Impact of Antidepressants on Cytokine Production of Depressed Patients in Vitro

Abstract
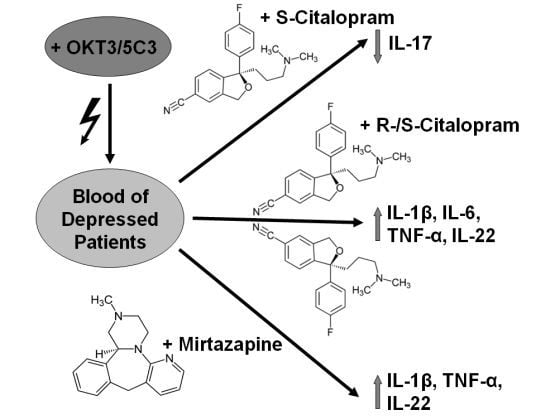
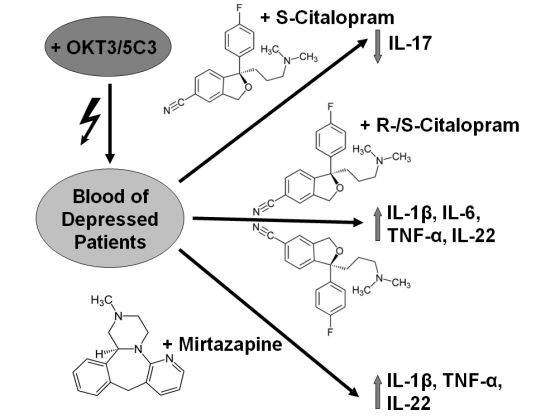
:
1. Introduction
2. Results and Discussion
2.1. Results
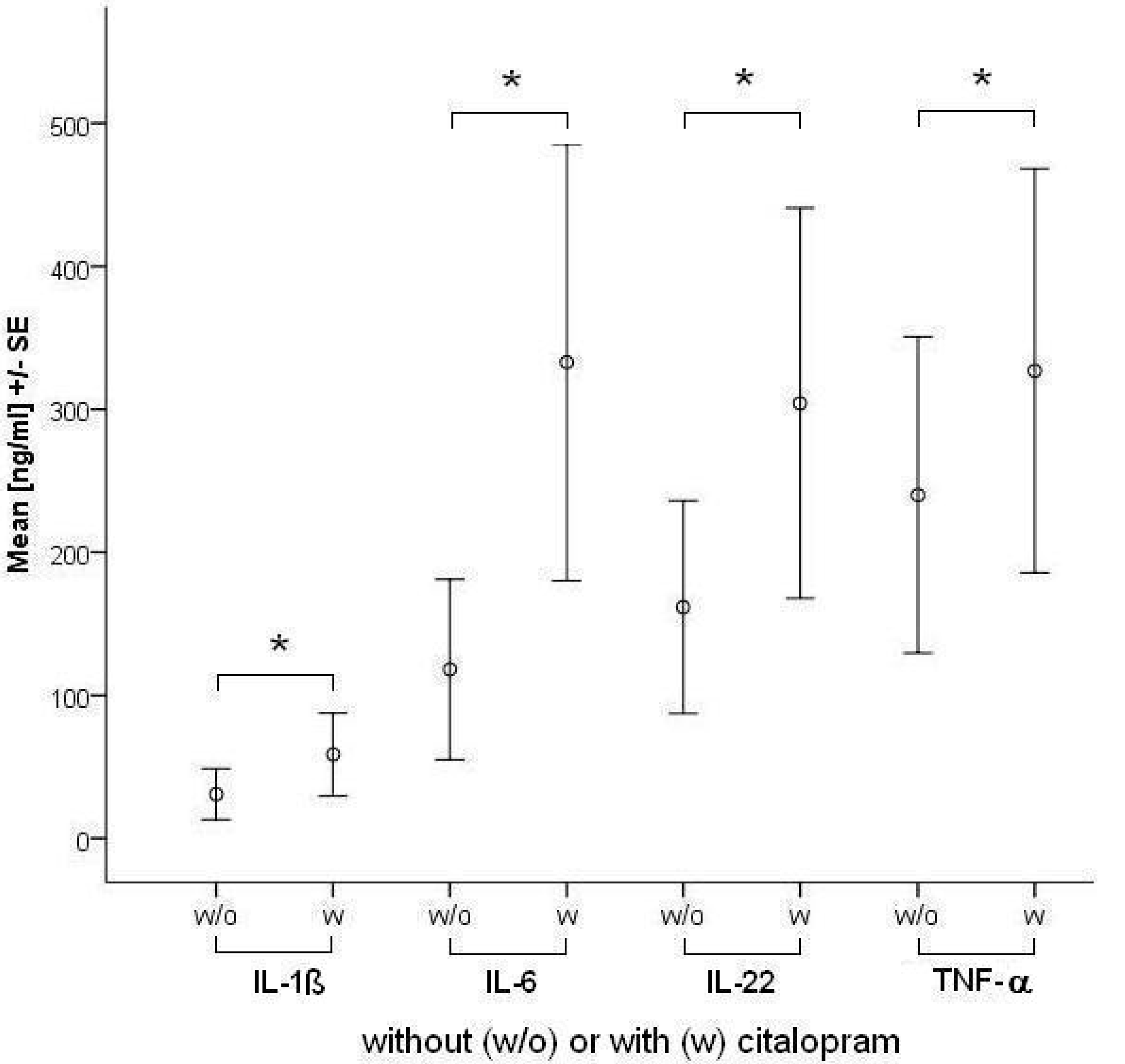
2.1.1. Influence of Antidepressants on Cytokine Production

{kind=link}
{kind=link}
| Concentration | 1-fold | 2-fold | ||||||
|---|---|---|---|---|---|---|---|---|
| Median | 1.Qu | 3.Qu | Median | 1.Qu | 3.Qu | |||
| IL-1β | Control | 1.6 | 0.1 | 4.3 | ||||
| Citalopram | 5.0* | 0.8 | 37.7 | 7.0* | 0.6 | 37.3 | ||
| Escitalopram | 1.3 | 0.0 | 4.5 | 1.2 | 0.0 | 3.6 | ||
| Mirtazapine | 3.1* | 0.0 | 37.7 | 5.0* | 0.0 | 45.2 | ||
| IL-2 | Control | 0.0 | 0.0 | 0.0 | ||||
| Citalopram | 0.0 | 0.0 | 2.4 | 0.0 | 0.0 | 1.9 | ||
| Escitalopram | 0.0 | 0.0 | 1.0 | 0.0 | 0.0 | 1.2 | ||
| Mirtazapine | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | ||
| IL-4 | Control | 0.1 | 0.0 | 7.2 | ||||
| Citalopram | 1.5 | 0.0 | 10.8 | 2.7 | 0.0 | 14.0 | ||
| Escitalopram | 0.6 | 0.0 | 8.5 | 0.6 | 0.0 | 6.5 | ||
| Mirtazapine | 0.8 | 0.0 | 4.0 | 1.9 | 0.0 | 9.5 | ||
| IL-6 | Control | 2.8 | 0.9 | 82.4 | ||||
| Citalopram | 49.8* | 2.4 | 428.0 | 58.1* | 4.6 | 405.6 | ||
| Escitalopram | 6.1 | 0.3 | 111.7 | 3.7 | 0.7 | 62.6 | ||
| Mirtazapin | 5.6 | 0.4 | 76.2 | 24.6 | 0.1 | 470.0 | ||
| IL-17 | Control | 3.4 | 0.1 | 9.4 | ||||
| Citalopram | 10.6 | 0.0 | 15.5 | 8.2 | 0.6 | 14.2 | ||
| Escitalopram | 3.2 | 0.0 | 7.9 | 1.3* | 0.1 | 3.1 | ||
| Mirtazapine | 4.7 | 0.0 | 8.3 | 9.0 | 0.0 | 10.4 | ||
| IL-22 | Control | 0.0 | 0.0 | 188.9 | ||||
| Citalopram | 42.0* | 0.0 | 366.1 | 96.1* | 0.0 | 575.0 | ||
| Escitalopram | 0.0 | 0.0 | 55.1 | 0.0 | 0.0 | 187.3 | ||
| Mirtazapine | 24.6 | 0.0 | 284.2 | 69.6* | 0.0 | 403.4 | ||
| TNF-α | Control | 3.8 | 0.0 | 298.0 | ||||
| Citalopram | 38.3 | 0.0 | 347.7 | 153.3* | 0.7 | 400.9 | ||
| Escitalopram | 4.1 | 0.0 | 32.7 | 2.8 | 0.0 | 167.4 | ||
| Mirtazapine | 41.9 | 0.0 | 365.6 | 205.3* | 0.0 | 659.2 |

2.1.2. Comparisons of antidepressants
2.2. Discussion
3. Experimental Section
3.1. Subjects
3.2. Procedure
3.3. Cytokine Measurement
3.4. Statistical Analysis
4. Conclusion
Acknowledgments
Conflicts of Interest
References
- Lichtblau, N.; Schmidt, F.M.; Schumann, R.; Kirkby, K.C.; Himmerich, H. Cytokines as biomarkers in depressive disorder: Current standing and prospects. Int. Rev. Psychiatry 2013, 25, 592–603. [Google Scholar] [CrossRef]
- Seidel, A.; Arolt, V.; Hunstiger, M.; Rink, L.; Behnisch, A.; Kirchner, H. Cytokine production and serum proteins in depression. Scand. J. Immunol. 1995, 41, 534–538. [Google Scholar] [CrossRef]
- Dantzer, R. Cytokine, sickness behavior, and depression. Immunol. Allergy Clin. North Am. 2009, 29, 247–264. [Google Scholar] [CrossRef]
- Himmerich, H.; Fulda, S.; Linseisen, J.; Seiler, H.; Wolfram, G.; Himmerich, S.; Gedrich, K.; Kloiber, S.; Lucae, S.; Ising, M.; Uhr, M.; Holsboer, F.; Pollmächer, T. Depression, comorbidities and the TNF-alpha system. Eur. Psychiatry 2008, 23, 421–429. [Google Scholar] [CrossRef]
- Heiser, P.; Lanquillon, S.; Krieg, J.C.; Vedder, H. Differential modulation of cytokine production in major depressive disorder by cortisol and dexamethasone. Eur. Neuropsychopharmacol. 2008, 18, 860–870. [Google Scholar] [CrossRef]
- Maes, M.; Berk, M.; Goehler, L.; Song, C.; Anderson, G.; Gałecki, P.; Leonard, B. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med. 2012, 10, 66. [Google Scholar] [CrossRef]
- Capuron, L.; Miller, A.H. Immune system to brain signaling: Neuropsychopharmacological implications. Pharmacol. Ther. 2011, 130, 226–238. [Google Scholar] [CrossRef]
- Himmerich, H.; Berthold-Losleben, M.; Pollmächer, T. The relevance of the TNF-alpha system in psychiatric disorders. Fortschr. Neurol. Psychiatr. 2009, 77, 334–345. [Google Scholar] [CrossRef]
- Berthold-Losleben, M.; Himmerich, H. The TNF-alpha system: functional aspects in depression, narcolepsy and psychopharmacology. Curr. Neuropharmacol. 2008, 6, 193–202. [Google Scholar] [CrossRef]
- Ramamoorthy, S.; Ramamoorthy, J.D.; Prasad, P.D.; Bhat, G.K.; Mahesh, V.B.; Leibach, F.H.; Ganapathy, V. Regulation of the human serotonin transporter by interleukin-1 beta. Biochem. Biophys. Res. Commun. 1995, 216, 560–567. [Google Scholar] [CrossRef]
- Müller, N.; Schwarz, M.J. Immunological aspects of depressive disorders. Nervenarzt 2007, 78, 1261–1273. [Google Scholar] [CrossRef]
- Catena-Dell'Osso, M.; Rotella, F.; Dell'Osso, A.; Fagiolini, A.; Marazziti, D. Inflammation, serotonin and major depression. Curr. Drug Targets 2013, 14, 571–577. [Google Scholar] [CrossRef]
- Besedovsky, H.O.; del Rey, A.; Klusman, I.; Furukawa, H.; Monge Arditi, G.; Kabiersch, A. Cytokines as modulators of the hypothalamus-pituitary-adrenal axis. J. Steroid Biochem. Mol. Biol. 1991, 40, 613–618. [Google Scholar] [CrossRef]
- Makhija, K.; Karunakaran, S. The role of inflammatory cytokines on the aetiopathogenesis of depression. Aust. N. Z. J. Psychiatry 2013, 47, 828–839. [Google Scholar] [CrossRef]
- Maes, M. The cytokine hypothesis of depression: inflammation, oxidative & nitrosative stress (IO&NS) and leaky gut as new targets for adjunctive treatments in depression. Neuro Endocrinol. Lett. 2008, 29, 287–291. [Google Scholar]
- Audet, M.C.; Anisman, H. Interplay between pro-inflammatory cytokines and growth factors in depressive illnesses. Front. Cell. Neurosci. 2013, 7, 68. [Google Scholar]
- Himmerich, H.; Milenović, S.; Fulda, S.; Plümäkers, B.; Sheldrick, A.J.; Michel, T.M.; Kircher, T.; Rink, L. Regulatory T cells increased while IL-1β decreased during antidepressant therapy. J. Psychiatr. Res. 2010, 44, 1052–1057. [Google Scholar] [CrossRef]
- Himmerich, H.; Fulda, S.; Sheldrick, A.J.; Plümäkers, B.; Rink, L. IFN-gamma reduction by tricyclic antidepressants. Int. J. Psychiatry Med. 2010, 40, 413–424. [Google Scholar] [CrossRef]
- Akhondzadeh, S.; Jafari, S.; Raisi, F.; Nasehi, A.A.; Ghoreishi, A.; Salehi, B.; Mohebbi-Rasa, S.; Raznahan, M.; Kamalipour, A. Clinical trial of adjunctive celecoxib treatment in patients with major depression: a double blind and placebo controlled trial. Depress. Anxiety 2009, 26, 607–611. [Google Scholar] [CrossRef]
- Müller, N.; Schwarz, M.J.; Dehning, S.; Douhet, A.; Cerovecki, A.; Goldstein-Müller, B.; Spellmann, I.; Hetzel, G.; Maino, K.; Kleindienst, N.; Möller, H.J.; Arolt, V.; Riedel, M. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol. Psychiatry 2006, 11, 680–684. [Google Scholar] [CrossRef]
- Müller, N. The role of anti-inflammatory treatment in psychiatric disorders. Psychiatr. Danub. 2013, 25, 292–298. [Google Scholar]
- Hemdan, N.Y.; Birkenmeier, G.; Wichmann, G.; Abu El-Saad, A.M.; Krieger, T.; Conrad, K.; Sack, U. Interleukin-17-producing T helper cells in autoimmunity. Autoimmun. Rev. 2010, 9, 785–792. [Google Scholar] [CrossRef]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; Dong, C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef]
- Murdaca, G.; Colombo, B.M.; Puppo, F. The role of Th17 lymphocytes in the autoimmune and chronic inflammatory diseases. Intern. Emerg. Med. 2011, 6, 487–495. [Google Scholar] [CrossRef]
- Pan, H.F.; Li, X.P.; Zheng, S.G.; Ye, D.Q. Emerging role of interleukin-22 in autoimmune diseases. Cytokine Growth Factor Rev. 2013, 24, 51–57. [Google Scholar] [CrossRef]
- Li, S.; Yu, M.; Li, H.; Zhang, H.; Jiang, Y. IL-17 and IL-22 in cerebrospinal fluid and plasma are elevated in Guillain-Barré syndrome. Mediators Inflamm. 2012, 2012, 260473. [Google Scholar]
- Wang, P.; Bai, F.; Zenewicz, L.A.; Dai, J.; Gate, D.; Cheng, G.; Yang, L.; Qian, F.; Yuan, X.; Montgomery, R.R.; Flavell, R.A.; Town, T.; Fikrig, E. IL-22 signaling contributes to West Nile encephalitis pathogenesis. PLoS One 2012, 7, e44153. [Google Scholar]
- Jadidi-Niaragh, F.; Mirshafiey, A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand. J. Immunol. 2011, 74, 1–13. [Google Scholar] [CrossRef]
- Adair, J.R.; Athwal, D.S.; Bodmer, M.W.; Bright, S.M.; Collins, A.M.; Pulito, V.L.; Rao, P.E.; Reedman, R.; Rothermel, A.L.; Xu, D.; Zivin, R.A.; Jolliffe, L.K. Humanization of the murine anti-human CD3 monoclonal antibody OKT3. Hum. Antibodies Hybridomas 1994, 5, 41–47. [Google Scholar]
- Pound, J.D.; Challa, A.; Holder, M.J.; Armitage, R.J.; Dower, S.K.; Fanslow, W.C.; Kikutani, H.; Paulie, S.; Gregory, C.D.; Gordon, J. Minimal crosslinking and epitope requirements for CD40-dependent suppression of apoptosis contrast with those for promotion of the cell cycle and homotypic adhesions in human B cells. Int. Immunol. 1999, 11, 11–20. [Google Scholar] [CrossRef]
- Montgomery, S.; Hansen, T.; Kasper, S. Efficacy of escitalopram compared to citalopram: a meta-analysis. Int. J. Neuropsychopharmacol. 2011, 14, 261–268. [Google Scholar] [CrossRef]
- Leonard, B.; Taylor, D. Escitalopram-translating molecular properties into clinical benefit: reviewing the evidence in major depression. J. Psychopharmacol. 2010, 24, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Cipriani, A.; Furukawa, T.A.; Salanti, G.; Geddes, J.R.; Higgins, J.P.; Churchill, R.; Watanabe, N.; Nakagawa, A.; Omori, I.M.; McGuire, H.; Tansella, M.; Barbui, C. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet 2009, 373, 746–758. [Google Scholar] [CrossRef]
- Sánchez, C.; Bergqvist, P.B.; Brennum, L.T.; Gupta, S.; Hogg, S.; Larsen, A.; Wiborg, O. Escitalopram, the S-(+)-enantiomer of citalopram, is a selective serotonin reuptake inhibitor with potent effects in animal models predictive of antidepressant and anxiolytic activities. Psychopharmacology 2003, 167, 353–362. [Google Scholar]
- Kennedy, S.H.; Andersen, H.F.; Thase, M.E. Escitalopram in the treatment of major depressive disorder: a meta-analysis. Curr. Med. Res. Opin. 2009, 25, 161–175. [Google Scholar] [CrossRef]
- Himmerich, H.; Wranik, D.W. Choice of treatment with antidepressants: influencing factors. Curr. Pharm. Des. 2012, 18, 5958–5975. [Google Scholar] [CrossRef]
- Himmerich, H.; Bartsch, S.; Hamer, H.; Mergl, R.; Schönherr, J.; Petersein, C.; Munzer, A.; Kirkby, K.C.; Bauer, K.; Sack, U. Impact of mood stabilizers and antiepileptic drugs on cytokine production in vitro. J. Psychiatr. Res. 2013, 47, 1751–1759. [Google Scholar] [CrossRef]
- Schilström, B.; Konradsson-Geuken, A.; Ivanov, V.; Gertow, J.; Feltmann, K.; Marcus, M.M.; Jardemark, K.; Svensson, T.H. Effects of S-citalopram, citalopram, and R-citalopram on the firing patterns of dopamine neurons in the ventral tegmental area, N-methyl-d-aspartate receptor-mediated transmission in the medial prefrontal cortex and cognitive function in the rat. Synapse 2011, 65, 357–367. [Google Scholar] [CrossRef]
- Friedman, E.S.; Wisniewski, S.R.; Gilmer, W.; Nierenberg, A.A.; Rush, A.J.; Fava, M.; Zisook, S.; Balasubramani, G.K.; Trivedi, M.H. Sociodemographic, clinical, and treatment characteristics associated with worsened depression during treatment with citalopram: results of the NIMH STAR(*)D trial. Depress. Anxiety 2009, 26, 612–621. [Google Scholar] [CrossRef]
- Vieweg, W.V.; Hasnain, M.; Howland, R.H.; Hettema, J.M.; Kogut, C.; Wood, M.A.; Pandurangi, A.K. Citalopram, QTc interval prolongation, and torsade de pointes. How should we apply the recent FDA ruling? Am. J. Med. 2012, 125, 859–868. [Google Scholar] [CrossRef]
- Adkins, D.E.; Clark, S.L.; Åberg, K.; Hettema, J.M.; Bukszár, J.; McClay, J.L.; Souza, R.P.; van den Oord, E.J. Genome-wide pharmacogenomic study of citalopram-induced side effects in STAR*D. Transl. Psychiatry 2012, 2, e129. [Google Scholar]
- Rozanski, G.J.; Witt, R.C. IL-1 inhibits b-adrenergic control of cardiac calcium current: role of l-arginine/nitric oxide pathway. Am. J. Physiol. 1994, 267, 1753–1758. [Google Scholar]
- Liu, S.J.; Zhou, W.; Kennedy, R.H. Suppression of b-adrenergic responsiveness of L-type Ca2+ current by IL-1b in rat ventricular myocytes. Am. J. Physiol. 1999, 276, 141–148. [Google Scholar]
- Kaprielian, R.; Wickenden, A.D.; Kassiri, Z.; Parker, T.G.; Liu, P.P.; Backx, P.H. Relationship between K+ channel down-regulation and [Ca2+]i in rat ventricular myocytes following myocardial infarction. J. Physiol. 1999, 517, 229–245. [Google Scholar] [CrossRef]
- Gazewood, J.D.; Richards, D.R.; Clebak, K. Parkinson disease: an update. Am. Fam. Physician 2013, 87, 267–273. [Google Scholar]
- Shneyder, N.; Harris, M.K.; Minagar, A. Movement disorders in patients with multiple sclerosis. Handb. Clin. Neurol. 2011, 100, 307–314. [Google Scholar] [CrossRef]
- Weschenfelder, J.; Sander, C.; Kluge, M.; Kirkby, K.C.; Himmerich, H. The influence of cytokines on wakefulness regulation: clinical relevance, mechanisms and methodological problems. Psychiatr. Danub. 2012, 24, 112–126. [Google Scholar]
- Avitsur, R.; Yirmiya, R. The immunobiology of sexual behavior: gender differences in the suppression of sexual activity during illness. Pharmacol. Biochem. Behav. 1999, 64, 787–796. [Google Scholar] [CrossRef]
- Kraus, T.; Haack, M.; Schuld, A.; Hinze-Selch, D.; Koethe, D.; Pollmächer, T. Body weight, the tumor necrosis factor system, and leptin production during treatment with mirtazapine or venlafaxine. Pharmacopsychiatry 2002, 35, 220–225. [Google Scholar] [CrossRef]
- Himmerich, H.; Schuld, A.; Pollmächer, T. Weight gain during treatment with antipsychotics: clinical relevance, pathophysiology, and therapeutical strategies. Psychiatr. Prax. 2004, 31 (Suppl. 2), 233–237. [Google Scholar] [CrossRef]
- Hannestad, J.; DellaGioia, N.; Bloch, M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis. Neuropsychopharmacology 2011, 36, 2452–2459. [Google Scholar] [CrossRef]
- Liberman, A.C.; Druker, J.; Refojo, D.; Holsboer, F.; Arzt, E. Glucocorticoids inhibit GATA-3 phosphorylation and activity in T cells. FASEB J. 2009, 23, 1558–1571. [Google Scholar] [CrossRef]
- Ising, M.; Lauer, C.J.; Holsboer, F.; Modell, S. The Munich vulnerability study on affective disorders: premorbid neuroendocrine profile of affected high-risk probands. J. Psychiatr. Res. 2005, 39, 21–28. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, B.; Qiu, W.; Yang, L.; Hu, B.; Tian, X.; Yang, H. Altered expression of CD4(+)CD25(+) regulatory T cells and its 5-HT(1a) receptor in patients with major depression disorder. J. Affect. Disord. 2010, 124, 68–75. [Google Scholar] [CrossRef]
- Wittchen, H.U.; Zaudig, M.; Fydrich, T. Strukturiertes Klinisches Interview für DSM-IV; Hogrefe: Göttingen, Germany, 1997. [Google Scholar]
- Beck, A.T.; Ward, C.H.; Mendelson, M.; Mock, J.; Erbaugh, J. An inventory for measuring depression. Arch. Gen. Psychiatry 1961, 4, 561–571. [Google Scholar] [CrossRef]
- Hamilton, M. A rating scale for depression. J. Neurol. Neurosurg. Psychiatry 1960, 23, 56–62. [Google Scholar] [CrossRef]
- Kirchner, H.; Kleinicke, C.; Digel, W. A whole-blood technique for testing production of human interferon by leukocytes. J. Immunol. Methods 1982, 48, 213–219. [Google Scholar]
- Seidel, A.; Arolt, V.; Hunstiger, M.; Rink, L.; Behnisch, A.; Kirchner, H. Cytokine production and serum proteins in depression. Scand. J. Immunol. 1995, 41, 534–538. [Google Scholar] [CrossRef]
- Baumann, P.; Hiemke, C.; Ulrich, S.; Eckermann, G.; Gaertner, I.; Gerlach, M.; Kuss, H.J.; Laux, G.; Müller-Oerlinghausen, B.; Rao, M.L.; Riederer, P.; Zernig, G.; Arbeitsgemeinschaft für Neuropsychopharmakologie und Pharmakopsychiatrie. The AGNP-TDM expert group consensus guidelines: therapeutic drug monitoring in psychiatry. Pharmacopsychiatry 2004, 37, 243–265. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Munzer, A.; Sack, U.; Mergl, R.; Schönherr, J.; Petersein, C.; Bartsch, S.; Kirkby, K.C.; Bauer, K.; Himmerich, H. Impact of Antidepressants on Cytokine Production of Depressed Patients in Vitro. Toxins 2013, 5, 2227-2240. https://doi.org/10.3390/toxins5112227
Munzer A, Sack U, Mergl R, Schönherr J, Petersein C, Bartsch S, Kirkby KC, Bauer K, Himmerich H. Impact of Antidepressants on Cytokine Production of Depressed Patients in Vitro. Toxins. 2013; 5(11):2227-2240. https://doi.org/10.3390/toxins5112227
Chicago/Turabian StyleMunzer, Alexander, Ulrich Sack, Roland Mergl, Jeremias Schönherr, Charlotte Petersein, Stefanie Bartsch, Kenneth C. Kirkby, Katrin Bauer, and Hubertus Himmerich. 2013. "Impact of Antidepressants on Cytokine Production of Depressed Patients in Vitro" Toxins 5, no. 11: 2227-2240. https://doi.org/10.3390/toxins5112227