Role of the Mannose Receptor (CD206) in Innate Immunity to Ricin Toxin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals, Reagents and Antibodies
2.2. Cytotoxicity and Apoptosis Assays
2.3. Toxin Binding Assays
2.4. Animals and Ricin Challenge Studies
2.5. Statistical Analysis and Software
3. Results
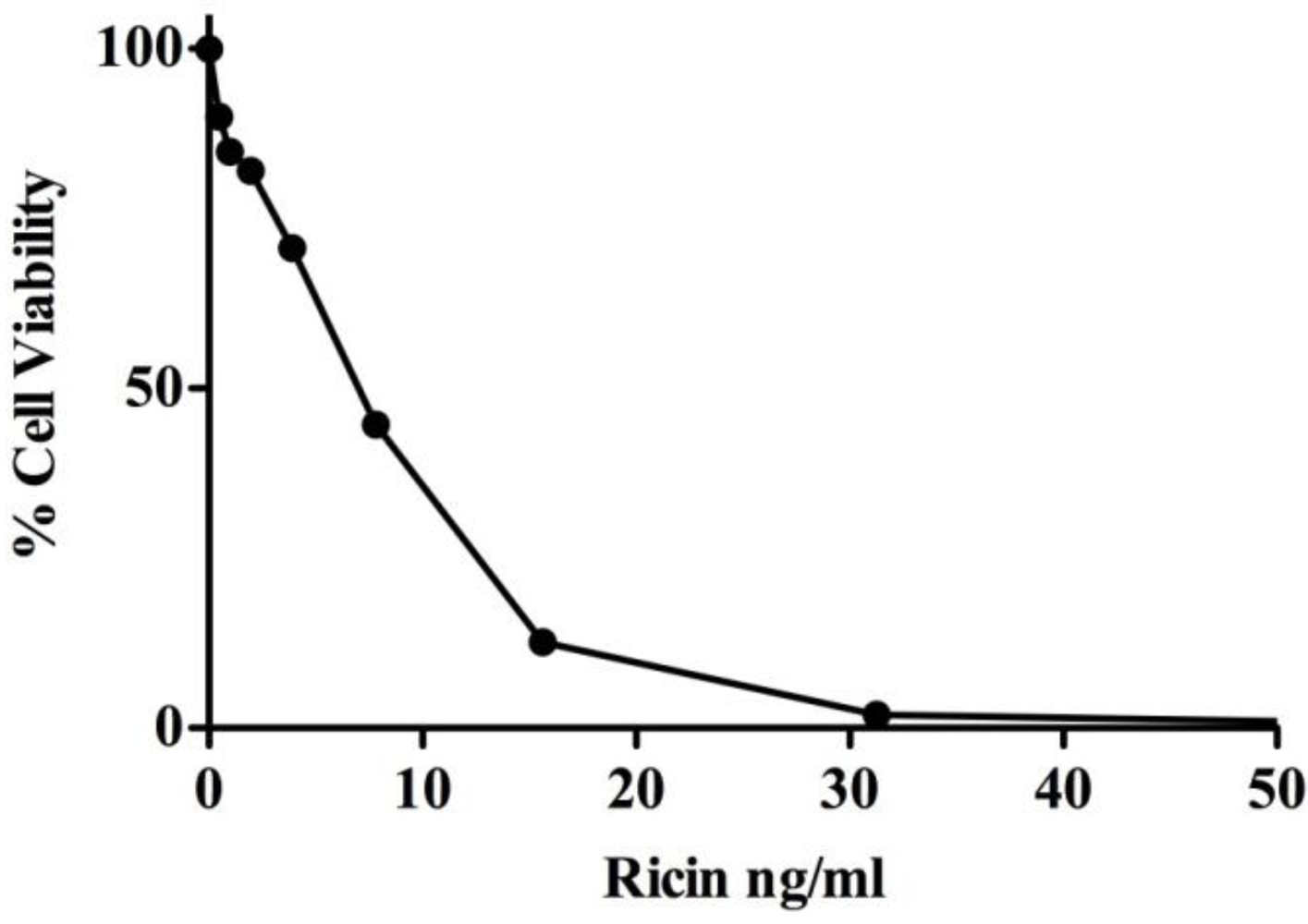
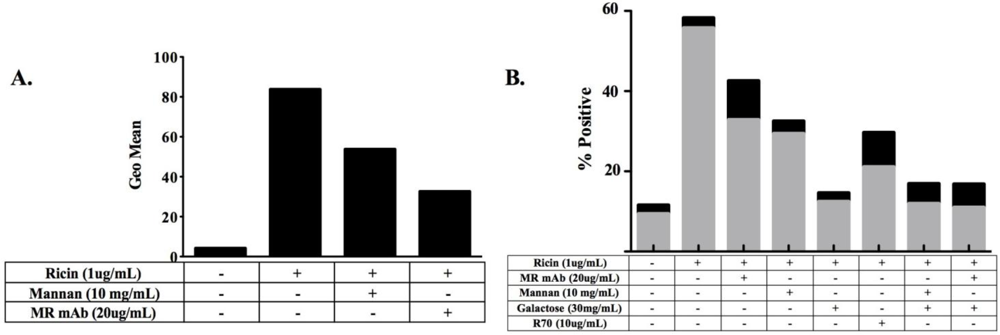
3.1. MR Contributes to Ricin-Induced Killing of Monocytes in Vitro


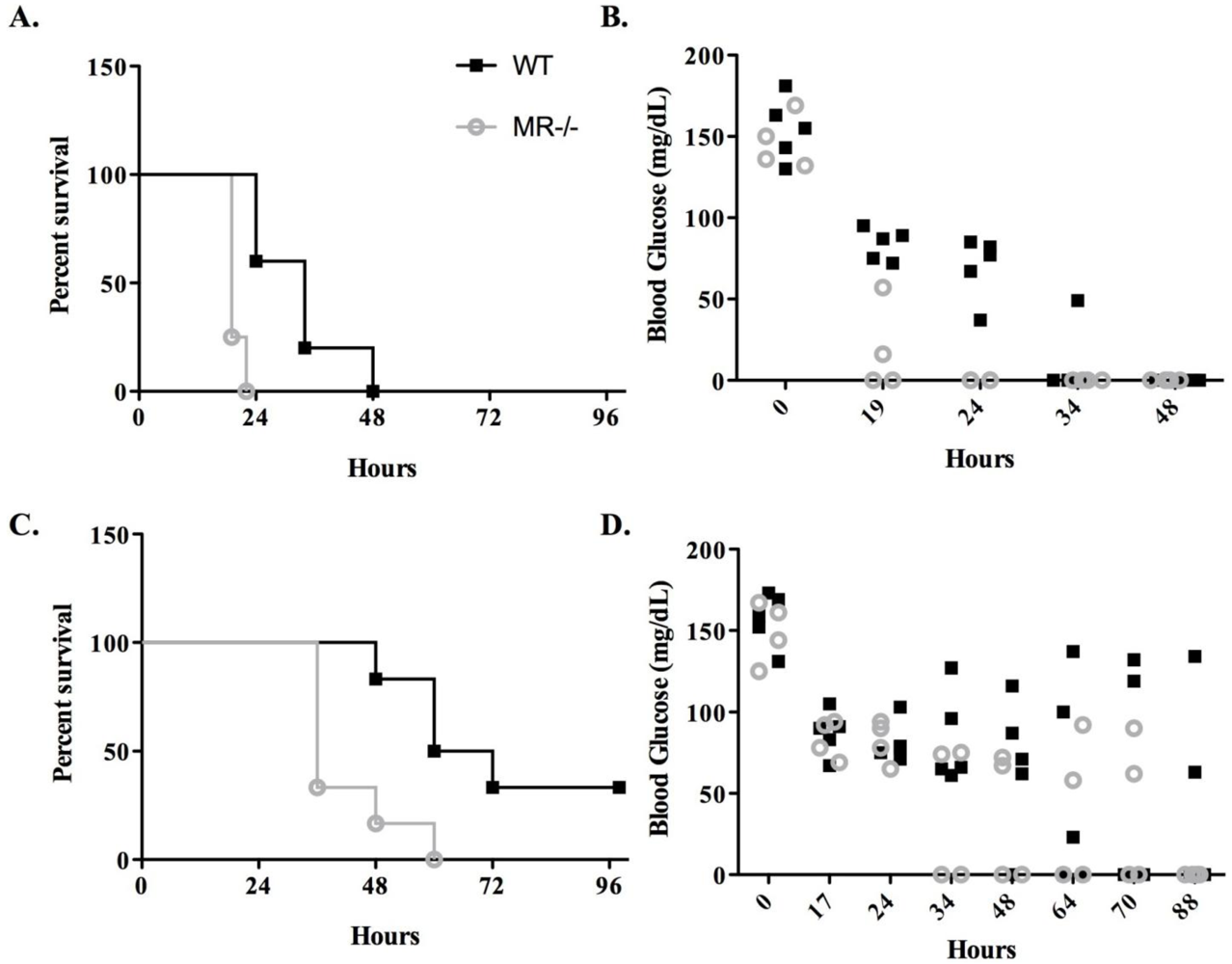
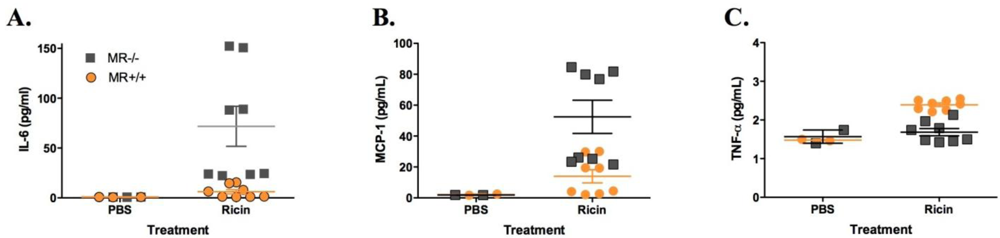
3.2. Sensitivity of MR−/− Mice to Ricin Intoxiction



4. Conclusions and Discussion
Acknowledgments
Conflict of Interest
References
- Audi, J.; Belson, M.; Patel, M.; Schier, J.; Osterloh, J. Ricin poisoning: A comprehensive review. J. Am. Med. Assoc. 2005, 294, 2342–2351. [Google Scholar]
- Olsnes, S. The history of ricin, abrin and related toxins. Toxicon 2004, 44, 361–370. [Google Scholar]
- Sandvig, K.; van Deurs, B. Delivery into cells: Lessons learned from plant and bacterial toxins. Gene. Ther. 2005, 12, 865–872. [Google Scholar]
- Franz, D.; Jaax, N. Ricin Toxin. In Textbook of Military Medicine; Zajtchuk, R.B., Ed.; Borden Institute: Washington, DC, USA, 1997; pp. 631–642. [Google Scholar]
- Maman, M.; Yehezkelli, Y. Ricin: A Possible, Non-Infectious Biological Weapon. In Bioterrorism and Infectious Agents; Fong, S., Alibek, K., Eds.; Springer Science and Business Media: New York, NY, USA, 2005; pp. 205–216. [Google Scholar]
- Hulse, C. Tests indicate poison in senate mail room of majority leader. N. Y. Times 2004, Sect. 12. [Google Scholar]
- Schier, J.G.; Patel, M.M.; Belson, M.G.; Patel, A.; Schwartz, M.; Fitzpatrick, N.; Drociuk, D.; Deitchman, S.; Meyer, R.; Litovitz, T.; Watson, W.A.; Rubin, C.H.; Kiefer, M. Public health investigation after the discovery of ricin in a South Carolina postal facility. Am. J. Public Health 2007, 97, S152–S157. [Google Scholar]
- Summary of the NIAID Ricin Expert Panel Workshop; National Institutes of Health: Bethesda, MD, USA, 2004.
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The mechanism of action of ricin and related toxins on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar]
- Eiklid, K.; Olsnes, S.; Pihl, A. Entry of lethal doses of abrin, ricin and modeccin into the cytosol of HeLa cells. Exp. Cell Res. 1980, 126, 321–326. [Google Scholar]
- Baenziger, J.U.; Fiete, D. Structural determinants of Ricinus communis agglutinin and toxin specificity for oligosaccharides. J. Biol. Chem. 1979, 254, 9795–9799. [Google Scholar]
- Foxwell, B.M.; Donovan, T.A.; Thorpe, P.E.; Wilson, G. The removal of carbohydrates from ricin with endoglycosidases H, F and D and alpha-mannosidase. Biochim. Biophys. Acta 1985, 840, 193–203. [Google Scholar]
- Kimura, Y.; Hase, S.; Kobayashi, Y.; Kyogoku, Y.; Ikenaka, T.; Funatsu, G. Structures of sugar chains of ricin D. J. Biochem. (Tokyo) 1988, 103, 944–949. [Google Scholar]
- Bradberry, S.M.; Dickers, K.J.; Rice, P.; Griffiths, G.D.; Vale, J.A. Ricin poisoning. Toxicol. Rev. 2003, 22, 65–70. [Google Scholar]
- Bingen, A.; Creppy, E.E.; Gut, J.P.; Dirheimer, G.; Kirn, A. The Kupffer cell is the first target in ricin-induced hepatitis. J. Submicrosc. Cytol. 1987, 19, 247–256. [Google Scholar]
- Derenzini, M.; Bonetti, E.; Marionozzi, V.; Stirpe, F. Toxic effects of ricin: Studies on the pathogenesis of liver lesions. Virchows Arch. B 1976, 20, 15–28. [Google Scholar]
- Fodstad, O.; Olsnes, S.; Pihl, A. Toxicity, distribution and elimination of the cancerostatic lectins abrin and ricin after parenteral injection into mice. Br. J. Cancer 1976, 34, 418–425. [Google Scholar]
- Skilleter, D.N.; Foxwell, B.M. Selective uptake of ricin A-chain by hepatic non-parenchymal cells in vitro. Importance of mannose oligosaccharides in the toxin. FEBS Lett. 1986, 196, 344–348. [Google Scholar]
- Thorpe, P.E.; Detre, S.I.; Foxwell, B.M.; Brown, A.N.; Skilleter, D.N.; Wilson, G.; Forrester, J.A.; Stirpe, F. Modification of the carbohydrate in ricin with metaperiodate-cyanoborohydride mixtures. Effects on toxicity and in vivo distribution. Eur. J. Biochem. 1985, 147, 197–206. [Google Scholar]
- Zenilman, M.E.; Fiani, M.; Stahl, P.; Brunt, E.; Flye, M.W. Use of ricin A-chain to selectively deplete Kupffer cells. J. Surg. Res. 1988, 45, 82–89. [Google Scholar]
- Brown, R.F.; White, D.E. Ultrastructure of rat lung following inhalation of ricin aerosol. Int. J. Exp. Path. 1997, 78, 267–276. [Google Scholar]
- Gonzalez, T.V.; Farrant, S.A.; Mantis, N.J. Ricin induces IL-8 secretion from human monocyte/macrophages by activating the p38 MAP kinase pathway. Mol. Immunol. 2006, 43, 1920–1923. [Google Scholar]
- Higuchi, S.; Tamura, T.; Oda, T. Cross-talk between the pathways leading to the induction of apoptosis and the secretion of tumor necrosis factor-alpha in ricin-treated RAW 264.7 cells. J. Biochem. (Tokyo) 2003, 134, 927–933. [Google Scholar]
- Korcheva, V.; Wong, J.; Lindauer, M.; Jacoby, D.B.; Iordanov, M.S.; Magun, B. Role of apoptotic signaling pathways in regulation of inflammatory responses to ricin in primary murine macrophages. Mol. Immunol. 2007, 44, 2761–2771. [Google Scholar]
- Licastro, F.; Morini, M.C.; Bolognesi, A.; Stirpe, F. Ricin induces the production of tumour necrosis factor-alpha and interleukin-1 beta by human peripheral-blood mononuclear cells. Biochem. J. 1993, 294, 517–520. [Google Scholar] [PubMed]
- Frankel, A.E.; Fu, T.; Burbage, C.; Tagge, E.; Harris, B.; Vesely, J.; Willingham, M.C. Lectin-deficient ricin toxin intoxicates cells bearing the D-mannose receptor. Carbohydr. Res. 1997, 300, 251–258. [Google Scholar]
- Simmons, B.M.; Stahl, P.D.; Russell, J.H. Mannose receptor-mediated uptake of ricin toxin and ricin A chain by macrophages. Multiple intracellular pathways for a chain translocation. J. Biol. Chem. 1986, 261, 7912–7920. [Google Scholar] [PubMed]
- Spooner, R.A.; Lord, J.M. How ricin and shiga toxin reach the cytosol of target cells: Retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol. 2011, in press. [Google Scholar]
- Van Deurs, B.; Sandvig, K.; Petersen, O.W.; Olsnes, S.; Simons, K.; Griffiths, G. Estimation of the amount of internalized ricin that reaches the trans-Golgi network. J. Cell Biol. 1988, 106, 253–267. [Google Scholar]
- Sandvig, K.; Olsnes, S.; Pihl, A. Kinetics of binding of the toxic lectins abrin and ricin to surface receptors of human cells. J. Biol. Chem. 1976, 251, 3977–3984. [Google Scholar]
- Magnusson, S.; Berg, T. Endocytosis of ricin by rat liver cells in vivo and in vitro is mainly mediated by mannose receptors on sinusoidal endothelial cells. Biochem. J. 1993, 291, 749–755. [Google Scholar]
- East, L.; Isacke, C.M. The mannose receptor family. Biochim. Biophys. Acta 2002, 1572, 364–386. [Google Scholar]
- Taylor, P.R.; Gordon, S.; Martinez-Pomares, L. The mannose receptor: Linking homeostasis and immunity through sugar recognition. Trends Immunol. 2005, 26, 104–110. [Google Scholar]
- Kerrigan, A.M.; Brown, G.D. C-type lectins and phagocytosis. Immunobiology 2009, 214, 562–575. [Google Scholar]
- Lee, S.J.; Evers, S.; Roeder, D.; Parlow, A.F.; Risteli, J.; Risteli, L.; Lee, Y.C.; Feizi, T.; Langen, H.; Nussenzweig, M.C. Mannose receptor-mediated regulation of serum glycoprotein homeostasis. Science 2002, 295, 1898–1901. [Google Scholar]
- Auwerx, J. The human leukemia cell line, THP-1: A multifacetted model for the study of monocyte-macrophage differentiation. Experientia 1991, 47, 22–31. [Google Scholar]
- Van Engeland, M.; Ramaekers, F.C.; Schutte, B.; Reutelingsperger, C.P. A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry 1996, 24, 131–139. [Google Scholar]
- McGuinness, C.R.; Mantis, N.J. Characterization of a novel high-affinity monoclonal immunoglobulin G antibody against the ricin B subunit. Infect. Immun. 2006, 74, 3463–3470. [Google Scholar]
- Baumann, J.; Park, C.G.; Mantis, N.J. Recognition of secretory IgA by DC-SIGN: Implications for immune surveillance in the intestine. Immunol. Lett. 2010, 131, 59–66. [Google Scholar]
- Diaz-Silvestre, H.; Espinosa-Cueto, P.; Sanchez-Gonzalez, A.; Esparza-Ceron, M.A.; Pereira-Suarez, A.L.; Bernal-Fernandez, G.; Espitia, C.; Mancilla, R. The 19-kDa antigen of Mycobacterium tuberculosis is a major adhesin that binds the mannose receptor of THP-1 monocytic cells and promotes phagocytosis of mycobacteria. Microb. Pathog. 2005, 39, 97–107. [Google Scholar]
- Puig-Kroger, A.; Serrano-Gomez, D.; Caparros, E.; Dominguez-Soto, A.; Relloso, M.; Colmenares, M.; Martínez-Muñoz, L.; Longo, N.; Sánchez-Sánchez, N.; Rincon, M.; Rivas, L.; Sánchez-Mateos, P.; Fernández-Ruiz, E.; Corbí, A.L. Regulated expression of the pathogen receptor dendritic cell-specific intercellular adhesion molecule 3 (ICAM-3)-grabbing nonintegrin in THP-1 human leukemic cells, monocytes, and macrophages. J. Biol. Chem. 2004, 279, 25680–25688. [Google Scholar]
- Sandvig, K.; Olsnes, S. Effect of temperature on the uptake, excretion and degradation of abrin and ricin by HeLa cells. Exp. Cell Res. 1979, 121, 15–25. [Google Scholar]
- Neal, L.M.; O’Hara, J.; Brey, R.N., 3rd.; Mantis, N.J. A monoclonal immunoglobulin G antibody directed against an immunodominant linear epitope on the ricin A chain confers systemic and mucosal immunity to ricin. Infect. Immun. 2010, 78, 552–561. [Google Scholar]
- Pincus, S.H.; Eng, L.; Cooke, C.L.; Maddaloni, M. Identification of hypoglycemia in mice as a surrogate marker of ricin toxicosis. Comp. Med. 2002, 52, 530–533. [Google Scholar]
- Yoder, J.M.; Aslam, R.U.; Mantis, N.J. Evidence for widespread epithelial damage and coincident production of monocyte chemotactic protein 1 in a murine model of intestinal ricin intoxication. Infect. Immun. 2007, 75, 1745–1750. [Google Scholar]
- Kang, B.K.; Schlesinger, L.S. Characterization of mannose receptor-dependent phagocytosis mediated by Mycobacterium tuberculosis lipoarabinomannan. Infect. Immun. 1998, 66, 2769–2777. [Google Scholar]
- Zamze, S.; Martinez-Pomares, L.; Jones, H.; Taylor, P.R.; Stillion, R.J.; Gordon, S.; Wong, S.Y.C. Recognition of bacterial capsular polysaccharides and lipopolysaccharides by the macrophage mannose receptor. J. Biol. Chem. 2002, 277, 41613–41623. [Google Scholar]
- Schulert, G.S.; Allen, L.A. Differential infection of mononuclear phagocytes by Francisella tularensis: Role of the macrophage mannose receptor. J. Leukoc. Biol. 2006, 80, 563–571. [Google Scholar]
- Ezekowitz, R.A.; Williams, D.J.; Koziel, H.; Armstrong, M.Y.; Warner, A.; Richards, F.F.; Rose, R.M. Uptake of Pneumocystis carinii mediated by the macrophage mannose receptor. Nature 1991, 351, 155–158. [Google Scholar]
- Lee, S.J.; Zheng, N.Y.; Clavijo, M.; Nussenzweig, M.C. Normal host defense during systemic candidiasis in mannose receptor-deficient mice. Infect. Immun. 2003, 71, 437–445. [Google Scholar]
- Dan, J.M.; Kelly, R.M.; Lee, C.K.; Levitz, S.M. Role of the mannose receptor in a murine model of Cryptococcus neoformans infection. Infect. Immun. 2008, 76, 2362–2367. [Google Scholar]
- Burgdorf, S.; Kautz, A.; Bohnert, V.; Knolle, P.A.; Kurts, C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 2007, 316, 612–616. [Google Scholar]
- Martinez-Pomares, L.; Mahoney, J.A.; Kaposzta, R.; Linehan, S.A.; Stahl, P.D.; Gordon, S. A functional soluble form of the murine mannose receptor is produced by macrophages in vitro and is present in mouse serum. J. Biol. Chem. 1998, 273, 23376–23380. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gage, E.; Hernandez, M.O.; O’Hara, J.M.; McCarthy, E.A.; Mantis, N.J. Role of the Mannose Receptor (CD206) in Innate Immunity to Ricin Toxin. Toxins 2011, 3, 1131-1145. https://doi.org/10.3390/toxins3091131
Gage E, Hernandez MO, O’Hara JM, McCarthy EA, Mantis NJ. Role of the Mannose Receptor (CD206) in Innate Immunity to Ricin Toxin. Toxins. 2011; 3(9):1131-1145. https://doi.org/10.3390/toxins3091131
Chicago/Turabian StyleGage, Emily, Maria O. Hernandez, Joanne M. O’Hara, Elizabeth A. McCarthy, and Nicholas J. Mantis. 2011. "Role of the Mannose Receptor (CD206) in Innate Immunity to Ricin Toxin" Toxins 3, no. 9: 1131-1145. https://doi.org/10.3390/toxins3091131