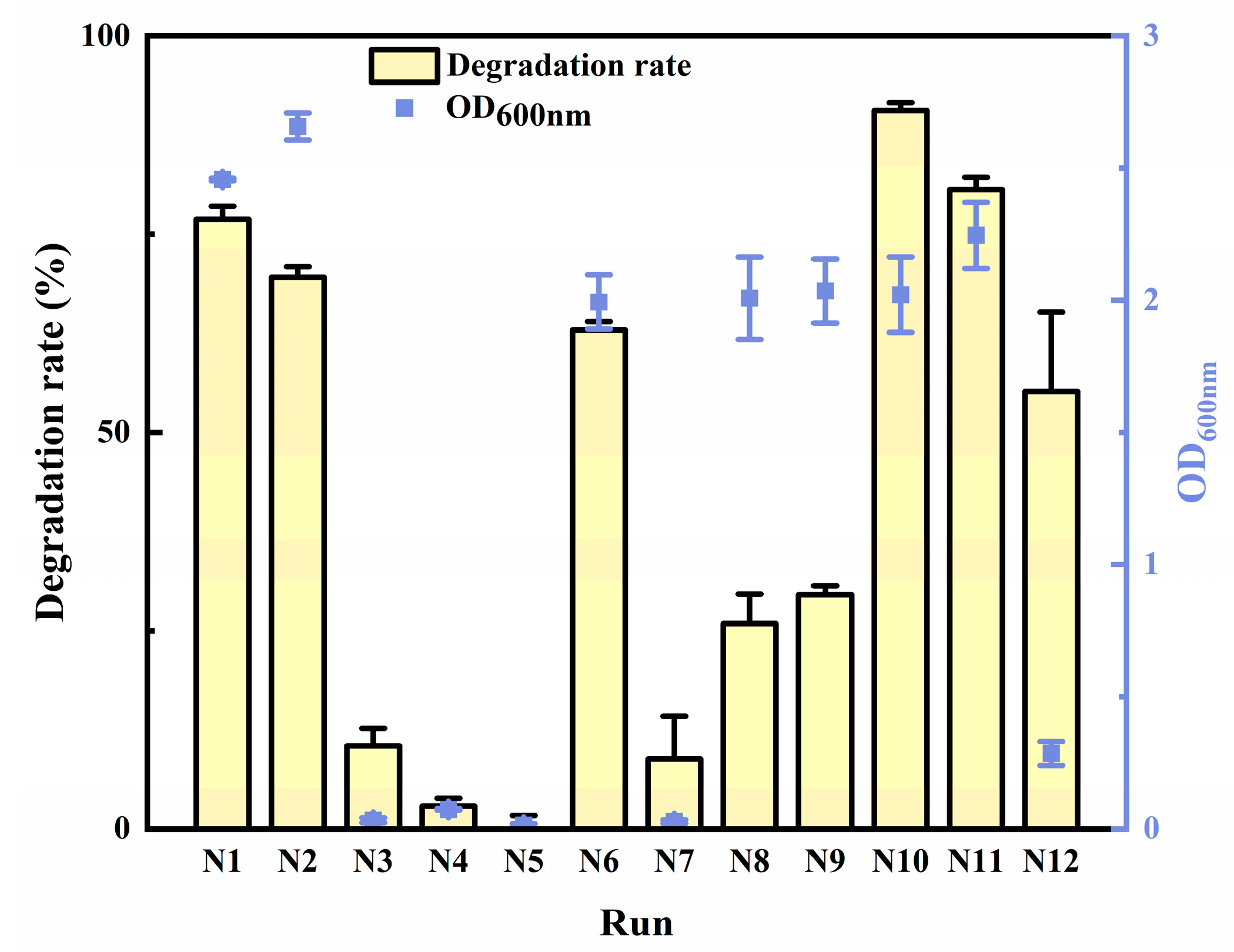
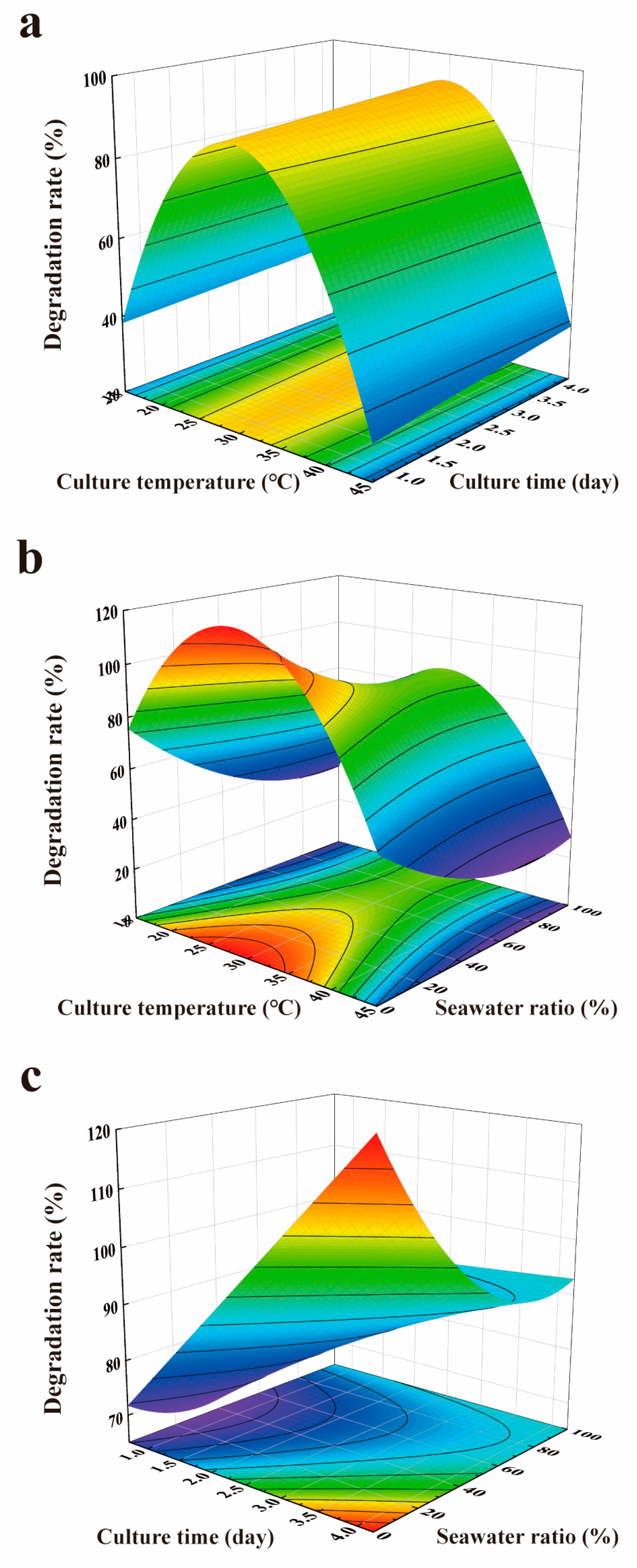
2.2. Optimization for AFB1 Degradation
0.0024620583619 × x1 × x1 + 0.00004951779435 × x3 × x3 + 0.009992701258 × x5 × x5 + 0.00023994501902 × x1
× x2 + 0.00005070750598 × x1 × x3 − 0.0014264859734 × x2 × x3
2.3. General Genomic Features of Strain 13
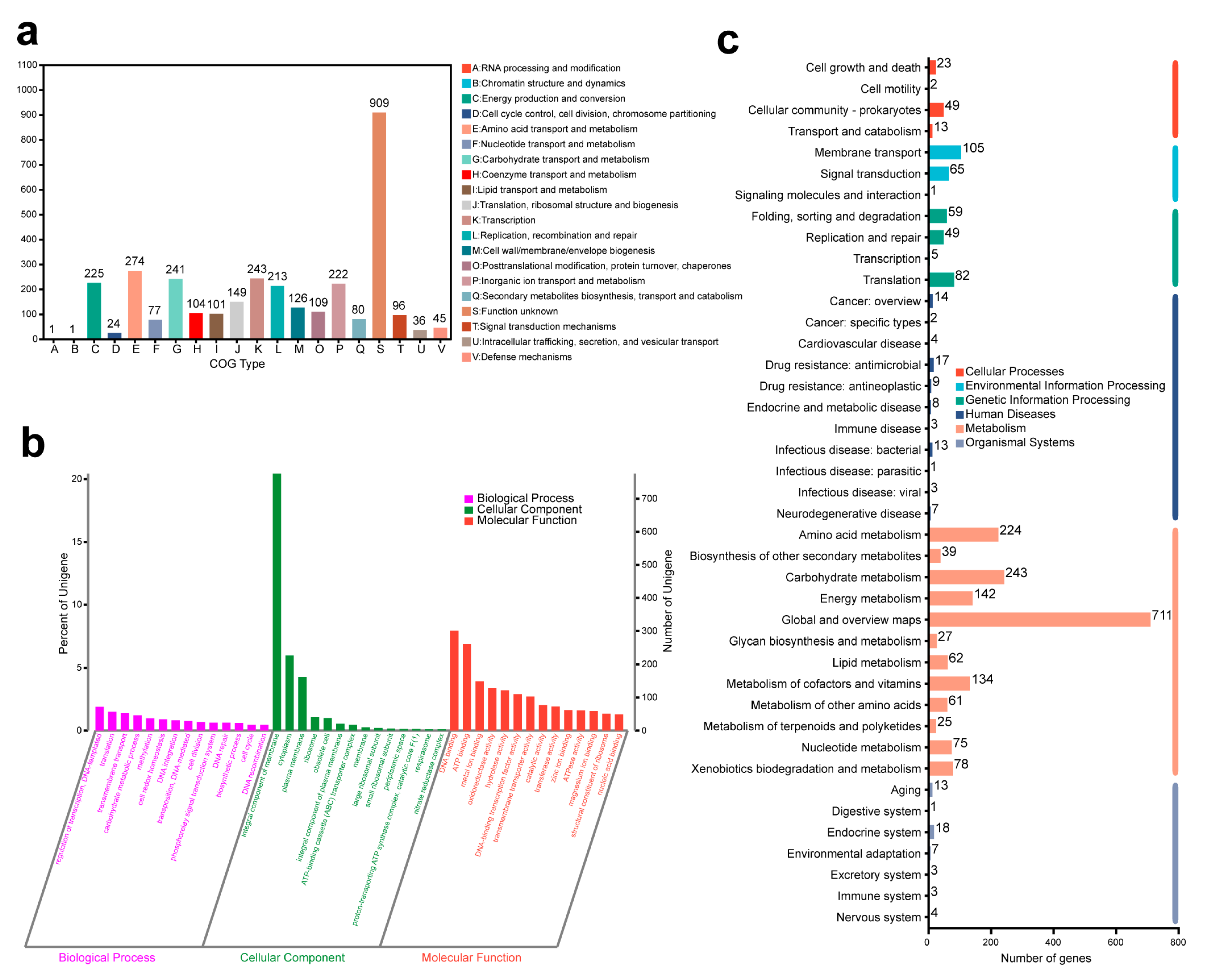
2.4. Gene Function Analysis
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Culture Media
4.2. Isolation of the Strain 13
4.3. Determination of Aflatoxin Degradation Rate
4.4. UD for Aflatoxin Degradation Optimization
4.5. DNA Extraction, Amplification, and 16S rRNA Gene Sequencing
4.6. Complete Genome Sequencing of Kocuria rosea
4.6.1. DNA Extraction
4.6.2. Genomic Library Construction and Sequencing
4.6.3. Genome Assembly and Plasmid Identification
4.6.4. Structural Genomics Analysis
4.6.5. Genome Function Annotation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kurtzman, C.P.; Horn, B.W.; Hesseltine, C.W. Aspergillus nomius, a new aflatoxin-producing species related to Aspergillus flavus and Aspergillus tamarii. Antonie Leeuwenhoek 1987, 53, 147–158. [Google Scholar] [CrossRef]
- IARC. Some Traditional Herbal Medicines, Some Mycotoxins, Naphthalene and Styrene; IARC Press; World Health Organization: Lyon, France, 2002; Volume 82. [Google Scholar]
- Ishikawa, A.T.; Hirooka, E.Y.; e Silva, P.L.A.; Bracarense, A.P.F.R.L.; Flaiban, K.K.M.d.C.; Akagi, C.Y.; Kawamura, O.; da Costa, M.C.; Itano, E.N. Impact of a Single Oral Acute Dose of Aflatoxin B-1 on Liver Function/Cytokines and the Lymphoproliferative Response in C57Bl/6 Mice. Toxins 2017, 9, 374. [Google Scholar] [CrossRef]
- Fletcher, M.T.; Netzel, G. Food Safety and Natural Toxins. Toxins 2020, 12, 236. [Google Scholar] [CrossRef]
- Wild, C.P.; Gong, Y.Y. Mycotoxins and human disease: A largely ignored global health issue. Carcinogenesis 2010, 31, 71–82. [Google Scholar] [CrossRef]
- McMillan, A.; Renaud, J.B.; Burgess, K.M.N.; Orimadegun, A.E.; Akinyinka, O.O.; Allen, S.J.; Miller, J.D.; Reid, G.; Sumarah, M.W. Aflatoxin exposure in Nigerian children with severe acute malnutrition. Food Chem. Toxicol. 2018, 111, 356–362. [Google Scholar] [CrossRef]
- Pukkasorn, P.; Ratphitagsanti, W.; Haruthaitanasan, V. Effect of ultra-superheated steam on aflatoxin reduction and roasted peanut properties. J. Sci. Food Agric. 2018, 98, 2935–2941. [Google Scholar] [CrossRef]
- Pasha, T.N.; Farooq, M.U.; Khattak, F.M.; Jabbar, M.A.; Khan, A.D. Effectiveness of sodium bentonite and two commercial products as aflatoxin absorbents in diets for broiler chickens. Anim. Feed Sci. Technol. 2007, 132, 103–110. [Google Scholar] [CrossRef]
- Alam, S.S.; Deng, Y. Protein interference on aflatoxin B1 adsorption by smectites in corn fermentation solution. Appl. Clay Sci. 2017, 144, 36–44. [Google Scholar] [CrossRef]
- Liu, R.; Jin, Q.; Huang, J.; Liu, Y.; Wang, X.; Mao, W.; Wang, S. Photodegradation of Aflatoxin B-1 in peanut oil. Eur. Food Res. Technol. 2011, 232, 843–849. [Google Scholar] [CrossRef]
- Jalili, M.; Jinap, S.; Noranizan, M.A. Aflatoxins and ochratoxin a reduction in black and white pepper by gamma radiation. Radiat. Phys. Chem. 2012, 81, 1786–1788. [Google Scholar] [CrossRef]
- Wang, B.; Mahoney, N.E.; Pan, Z.; Khir, R.; Wu, B.; Ma, H.; Zhao, L. Effectiveness of pulsed light treatment for degradation and detoxification of aflatoxin B1 and B2 in rough rice and rice bran. Food Control 2016, 59, 461–467. [Google Scholar] [CrossRef]
- Aiko, V.; Edamana, P.; Mehta, A. Decomposition and detoxification of aflatoxin B1 by lactic acid. J. Sci. Food Agric. 2016, 96, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Kamber, U.; Gülbaz, G.; Aksu, P.; Doğan, A. Detoxification of Aflatoxin B-1 in Red Pepper (Capsicum annuum L.) by Ozone Treatment and Its Effect on Microbiological and Sensory Quality. J. Food Process. Preserv. 2017, 41, e13102. [Google Scholar] [CrossRef]
- Ji, N.; Diao, E.; Li, X.; Zhang, Z.; Dong, H. Detoxification and safety evaluation of aflatoxin B1 in peanut oil using alkali refining. J. Sci. Food Agric. 2016, 96, 4009–4014. [Google Scholar] [CrossRef]
- Das, C.; Mishra, H.N. In vitro degradation of aflatoxin B1 in groundnut (Arachis hypogea) meal by horse radish peroxidase. LWT-Food Sci. Technol. 2000, 33, 308–312. [Google Scholar] [CrossRef]
- Velazhahan, R.; Vijayanandraj, S.; Vijayasamundeeswari, A.; Paranidharan, V.; Samiyappan, R.; Iwamoto, T.; Friebe, B.; Muthukrishnan, S. Detoxification of aflatoxins by seed extracts of the medicinal plant, Trachyspermum ammi (L.) Sprague ex Turrill—Structural analysis and biological toxicity of degradation product of aflatoxin G1. Food Control 2010, 21, 719–725. [Google Scholar] [CrossRef]
- Vijayanandraj, S.; Brinda, R.; Kannan, K.; Adhithya, R.; Vinothini, S.; Senthil, K.; Chinta, R.R.; Paranidharan, V.; Velazhahan, R. Detoxification of aflatoxin B1 by an aqueous extract from leaves of Adhatoda vasica Nees. Microbiol. Res. 2014, 169, 294–300. [Google Scholar] [CrossRef]
- Peltonen, K.; El-Nezami, H.; Haskard, C.; Ahokas, J.; Salminen, S. Aflatoxin B1 binding by dairy strains of lactic acid bacteria and bifidobacteria. J. Dairy Sci. 2001, 84, 2152–2156. [Google Scholar] [CrossRef]
- Shetty, P.H.; Hald, B.; Jespersen, L. Surface binding of aflatoxin B1 by Saccharomyces cerevisiae strains with potential decontaminating abilities in indigenous fermented foods. Int. J. Food Microbiol. 2007, 113, 41–46. [Google Scholar] [CrossRef]
- Wu, Q.; Jezkova, A.; Yuan, Z.; Pavlikova, L.; Dohnal, V.; Kuca, K. Biological degradation of aflatoxins. Drug Metab. Rev. 2009, 41, 1–7. [Google Scholar] [CrossRef]
- Gu, X.; Sun, J.; Cui, Y.; Wang, X.; Sang, Y. Biological degradation of aflatoxin M-1 by Bacillus pumilus E-1-1-1. MicrobiologyOpen 2019, 8, e00663. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Qu, Y.; Wu, D.; Huang, S.; Che, Y.; Yu, J.; Song, P. Biodegradation of Aflatoxin B1 in the Baijiu Brewing Process by Bacillus cereus. Toxins 2023, 15, 65. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, J.; Liu, Z.; Shi, Y.; Liu, J.; Xu, X.; Hao, S.; Mu, P.; Deng, F.; Deng, Y. Aflatoxin B1 Degradation and Detoxification by Escherichia coli CG1061 Isolated from Chicken Cecum. Front. Pharmacol. 2019, 9, 1548. [Google Scholar] [CrossRef]
- Smiley, R.D.; Draughon, F.A. Preliminary evidence that degradation of aflatoxin B1 by Flavobacterium aurantiacum is enzymatic. J. Food Prot. 2000, 63, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.H.; Guan, S.; Gao, X.; Ma, Q.G.; Lei, Y.P.; Bai, X.M.; Ji, C. Preparation, purification and characteristics of an aflatoxin degradation enzyme from Myxococcus fulvus ANSM068. J. Appl. Microbiol. 2011, 110, 147–155. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, C.; Zhang, D.; Zhao, M.; Zheng, D.; Lyu, Y.; Cheng, W.; Guo, P.; Cui, Z. Effective degradation of aflatoxin B1 using a novel thermophilic microbial consortium TADC7. Bioresour. Technol. 2017, 224, 166–173. [Google Scholar] [CrossRef]
- Taylor, M.C.; Jackson, C.J.; Tattersall, D.B.; French, N.; Peat, T.S.; Newman, J.; Briggs, L.J.; Lapalikar, G.V.; Campbell, P.M.; Scott, C.; et al. Identification and characterization of two families of F420H2-dependent reductases from Mycobacteria that catalyse aflatoxin degradation. Mol. Microbiol. 2010, 78, 561–575. [Google Scholar] [CrossRef]
- Farzaneh, M.; Shi, Z.-Q.; Ghassempour, A.; Sedaghat, N.; Ahmadzadeh, M.; Mirabolfathy, M.; Javan-Nikkhah, M. Aflatoxin B1 degradation by Bacillus subtilis UTBSP1 isolated from pistachio nuts of Iran. Food Control 2012, 23, 100–106. [Google Scholar] [CrossRef]
- Risa, A.; Divinyi, D.M.; Baka, E.; Krifaton, C. Aflatoxin B1 detoxification by cell-free extracts of Rhodococcus strains. Acta Microbiol. Immunol. Hung. 2017, 64, 423–438. [Google Scholar] [CrossRef]
- Branà, M.T.; Sergio, L.; Haidukowski, M.; Logrieco, A.F.; Altomare, C. Degradation of Aflatoxin B1 by a Sustainable Enzymatic Extract from Spent Mushroom Substrate of Pleurotus eryngii. Toxins 2020, 12, 49. [Google Scholar] [CrossRef]
- Alberts, J.F.; Gelderblom, W.C.A.; Botha, A.; van Zyl, W.H. Degradation of aflatoxin B-1 by fungal laccase enzymes. Int. J. Food Microbiol. 2009, 135, 47–52. [Google Scholar] [CrossRef]
- Yehia, R.S. Aflatoxin detoxification by manganese peroxidase purified from Pleurotus ostreatus. Braz. J. Microbiol. 2014, 45, 127–133. [Google Scholar] [CrossRef]
- Wang, J.; Ogata, M.; Hirai, H.; Kawagishi, H. Detoxification of aflatoxin B-1 by manganese peroxidase from the white-rot fungus Phanerochaete sordida YK-624. FEMS Microbiol. Lett. 2011, 314, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Cai, J.; Du, X.; Wang, C.; Lin, J.; Dai, J. Biotransformation and detoxification of aflatoxin B1 by extracellular extract of Cladosporium uredinicola. Food Sci. Biotechnol. 2016, 25, 1789–1794. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Du, M.; Chen, J.; Liu, T.; Zheng, Y.; Liao, Z.; Zhong, Q.; Wang, L.; Fang, X.; Wang, J. Degradation and Detoxification of Aflatoxin B1 by Tea-Derived Aspergillus niger RAF106. Toxins 2020, 12, 777. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.S.T.; Sarreal, S.B.L.; Chang, P.-K.; Yu, J. Transcriptional Regulation of Aflatoxin Biosynthesis and Conidiation in Aspergillus flavus by Wickerhamomyces anomalus WRL-076 for Reduction of Aflatoxin Contamination. Toxins 2019, 11, 81. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, X.; Chen, H.; Huang, W.; Jiang, H.; Wang, C.; Xiao, Z.; Zhang, Y.; Xu, J. Isolation and Aflatoxin B1-Degradation Characteristics of a Microbacterium proteolyticum B204 Strain from Bovine Faeces. Toxins 2022, 14, 525. [Google Scholar] [CrossRef]
- Frisvad, J.C.; Samson, R.A. Emericella venezuelensis, a new species with stellate ascospores producing sterigmatocystin and aflatoxin B1. Syst. Appl. Microbiol. 2004, 27, 672–680. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gao, X.; Wang, K.; Wang, W.; Wang, Q.; Yan, P. Isolation of a novel deep-sea Bacillus circulus strain and uniform design for optimization of its anti-aflatoxigenic bioactive metabolites production. Bioengineered 2019, 10, 13–22. [Google Scholar] [CrossRef]
- Zhou, L.; Dong, F.; Zhang, W.; Chen, Y.; Zhou, L.; Zheng, F.; Lv, Z.; Xue, J.; He, D. Biosorption and biomineralization of U(VI) by Kocuria rosea: Involvement of phosphorus and formation of U-P minerals. Chemosphere 2022, 288, 132659. [Google Scholar] [CrossRef]
- Parshetti, G.K.; Parshetti, S.; Kalyani, D.C.; Doong, R.-A.; Govindwar, S.P. Industrial dye decolorizing lignin peroxidase from Kocuria rosea MTCC 1532. Ann. Microbiol. 2012, 62, 217–223. [Google Scholar] [CrossRef]
- Parshetti, G.; Telke, A.; Kalyani, D.; Govindwar, S. Decolorization and detoxification of sulfonated azo dye methyl orange by Kocuria rosea MTCC 1532. J. Hazard. Mater. 2010, 176, 503–509. [Google Scholar] [CrossRef]
- Laxmi, M.V.; Chari, M.A. Isolation of novel bacterial strains from contaminated soils for phenol biodegradation. BioTechnology 2009, 3, 149–156. [Google Scholar]
- Bernal, C.; Cairó, J.; Coello, N. Purification and characterization of a novel exocellular keratinase from Kocuria rosea. Enzym. Microb. Technol. 2006, 38, 49–54. [Google Scholar] [CrossRef]
- Solyanikova, I.P.; Baskunov, B.P.; Baboshin, M.A.; Saralov, A.I.; Golovleva, L.A. Detoxification of high concentrations of trinitrotoluene by bacteria. Appl. Biochem. Microbiol. 2012, 48, 21–27. [Google Scholar] [CrossRef]
- Ahmed, R.Z.; Ahmed, N.; Gadd, G.M. Isolation of two Kocuria species capable of growing on various polycyclic aromatic hydrocarbons. Afr. J. Biotechnol. 2010, 9, 3611–3617. [Google Scholar]
- Khandelwal, A.; Sugavanam, R.; Ramakrishnan, B.; Dutta, A.; Varghese, E.; Banerjee, T.; Nain, L.; Singh, S.B.; Singh, N. Bio-polysaccharide composites mediated degradation of polyaromatic hydrocarbons in a sandy soil using free and immobilized consortium of Kocuria rosea and Aspergillus sydowii. Environ. Sci. Pollut. Res. 2022, 29, 80005–80020. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Zaveri, P.; Patel, R.; Shah, M.T.; Munshi, N.S. Optimization of microbial fuel cell process using a novel consortium for aromatic hydrocarbon bioremediation and bioelectricity generation. J. Environ. Manag. 2021, 298, 113546. [Google Scholar] [CrossRef]
- Kayan, B.; Gözmen, B. Degradation of Acid Red 274 using H2O2 in subcritical water: Application of response surface methodology. J. Hazard. Mater. 2012, 201, 100–106. [Google Scholar] [CrossRef]
- Ross, T. Indices for performance evaluation of predictive models in food microbiology. J. Appl. Bacteriol. 1996, 81, 501–508. [Google Scholar] [CrossRef]
- Sant’Ana, A.S.; Franco, B.D.G.M.; Schaffner, D.W. Modeling the growth rate and lag time of different strains of Salmonella enterica and Listeria monocytogenes in ready-to-eat lettuce. Food Microbiol. 2012, 30, 267–273. [Google Scholar] [CrossRef] [PubMed]
- McGibbon, R.T.; Schwantes, C.R.; Pande, V.S. Statistical Model Selection for Markov Models of Biomolecular Dynamics. J. Phys. Chem. B 2014, 118, 6475–6481. [Google Scholar] [CrossRef] [PubMed]
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control. 1974, 19, 716–723. [Google Scholar] [CrossRef]
- Schwarz, G. Estimating the Dimension of a Model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Stylianou, C.; Pickles, A.; Roberts, S.A. Using Bonferroni, BIC and AIC to assess evidence for alternative biological pathways: Covariate selection for the multilevel Embryo-Uterus model. BMC Med. Res. Methodol. 2013, 13, 73. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Ji, C.; Zhou, T.; Li, J.; Ma, Q.; Niu, T. Aflatoxin B1 degradation by Stenotrophomonas maltophilia and other microbes selected using coumarin medium. Int. J. Mol. Sci. 2008, 9, 1489–1503. [Google Scholar] [CrossRef]
- Suresh, G.; Cabezudo, I.; Pulicharla, R.; Cuprys, A.; Rouissi, T.; Brar, S.K. Biodegradation of aflatoxin B-1 with cell-free extracts of Trametes versicolor and Bacillus subtilis. Res. Vet. Sci. 2020, 133, 85–91. [Google Scholar] [CrossRef]
- Fang, K.-T.; Lin, D.K.J.; Winker, P.; Zhang, Y. Uniform design: Theory and application. Technometrics 2000, 42, 237–248. [Google Scholar] [CrossRef]
- Tang, Q.-Y.; Zhang, C.-X. Data Processing System (DPS) software with experimental design, statistical analysis and data mining developed for use in entomological research. Insect Sci. 2013, 20, 254–260. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. In Gene Prediction; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2019; Volume 1962, pp. 1–14. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Julien, P.; Kuhn, M.; von Mering, C.; Muller, J.; Doerks, T.; Bork, P. eggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2008, 36, D250–D254. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | R2 | RMSE | nAIC | BIC |
|---|---|---|---|---|
| The quadratic polynomial stepwise regression model | 1.000 | 0.000 | −19.728 | 25.918 |
| The stepwise regression model with multiple factors and interaction terms | 1.000 | 0.000 | −18.198 | 27.448 |
| The multivariate and squared stepwise regression model | 0.954 | 0.055 | −4.978 | 40.667 |
| Source of Variation | Sum of Squares | Degree of Freedom | Mean Square | F-Value | p-Value |
|---|---|---|---|---|---|
| Regression | 1.232811 | 10 | 0.123281 | 8,738,194 | 0.000263 |
| Residual | 1.41 × 10−8 | 1 | 1.41 × 10−8 | ||
| Total variation | 1.232811 | 11 |
| Factor | Regression Coefficient | Standard Regression Coefficient | Partial Correlation Coefficient | t-Value | p-Value |
|---|---|---|---|---|---|
| x1 | 0.139448 | 4.864194 | 1 | 1502.222 | 0.000424 |
| x2 | 0.146648 | 0.546966 | 1 | 1178.142 | 0.00054 |
| x3 | −0.00375 | −0.39932 | −1 | 390.2854 | 0.001631 |
| x5 | −0.09324 | −0.34776 | −0.99999 | 264.1455 | 0.00241 |
| x1 × x1 | −0.00246 | −5.20978 | −1 | 2130.127 | 0.000299 |
| x3 × x3 | 4.95 × 10−5 | 0.549742 | 1 | 1308.839 | 0.000486 |
| x5 × x5 | 0.009993 | 0.186559 | 0.999963 | 116.7742 | 0.005452 |
| x1 × x2 | 0.00024 | 0.031926 | 0.999878 | 63.88512 | 0.009964 |
| x1 × x3 | 5.07 × 10−5 | 0.195935 | 0.999978 | 149.4907 | 0.004259 |
| x2 × x3 | −0.00143 | −0.49983 | −1 | 1134.843 | 0.000561 |
| Independent Variables | 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|---|
| Temperature (°C) | x1 | 15 | 25 | 35 | 45 | ||
| Time (Days) | x2 | 0.7 | 1.4 | 2.1 | 2.8 | 3.5 | 4.2 |
| Seawater ratio (%) | x3 | 0 | 20 | 40 | 60 | 80 | 100 |
| pH | x4 | 6 | 7 | 8 | 9 | 10 | 11 |
| Inoculation dosage (%) | x5 | 0.7 | 1.4 | 2.1 | 2.8 | 3.5 | 4.2 |
| Run | x1 | x2 | x3 | x4 | x5 | Uniform Design Matrix Performance Parameters |
|---|---|---|---|---|---|---|
| N1 | 2 | 3 | 2 | 3 | 1 | Centered discrepancy = 0.18836 |
| N2 | 2 | 6 | 5 | 1 | 4 | L2-discrepancy = 0.03101 |
| N3 | 4 | 3 | 6 | 3 | 6 | Modified discrepancy = 0.26946 |
| N4 | 1 | 1 | 4 | 2 | 5 | Symmetric discrepancy = 1.06848 |
| N5 | 4 | 2 | 3 | 1 | 2 | Wrap-around discrepancy = 0.37733 |
| N6 | 2 | 2 | 1 | 5 | 4 | Design matrix condition number = 1.5899 |
| N7 | 4 | 4 | 2 | 4 | 5 | D-optimal = 0.0000 |
| N8 | 1 | 5 | 3 | 6 | 6 | |
| N9 | 1 | 4 | 6 | 4 | 2 | |
| N10 | 3 | 5 | 1 | 2 | 3 | |
| N11 | 3 | 6 | 4 | 5 | 1 | |
| N12 | 3 | 1 | 5 | 6 | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Chen, Q.; Yan, P.; Dong, C.; Shao, Z. Isolation and Optimization of Aflatoxin B1 Degradation by Uniform Design and Complete Genome Sequencing of Novel Deep-Sea Kocuria rosea Strain 13. Toxins 2023, 15, 520. https://doi.org/10.3390/toxins15090520
Wang J, Chen Q, Yan P, Dong C, Shao Z. Isolation and Optimization of Aflatoxin B1 Degradation by Uniform Design and Complete Genome Sequencing of Novel Deep-Sea Kocuria rosea Strain 13. Toxins. 2023; 15(9):520. https://doi.org/10.3390/toxins15090520
Chicago/Turabian StyleWang, Jingying, Qiqi Chen, Peisheng Yan, Chunming Dong, and Zongze Shao. 2023. "Isolation and Optimization of Aflatoxin B1 Degradation by Uniform Design and Complete Genome Sequencing of Novel Deep-Sea Kocuria rosea Strain 13" Toxins 15, no. 9: 520. https://doi.org/10.3390/toxins15090520