Bacterial Toxins Fuel Disease Progression in Cutaneous T-Cell Lymphoma


Abstract
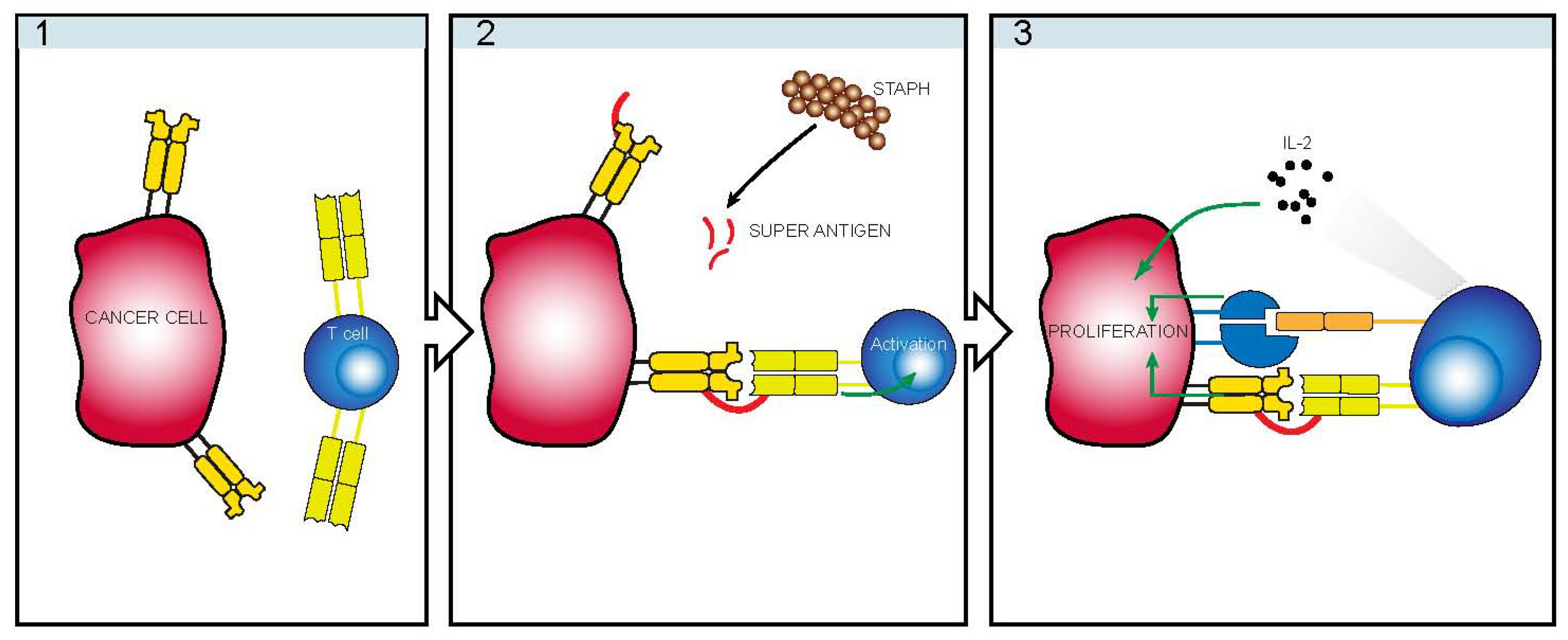
:1. Introduction
2. High Prevalence of Infections
3. Immunopathogenesis
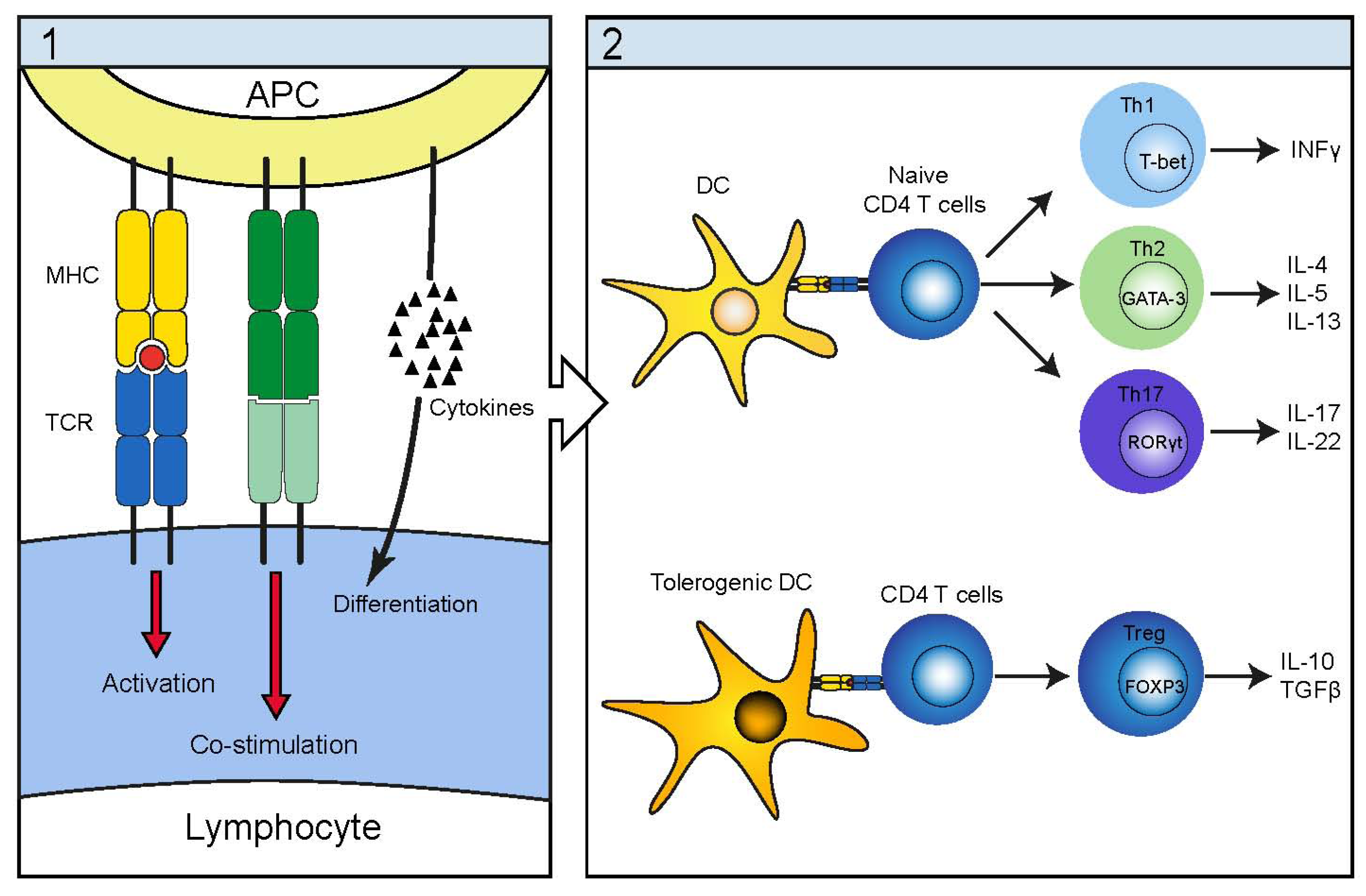
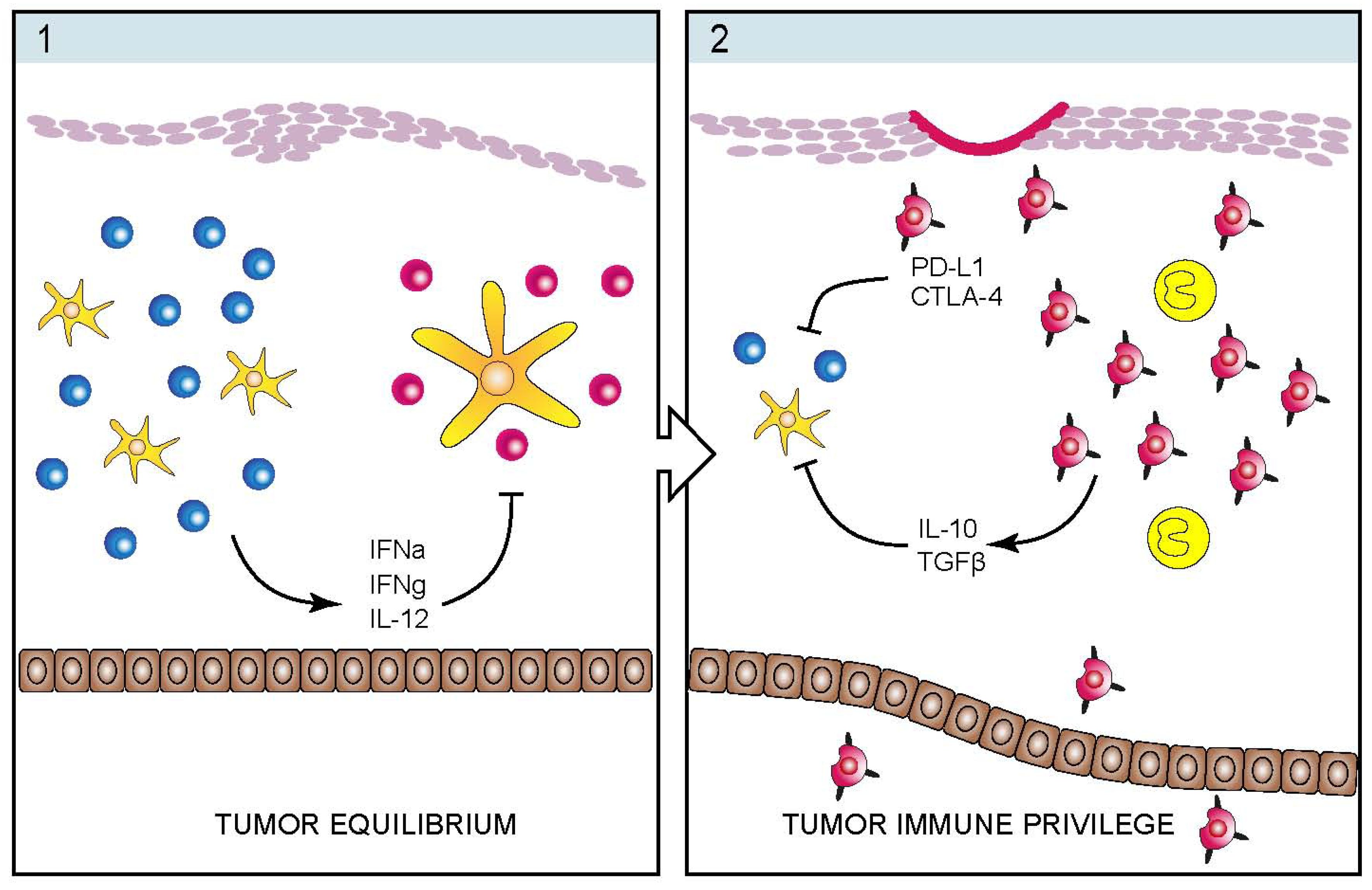
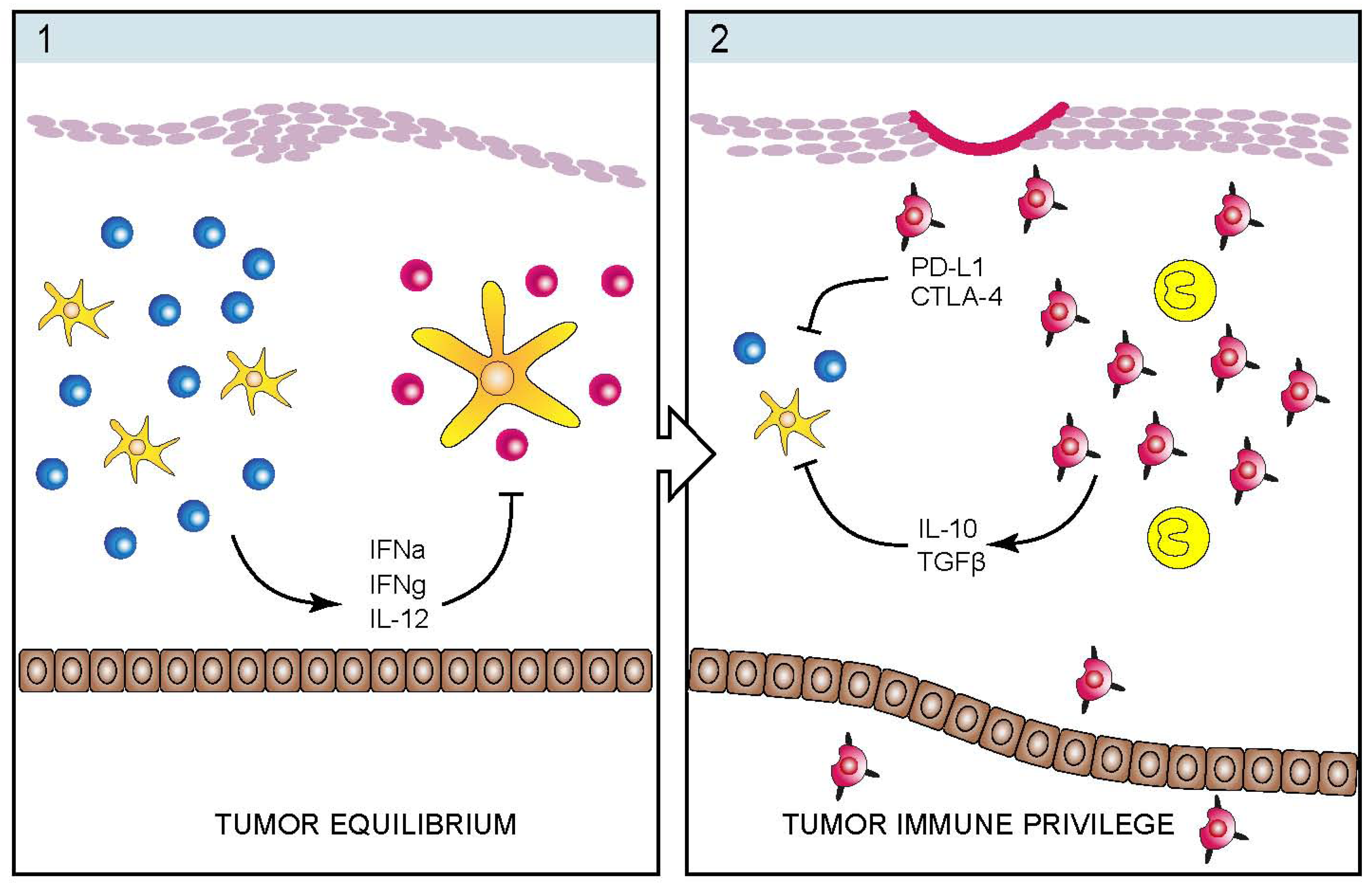
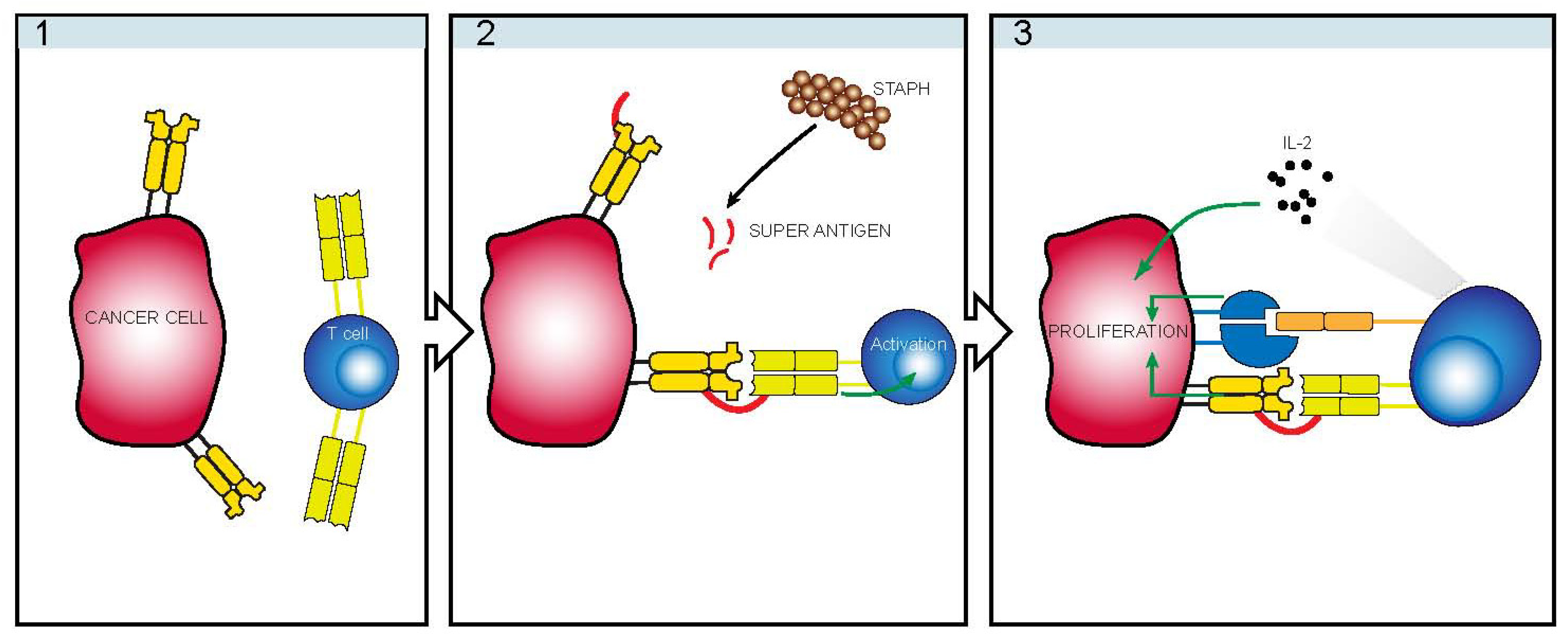
4. Infectious Etiology

{kind=link}
{kind=link}
{kind=link}
| Bacteria | Number of infections | Frequency |
|---|---|---|
| Staphylococcus aureus | 117 | 33%–38% * |
| Enterobacteriaceae | 38 | 10.7% |
| Beta-hemolytic streptococci | 35 | 9.8% |
| Pseudomonas aeruginosa | 12 | 3.4% |
| Viruses | ||
| Herpes zoster | 34 | 9.6% |
| Herpes simplex | 30 | 8.4% |
| Co-morbidity from infections | |
|---|---|
| Bacterial infections | bacteremia, pneumonia, intra-abdominal infections |
| Viral infections | ulcerative skin lesions, dissemination (Kaposi varicelliform eruption) |
5. Staphylococcus
6. TCRVβ Restriction
7. Indirect Mechanism of Action
8. Clinical Improvement after Antibiotic Treatment
9. Conclusions
Acknowledgements
Conflict of Interest
References
- Girardi, M.; Heald, P.W.; Wilson, L.D. The pathogenesis of mycosis fungoides. N. Engl. J. Med. 2004, 350, 1978–1988. [Google Scholar] [CrossRef]
- Hwang, S.T.; Janik, J.E.; Jaffe, E.S.; Wilson, W.H. Mycosis fungoides and sezary syndrome. Lancet 2008, 371, 945–957. [Google Scholar] [CrossRef]
- Weinstock, M.A.; Gardstein, B. Twenty-year trends in the reported incidence of mycosis fungoides and associated mortality. Am. J. Public Health 1999, 89, 1240–1244. [Google Scholar] [CrossRef]
- Willemze, R.; Jaffe, E.S.; Burg, G.; Cerroni, L.; Berti, E.; Swerdlow, S.H.; Ralfkiaer, E.; Chimenti, S.; Diaz-Perez, J.L.; Duncan, L.M.; et al. Who-eortc classification for cutaneous lymphomas. Blood 2005, 105, 3768–3785. [Google Scholar] [CrossRef]
- Willemze, R.; Meijer, C.J. Classification of cutaneous t-cell lymphoma: From alibert to who-eortc. J. Cutan. Pathol. 2006, 33, 18–26. [Google Scholar] [CrossRef]
- Wong, H.K.; Mishra, A.; Hake, T.; Porcu, P. Evolving insights in the pathogenesis and therapy of cutaneous t-cell lymphoma (mycosis fungoides and sezary syndrome). Br. J. Haematol. 2011, 155, 150–166. [Google Scholar] [CrossRef]
- Ralfkiaer, U.; Hagedorn, P.H.; Bangsgaard, N.; Lovendorf, M.B.; Ahler, C.B.; Svensson, L.; Kopp, K.L.; Vennegaard, M.T.; Lauenborg, B.; Zibert, J.R.; et al. Diagnostic microrna profiling in cutaneous t-cell lymphoma (ctcl). Blood 2011, 118, 5891–5900. [Google Scholar] [CrossRef]
- Marstrand, T.; Ahler, C.B.; Ralfkiaer, U.; Clemmensen, A.; Kopp, K.L.; Sibbesen, N.A.; Krejsgaard, T.; Litman, T.; Wasik, M.A.; Bonefeld, C.M.; et al. Validation of a diagnostic mirna classifier in cutaneous t-cell lymphomas. Leuk. Lymphoma 2013. [Google Scholar] [CrossRef]
- Krejsgaard, T.; Odum, N.; Geisler, C.; Wasik, M.A.; Woetmann, A. Regulatory t cells and immunodeficiency in mycosis fungoides and sezary syndrome. Leukemia 2012, 26, 424–432. [Google Scholar] [CrossRef]
- Kim, E.J.; Hess, S.; Richardson, S.K.; Newton, S.; Showe, L.C.; Benoit, B.M.; Ubriani, R.; Vittorio, C.C.; Junkins-Hopkins, J.M.; Wysocka, M.; et al. Immunopathogenesis and therapy of cutaneous t cell lymphoma. J. Clin. Investig. 2005, 115, 798–812. [Google Scholar]
- Izban, K.F.; Ergin, M.; Qin, J.Z.; Martinez, R.L.; Pooley, R.J.; Saeed, S.; Alkan, S. Constitutive expression of nf-kappa b is a characteristic feature of mycosis fungoides: Implications for apoptosis resistance and pathogenesis. Hum. Pathol. 2000, 31, 1482–1490. [Google Scholar] [CrossRef]
- Mao, X.; Orchard, G.; Mitchell, T.J.; Oyama, N.; Russell-Jones, R.; Vermeer, M.H.; Willemze, R.; van Doorn, R.; Tensen, C.P.; Young, B.D.; et al. A genomic and expression study of ap-1 in primary cutaneous t-cell lymphoma: Evidence for dysregulated expression of junb and jund in mf and ss. J. Cutan. Pathol. 2008, 35, 899–910. [Google Scholar]
- Van Kester, M.S.; Borg, M.K.; Zoutman, W.H.; Out-Luiting, J.J.; Jansen, P.M.; Dreef, E.J.; Vermeer, M.H.; van Doorn, R.; Willemze, R.; Tensen, C.P. A meta-analysis of gene expression data identifies a molecular signature characteristic for tumor-stage mycosis fungoides. J. Investig. Dermatol. 2012, 132, 2050–2059. [Google Scholar] [CrossRef]
- Tuyp, E.; Burgoyne, A.; Aitchison, T.; MacKie, R. A case-control study of possible causative factors in mycosis fungoides. Arch. Dermatol. 1987, 123, 196–200. [Google Scholar] [CrossRef]
- Lynge, E.; Afonso, N.; Kaerlev, L.; Olsen, J.; Sabroe, S.; Ahrens, W.; Eriksson, M.; Guenel, P.; Merletti, F.; Stengrevics, A.; et al. European multi-centre case-control study on risk factors for rare cancers of unknown aetiology. Eur. J. Cancer 2005, 41, 601–612. [Google Scholar] [CrossRef]
- Morales-Suarez-Varela, M.M.; Olsen, J.; Johansen, P.; Kaerlev, L.; Guenel, P.; Arveux, P.; Wingren, G.; Hardell, L.; Ahrens, W.; Stang, A.; et al. Occupational risk factors for mycosis fungoides: A European multicenter case-control study. J. Occup. Environ. Med. 2004, 46, 205–211. [Google Scholar] [CrossRef]
- Jahan-Tigh, R.R.; Huen, A.O.; Lee, G.L.; Pozadzides, J.V.; Liu, P.; Duvic, M. Hydrochlorothiazide and cutaneous t cell lymphoma: Prospective analysis and case series. Cancer 2013, 119, 825–831. [Google Scholar] [CrossRef]
- Hodak, E.; Klein, T.; Gabay, B.; Ben-Amitai, D.; Bergman, R.; Gdalevich, M.; Feinmesser, M.; Maron, L.; David, M. Familial mycosis fungoides: Report of 6 kindreds and a study of the hla system. J. Am. Acad. Dermatol. 2005, 52, 393–402. [Google Scholar] [CrossRef]
- Jackow, C.M.; McHam, J.B.; Friss, A.; Alvear, J.; Reveille, J.R.; Duvic, M. Hla-dr5 and dqb1*03 class ii alleles are associated with cutaneous t-cell lymphoma. J. Investig. Dermatol. 1996, 107, 373–376. [Google Scholar]
- Bonin, S.; Tothova, S.M.; Barbazza, R.; Brunetti, D.; Stanta, G.; Trevisan, G. Evidence of multiple infectious agents in mycosis fungoides lesions. Exp. Mol. Pathol. 2010, 89, 46–50. [Google Scholar] [CrossRef]
- Axelrod, P.I.; Lorber, B.; Vonderheid, E.C. Infections complicating mycosis fungoides and sezary syndrome. JAMA 1992, 267, 1354–1358. [Google Scholar] [CrossRef]
- Mirvish, E.D.; Pomerantz, R.G.; Geskin, L.J. Infectious agents in cutaneous t-cell lymphoma. J. Am. Acad. Dermatol. 2011, 64, 423–431. [Google Scholar] [CrossRef]
- Posner, L.E.; Fossieck, B.E., Jr.; Eddy, J.L.; Bunn, P.A., Jr. Septicemic complications of the cutaneous t-cell lymphomas. Am. J. Med. 1981, 71, 210–216. [Google Scholar] [CrossRef]
- Li, J.Y.; Horwitz, S.; Moskowitz, A.; Myskowski, P.L.; Pulitzer, M.; Querfeld, C. Management of cutaneous t cell lymphoma: New and emerging targets and treatment options. Cancer Manag. Res. 2012, 4, 75–89. [Google Scholar]
- Vermeer, M.H.; van Doorn, R.; Dukers, D.; Bekkenk, M.W.; Meijer, C.J.; Willemze, R. Cd8+ t cells in cutaneous t-cell lymphoma: Expression of cytotoxic proteins, fas ligand, and killing inhibitory receptors and their relationship with clinical behavior. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2001, 19, 4322–4329. [Google Scholar]
- Rook, A.H.; Kuzel, T.M.; Olsen, E.A. Cytokine therapy of cutaneous t-cell lymphoma: Interferons, interleukin-12, and interleukin-2. Hematol. Oncol. Clin. North Am. 2003, 17, 1435–1448. [Google Scholar] [CrossRef]
- Munir, S.; Andersen, G.H.; Woetmann, A.; Odum, N.; Becker, J.C.; Andersen, M.H. Cutaneous T cell lymphoma cells are targets for immune checkpoint ligand pd-l1-specific, cytotoxic T cells. Leukemia 2013. [Google Scholar] [CrossRef]
- Larsen, S.; Munir, S.; Woetmann, A.; Frøsig, T.; Odum, N.; Svane, I.; Becker, J.C.; Andersen, M.H. Functional characterization of Foxp3-specific spontaneous immune responses. Leukemi 2013. [Google Scholar] [CrossRef]
- Zackheim, H.S.; Koo, J.; LeBoit, P.E.; McCalmont, T.H.; Bowman, P.H.; Kashani-Sabet, M.; Jones, C.; Zehnder, J. Psoriasiform mycosis fungoides with fatal outcome after treatment with cyclosporine. J. Am. Acad. Dermatol. 2002, 47, 155–157. [Google Scholar] [CrossRef]
- Pielop, J.A.; Jones, D.; Duvic, M. Transient cd30+ nodal transformation of cutaneous t-cell lymphoma associated with cyclosporine treatment. Int. J. Dermatol. 2001, 40, 505–511. [Google Scholar] [CrossRef]
- Leroy, S.; Dubois, S.; Tenaud, I.; Chebassier, N.; Godard, A.; Jacques, Y.; Dreno, B. Interleukin-15 expression in cutaneous t-cell lymphoma (mycosis fungoides and sezary syndrome). Br. J. Dermatol. 2001, 144, 1016–1023. [Google Scholar] [CrossRef]
- Guenova, E.; Watanabe, R.; Teague, J.E.; Desimone, J.A.; Jiang, Y.; Dowlatshahi, M.; Schlapbach, C.; Schaekel, K.; Rook, A.H.; Tawa, M.; et al. Th2 cytokines from malignant cells suppress th1 responses and enforce a global th2 bias in leukemic cutaneous t-cell lymphoma. Clin. Cancer Res. 2013. [Google Scholar] [CrossRef]
- Krejsgaard, T.; Vetter-Kauczok, C.S.; Woetmann, A.; Lovato, P.; Labuda, T.; Eriksen, K.W.; Zhang, Q.; Becker, J.C.; Odum, N. Jak3- and jnk-dependent vascular endothelial growth factor expression in cutaneous t-cell lymphoma. Leukemia 2006, 20, 1759–1766. [Google Scholar] [CrossRef]
- Pedersen, I.H.; Willerslev-Olsen, A.; Vetter-Kauczok, C.; Krejsgaard, T.; Lauenborg, B.; Kopp, K.L.; Geisler, C.; Bonefeld, C.M.; Zhang, Q.; Wasik, M.A.; et al. Vascular endothelial growth factor receptor-3 expression in mycosis fungoides. Leuk. Lymphoma 2013, 54, 819–826. [Google Scholar] [CrossRef]
- Nielsen, M.; Nissen, M.H.; Gerwien, J.; Zocca, M.B.; Rasmussen, H.M.; Nakajima, K.; Ropke, C.; Geisler, C.; Kaltoft, K.; Odum, N. Spontaneous interleukin-5 production in cutaneous t-cell lymphoma lines is mediated by constitutively activated stat3. Blood 2002, 99, 973–977. [Google Scholar] [CrossRef]
- Sommer, V.H.; Clemmensen, O.J.; Nielsen, O.; Wasik, M.; Lovato, P.; Brender, C.; Eriksen, K.W.; Woetmann, A.; Kaestel, C.G.; Nissen, M.H.; et al. In vivo activation of stat3 in cutaneous t-cell lymphoma. Evidence for an antiapoptotic function of stat3. Leukemia 2004, 18, 1288–1295. [Google Scholar] [CrossRef]
- Kopp, K.L.; Ralfkiaer, U.; Gjerdrum, L.M.; Helvad, R.; Pedersen, I.H.; Litman, T.; Jonson, L.; Hagedorn, P.H.; Krejsgaard, T.; Gniadecki, R.; et al. Stat5-mediated expression of oncogenic mir-155 in cutaneous t-cell lymphoma. Cell Cycle 2013, 12, 1939–1937. [Google Scholar] [CrossRef]
- Krejsgaard, T.; Vetter-Kauczok, C.S.; Woetmann, A.; Kneitz, H.; Eriksen, K.W.; Lovato, P.; Zhang, Q.; Wasik, M.A.; Geisler, C.; Ralfkiaer, E.; et al. Ectopic expression of b-lymphoid kinase in cutaneous t-cell lymphoma. Blood 2009, 113, 5896–5904. [Google Scholar] [CrossRef]
- Kopp, K.L.; Kauczok, C.S.; Lauenborg, B.; Krejsgaard, T.; Eriksen, K.W.; Zhang, Q.; Wasik, M.A.; Geisler, C.; Ralfkiaer, E.; Becker, J.C.; et al. Cox-2-dependent pge(2) acts as a growth factor in mycosis fungoides (mf). Leukemia 2010, 24, 1179–1185. [Google Scholar] [CrossRef]
- Brender, C.; Nielsen, M.; Kaltoft, K.; Mikkelsen, G.; Zhang, Q.; Wasik, M.; Billestrup, N.; Odum, N. Stat3-mediated constitutive expression of socs-3 in cutaneous t-cell lymphoma. Blood 2001, 97, 1056–1062. [Google Scholar] [CrossRef]
- Brender, C.; Lovato, P.; Sommer, V.H.; Woetmann, A.; Mathiesen, A.M.; Geisler, C.; Wasik, M.; Odum, N. Constitutive socs-3 expression protects t-cell lymphoma against growth inhibition by ifnalpha. Leukemia 2005, 19, 209–213. [Google Scholar] [CrossRef]
- Gjerdrum, L.M.; Woetmann, A.; Odum, N.; Burton, C.M.; Rossen, K.; Skovgaard, G.L.; Ryder, L.P.; Ralfkiaer, E. Foxp3+ regulatory t cells in cutaneous t-cell lymphomas: Association with disease stage and survival. Leukemia 2007, 21, 2512–2518. [Google Scholar] [CrossRef]
- Hallermann, C.; Niermann, C.; Schulze, H.J. Regulatory t-cell phenotype in association with large cell transformation of mycosis fungoides. Eur. J. Haematol. 2007, 78, 260–263. [Google Scholar] [CrossRef]
- Kasprzycka, M.; Zhang, Q.; Witkiewicz, A.; Marzec, M.; Potoczek, M.; Liu, X.; Wang, H.Y.; Milone, M.; Basu, S.; Mauger, J.; et al. Gamma c-signaling cytokines induce a regulatory t cell phenotype in malignant cd4+ t lymphocytes. J. Immunol. 2008, 181, 2506–2512. [Google Scholar]
- Krejsgaard, T.; Gjerdrum, L.M.; Ralfkiaer, E.; Lauenborg, B.; Eriksen, K.W.; Mathiesen, A.M.; Bovin, L.F.; Gniadecki, R.; Geisler, C.; Ryder, L.P.; et al. Malignant tregs express low molecular splice forms of foxp3 in sezary syndrome. Leukemia 2008, 22, 2230–2239. [Google Scholar] [CrossRef]
- Wong, H.K.; Wilson, A.J.; Gibson, H.M.; Hafner, M.S.; Hedgcock, C.J.; Berger, C.L.; Edelson, R.L.; Lim, H.W. Increased expression of ctla-4 in malignant t-cells from patients with mycosis fungoides—Cutaneous t cell lymphoma. J. Investig. Dermatol. 2006, 126, 212–219. [Google Scholar] [CrossRef]
- Samimi, S.; Benoit, B.; Evans, K.; Wherry, E.J.; Showe, L.; Wysocka, M.; Rook, A.H. Increased programmed death-1 expression on cd4+ t cells in cutaneous t-cell lymphoma: Implications for immune suppression. Arch. Dermatol. 2010, 146, 1382–1388. [Google Scholar] [CrossRef]
- Kantekure, K.; Yang, Y.; Raghunath, P.; Schaffer, A.; Woetmann, A.; Zhang, Q.; Odum, N.; Wasik, M. Expression patterns of the immunosuppressive proteins pd-1/cd279 and pd-l1/cd274 at different stages of cutaneous t-cell lymphoma/mycosis fungoides. Am. J. Dermatopathol. 2012, 34, 126–128. [Google Scholar] [CrossRef]
- Abraham, R.M.; Zhang, Q.; Odum, N.; Wasik, M.A. The role of cytokine signaling in the pathogenesis of cutaneous t-cell lymphoma. Cancer Biol. Therapy 2011, 12, 1019–1022. [Google Scholar] [CrossRef]
- Berger, C.L.; Tigelaar, R.; Cohen, J.; Mariwalla, K.; Trinh, J.; Wang, N.; Edelson, R.L. Cutaneous t-cell lymphoma: Malignant proliferation of t-regulatory cells. Blood 2005, 105, 1640–1647. [Google Scholar] [CrossRef]
- Berger, C.L.; Hanlon, D.; Kanada, D.; Dhodapkar, M.; Lombillo, V.; Wang, N.; Christensen, I.; Howe, G.; Crouch, J.; El-Fishawy, P.; et al. The growth of cutaneous t-cell lymphoma is stimulated by immature dendritic cells. Blood 2002, 99, 2929–2939. [Google Scholar]
- Hofmann, B.; Odum, N.; Platz, P.; Ryder, L.P.; Svejgaard, A.; Neilsen, J.O. Immunological studies in acquired immunodeficiency syndrome. Functional studies of lymphocyte subpopulations. Scand. J. Immunol. 1985, 21, 235–243. [Google Scholar] [CrossRef]
- Gardner, J.M.; Evans, K.G.; Musiek, A.; Rook, A.H.; Kim, E.J. Update on treatment of cutaneous t-cell lymphoma. Curr. Opin. Oncol. 2009, 21, 131–137. [Google Scholar] [CrossRef]
- Ciree, A.; Michel, L.; Camilleri-Broet, S.; Jean Louis, F.; Oster, M.; Flageul, B.; Senet, P.; Fossiez, F.; Fridman, W.H.; Bachelez, H.; et al. Expression and activity of il-17 in cutaneous t-cell lymphomas (mycosis fungoides and sezary syndrome). Int. J. Cancer. 2004, 112, 113–120. [Google Scholar] [CrossRef]
- Krejsgaard, T.; Litvinov, I.V.; Wang, Y.; Xia, L.; Willerslev-Olsen, A.; Koralov, S.B.; Kopp, K.L.; Bonefeld, C.M.; Wasik, M.A.; Geisler, C.; et al. Elucidating the role of interleukin-17f in cutaneous t-cell lymphoma. Blood 2013. [Google Scholar] [CrossRef]
- Krejsgaard, T.; Ralfkiaer, U.; Clasen-Linde, E.; Eriksen, K.W.; Kopp, K.L.; Bonefeld, C.M.; Geisler, C.; Dabelsteen, S.; Wasik, M.A.; Ralfkiaer, E.; et al. Malignant cutaneous t-cell lymphoma cells express il-17 utilizing the jak3/stat3 signaling pathway. J. Investig. Dermatol. 2011, 131, 1331–1338. [Google Scholar] [CrossRef]
- MacKie, R.M. Initial event in mycosis fungoides of the skin is viral infection of epidermal langerhans cells. Lancet 1981, 2, 283–285. [Google Scholar] [CrossRef]
- van der Loo, E.M.; van Muijen, G.N.; van Vloten, W.A.; Beens, W.; Scheffer, E.; Meijer, C.J. C-type virus-like particles specifically localized in langerhans cells and related cells of skin and lymph nodes of patients with mycosis fungoides and sezary’s syndrome. A morphological and biochemical study. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1979, 31, 193–203. [Google Scholar] [CrossRef]
- Potoczna, N.; Boehncke, W.H.; Nestle, F.O.; Kuenzlen, C.; Sterry, W.; Burg, G.; Dummer, R. T-cell receptor beta variable region (v beta) usage in cutaneous t-cell lymphomas (ctcl) in comparison to normal and eczematous skin. J. Cutan. Pathol. 1996, 23, 298–305. [Google Scholar] [CrossRef]
- Yawalkar, N.; Ferenczi, K.; Jones, D.A.; Yamanaka, K.; Suh, K.Y.; Sadat, S.; Kupper, T.S. Profound loss of t-cell receptor repertoire complexity in cutaneous t-cell lymphoma. Blood 2003, 102, 4059–4066. [Google Scholar] [CrossRef]
- Cornberg, M.; Chen, A.T.; Wilkinson, L.A.; Brehm, M.A.; Kim, S.K.; Calcagno, C.; Ghersi, D.; Puzone, R.; Celada, F.; Welsh, R.M.; et al. Narrowed tcr repertoire and viral escape as a consequence of heterologous immunity. J. Clin. Investig. 2006, 116, 1443–1456. [Google Scholar] [CrossRef]
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type c retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous t-cell lymphoma. Proc. Natl. Acad. Sci. USA 1980, 77, 7415–7419. [Google Scholar] [CrossRef]
- Yoshida, M.; Miyoshi, I.; Hinuma, Y. Isolation and characterization of retrovirus from cell lines of human adult t-cell leukemia and its implication in the disease. Proc. Natl. Acad. Sci. USA 1982, 79, 2031–2035. [Google Scholar] [CrossRef]
- Barre-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; et al. Isolation of a t-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (aids). Science 1983, 220, 868–871. [Google Scholar]
- Zucker-Franklin, D.; Pancake, B.A. The role of human t-cell lymphotropic viruses (htlv-i and ii) in cutaneous t-cell lymphomas. Semin. Dermatol. 1994, 13, 160–165. [Google Scholar]
- Pancake, B.A.; Zucker-Franklin, D.; Coutavas, E.E. The cutaneous T cell lymphoma, mycosis fungoides, is a human T cell lymphotropic virus-associated disease. A study of 50 patients. J. Clin. Investig. 1995, 95, 547–554. [Google Scholar] [CrossRef]
- Lessin, S.R.; Rook, A.H.; Li, G.; Wood, G.S. Htlv-i and ctcl: The link is missing. J. Investig. Dermatol. 1996, 107, 783–784. [Google Scholar]
- Wood, G.S.; Salvekar, A.; Schaffer, J.; Crooks, C.F.; Henghold, W.; Fivenson, D.P.; Kim, Y.H.; Smoller, B.R. Evidence against a role for human t-cell lymphotrophic virus type i (htlv-i) in the pathogenesis of American cutaneous t-cell lymphoma. J. Investig. Dermatol. 1996, 107, 301–307. [Google Scholar]
- Bazarbachi, A.; Soriano, V.; Pawson, R.; Vallejo, A.; Moudgil, T.; Matutes, E.; Peries, J.; Molina, A.; de The, H.; Schulz, T.F.; et al. Mycosis fungoides and sezary syndrome are not associated with htlv-i infection: An international study. Br. J. Haematol. 1997, 98, 927–933. [Google Scholar] [CrossRef]
- Erkek, E.; Sahin, S.; Atakan, N.; Kocagoz, T.; Olut, A.; Gokoz, A. Examination of mycosis fungoides for the presence of epstein-barr virus and human herpesvirus-6 by polymerase chain reaction. J. Eur. Acad. Dermatol. Venereol 2001, 15, 422–426. [Google Scholar] [CrossRef]
- Kreuter, A.; Bischoff, S.; Skrygan, M.; Wieland, U.; Brockmeyer, N.H.; Stucker, M.; Altmeyer, P.; Gambichler, T. High association of human herpesvirus 8 in large-plaque parapsoriasis and mycosis fungoides. Arch. Dermatol. 2008, 144, 1011–1016. [Google Scholar] [CrossRef]
- Gupta, R.K.; Ramble, J.; Tong, C.Y.; Whittaker, S.; MacMahon, E. Cytomegalovirus seroprevalence is not higher in patients with mycosis fungoides/sezary syndrome. Blood 2006, 107, 1241–1242. [Google Scholar]
- Herne, K.L.; Talpur, R.; Breuer-McHam, J.; Champlin, R.; Duvic, M. Cytomegalovirus seropositivity is significantly associated with mycosis fungoides and sezary syndrome. Blood 2003, 101, 2132–2136. [Google Scholar] [CrossRef]
- Nagore, E.; Ledesma, E.; Collado, C.; Oliver, V.; Perez-Perez, A.; Aliaga, A. Detection of epstein-barr virus and human herpesvirus 7 and 8 genomes in primary cutaneous t- and b-cell lymphomas. Br. J. Dermatol. 2000, 143, 320–323. [Google Scholar] [CrossRef]
- Abrams, J.T.; Vonderheid, E.C.; Kolbe, S.; Appelt, D.M.; Arking, E.J.; Balin, B.J. Sezary t-cell activating factor is a chlamydia pneumoniae-associated protein. Clin. Diagn. Lab. Immunol. 1999, 6, 895–905. [Google Scholar]
- Abrams, J.T.; Balin, B.J.; Vonderheid, E.C. Association between sezary T cell-activating factor, chlamydia pneumoniae, and cutaneous T cell lymphoma. Ann. N.Y. Acad. Sci. 2001, 941, 69–85. [Google Scholar]
- Tothova, S.M.; Bonin, S.; Trevisan, G.; Stanta, G. Mycosis fungoides: Is it a borrelia burgdorferi-associated disease? Br. J. Cancer 2006, 94, 879–883. [Google Scholar] [CrossRef]
- Rossler, M.J.; Rappl, G.; Muche, M.; Hasselmann, D.O.; Sterry, W.; Tilgen, W.; Reinhold, U. No evidence of skin infection with chlamydia pneumoniae in patients with cutaneous T cell lymphoma. Clin. Microbiol. Infect. Off. Public. Eur. Soc. Clin. Microbiol. Infect. Dis. 2003, 9, 721–723. [Google Scholar]
- Ponzoni, M.; Ferreri, A.J.; Mappa, S.; Pasini, E.; Govi, S.; Facchetti, F.; Fanoni, D.; Tucci, A.; Vino, A.; Doglioni, C.; et al. Prevalence of borrelia burgdorferi infection in a series of 98 primary cutaneous lymphomas. Oncologist 2011, 16, 1582–1588. [Google Scholar] [CrossRef]
- Hotz, C.; Ingen-Housz-Oro, S.; tran van Nhieu, J.; Charlier, C.; Foulet, F.; Rahmouni, A.; Zegai, B.; Duong, T.A.; Wolkenstein, P.; Bagot, M.; et al. Pulmonary cryptococcoma in a patient with sezary syndrome treated with alemtuzumab. Eur. J. Dermatol. 2011, 21, 1018–1020. [Google Scholar]
- Poonawalla, T.; Diwan, H.; Duvic, M. Mycosis fungoides with coccidioidomycosis. Clin. Lymphoma Myeloma 2006, 7, 148–150. [Google Scholar] [CrossRef]
- Duvic, M.; Feasel, A.M.; Schwartz, C.A.; Cather, J.C. Enterococcal eschars in cutaneous t-cell lymphoma tumors: A distinct clinical entity. Clin. Lymphoma 2000, 1, 141–145. [Google Scholar] [CrossRef]
- Jackow, C.M.; Cather, J.C.; Hearne, V.; Asano, A.T.; Musser, J.M.; Duvic, M. Association of erythrodermic cutaneous t-cell lymphoma, superantigen-positive staphylococcus aureus, and oligoclonal t-cell receptor v beta gene expansion. Blood 1997, 89, 32–40. [Google Scholar]
- Baser, S.; Onn, A.; Lin, E.; Morice, R.C.; Duvic, M. Pulmonary manifestations in patients with cutaneous t-cell lymphomas. Cancer 2007, 109, 1550–1555. [Google Scholar] [CrossRef]
- Ortega, E.; Abriouel, H.; Lucas, R.; Galvez, A. Multiple roles of staphylococcus aureus enterotoxins: Pathogenicity, superantigenic activity, and correlation to antibiotic resistance. Toxins 2010, 2, 2117–2131. [Google Scholar] [CrossRef]
- Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Staphylococcal enterotoxins. Toxins 2010, 2, 2177–2197. [Google Scholar] [CrossRef]
- Tokura, Y.; Heald, P.W.; Yan, S.L.; Edelson, R.L. Stimulation of cutaneous t-cell lymphoma cells with superantigenic staphylococcal toxins. J. Investig. Dermatol. 1992, 98, 33–37. [Google Scholar]
- Irwin, M.J.; Hudson, K.R.; Ames, K.T.; Fraser, J.D.; Gascoigne, N.R. T-cell receptor beta-chain binding to enterotoxin superantigens. Immunol. Rev. 1993, 131, 61–78. [Google Scholar] [CrossRef]
- Linnemann, T.; Gellrich, S.; Lukowsky, A.; Mielke, A.; Audring, H.; Sterry, W.; Walden, P. Polyclonal expansion of T cells with the tcr v beta type of the tumour cell in lesions of cutaneous t-cell lymphoma: Evidence for possible superantigen involvement. Br. J. Dermatol. 2004, 150, 1013–1017. [Google Scholar] [CrossRef]
- McCormack, J.E.; Callahan, J.E.; Kappler, J.; Marrack, P.C. Profound deletion of mature T cells in vivo by chronic exposure to exogenous superantigen. J. Immunol. 1993, 150, 3785–3792. [Google Scholar]
- Vonderheid, E.C.; Boselli, C.M.; Conroy, M.; Casaus, L.; Espinoza, L.C.; Venkataramani, P.; Bigler, R.D.; Hou, J.S. Evidence for restricted vbeta usage in the leukemic phase of cutaneous T cell lymphoma. J. Investig. Dermatol. 2005, 124, 651–661. [Google Scholar] [CrossRef]
- Van der Fits, L.; Sandberg, Y.; Darzentas, N.; Zoutman, W.H.; Tielemans, D.; Wolvers-Tettero, I.L.; Vermeer, M.H.; Langerak, A.W. A restricted clonal t-cell receptor alphabeta repertoire in sezary syndrome is indicative of superantigenic stimulation. Br. J. Dermatol. 2011, 165, 78–84. [Google Scholar] [CrossRef]
- Ni, X.; Hazarika, P.; Zhang, C.; Talpur, R.; Duvic, M. Fas ligand expression by neoplastic t lymphocytes mediates elimination of cd8+ cytotoxic t lymphocytes in mycosis fungoides: A potential mechanism of tumor immune escape? Clin. Cancer Res. 2001, 7, 2682–2692. [Google Scholar]
- Wu, J.; Nihal, M.; Siddiqui, J.; Vonderheid, E.C.; Wood, G.S. Low fas/cd95 expression by ctcl correlates with reduced sensitivity to apoptosis that can be restored by fas upregulation. J. Investig. Dermatol. 2009, 129, 1165–1173. [Google Scholar] [CrossRef]
- Lauenborg, B.; Kopp, K.; Krejsgaard, T.; Eriksen, K.W.; Geisler, C.; Dabelsteen, S.; Gniadecki, R.; Zhang, Q.; Wasik, M.A.; Woetmann, A.; et al. Programmed cell death-10 enhances proliferation and protects malignant T cells from apoptosis. APMIS 2010, 118, 719–728. [Google Scholar] [CrossRef]
- Nielsen, M.; Kaestel, C.G.; Eriksen, K.W.; Woetmann, A.; Stokkedal, T.; Kaltoft, K.; Geisler, C.; Ropke, C.; Odum, N. Inhibition of constitutively activated stat3 correlates with altered bcl-2/bax expression and induction of apoptosis in mycosis fungoides tumor cells. Leukemia 1999, 13, 735–738. [Google Scholar] [CrossRef]
- Thurber, S.E.; Zhang, B.; Kim, Y.H.; Schrijver, I.; Zehnder, J.; Kohler, S. T-cell clonality analysis in biopsy specimens from two different skin sites shows high specificity in the diagnosis of patients with suggested mycosis fungoides. J. Am. Acad. Dermatol. 2007, 57, 782–790. [Google Scholar] [CrossRef]
- Gorochov, G.; Bachelez, H.; Cayuela, J.M.; Legac, E.; Laroche, L.; Dubertret, L.; Sigaux, F. Expression of v beta gene segments by sezary cells. J. Investig. Dermatol. 1995, 105, 56–61. [Google Scholar]
- Longley, J.; Tyrrell, L.; Lu, S.Z.; Farrell, J.; Ding, T.G.; Yan, S.; Sallee, D.; Heald, P.; Berger, C.; Tigelaar, R.; et al. Malignant and normal T cells show random use of t-cell receptor alpha chain variable regions in patients with cutaneous t-cell lymphoma. J. Investig. Dermatol. 1995, 105, 62–64. [Google Scholar] [CrossRef]
- Morgan, S.M.; Hodges, E.; Mitchell, T.J.; Harris, S.; Whittaker, S.J.; Smith, J.L. Molecular analysis of t-cell receptor beta genes in cutaneous t-cell lymphoma reveals jbeta1 bias. J. Investig. Dermatol. 2006, 126, 1893–1899. [Google Scholar] [CrossRef]
- Klemke, C.D.; Brenner, D.; Weiss, E.M.; Schmidt, M.; Leverkus, M.; Gulow, K.; Krammer, P.H. Lack of t-cell receptor-induced signaling is crucial for cd95 ligand up-regulation and protects cutaneous t-cell lymphoma cells from activation-induced cell death. Cancer Res. 2009, 69, 4175–4183. [Google Scholar] [CrossRef]
- Edelman, J.; Meyerson, H.J. Diminished cd3 expression is useful for detecting and enumerating sezary cells. Am. J. Clin. Pathol. 2000, 114, 467–477. [Google Scholar]
- Morice, W.G.; Katzmann, J.A.; Pittelkow, M.R.; El-Azhary, R.A.; Gibson, L.E.; Hanson, C.A. A comparison of morphologic features, flow cytometry, tcr-vbeta analysis, and tcr-pcr in qualitative and quantitative assessment of peripheral blood involvement by sezary syndrome. Am. J. Clin. Pathol. 2006, 125, 364–374. [Google Scholar]
- Woetmann, A.; Lovato, P.; Eriksen, K.W.; Krejsgaard, T.; Labuda, T.; Zhang, Q.; Mathiesen, A.M.; Geisler, C.; Svejgaard, A.; Wasik, M.A.; et al. Nonmalignant T cells stimulate growth of t-cell lymphoma cells in the presence of bacterial toxins. Blood 2007, 109, 3325–3332. [Google Scholar] [CrossRef]
- Fraser, J.D.; Proft, T. The bacterial superantigen and superantigen-like proteins. Immunol. Rev. 2008, 225, 226–243. [Google Scholar] [CrossRef]
- Odum, N.; Ledbetter, J.A.; Martin, P.; Geraghty, D.; Tsu, T.; Hansen, J.A.; Gladstone, P. Homotypic aggregation of human cell lines by hla class ii-, class ia- and hla-g-specific monoclonal antibodies. Eur. J. Immunol. 1991, 21, 2121–2131. [Google Scholar] [CrossRef]
- Nielsen, M.; Odum, N.; Bendtzen, K.; Ryder, L.P.; Jakobsen, B.K.; Svejgaard, A. Mhc class ii molecules regulate growth in human T cells. Exp. Clin. Immunogenet. 1994, 11, 23–32. [Google Scholar]
- Odum, N.; Kanner, S.B.; Ledbetter, J.A.; Svejgaard, A. Mhc class ii molecules deliver costimulatory signals in human T cells through a functional linkage with il-2-receptors. J. Immunol. 1993, 150, 5289–5298. [Google Scholar]
- Odum, N.; Martin, P.J.; Schieven, G.L.; Hansen, J.A.; Ledbetter, J.A. Signal transduction by hla class ii antigens expressed on activated T cells. Eur. J. Immunol. 1991, 21, 123–129. [Google Scholar] [CrossRef]
- Kanner, S.B.; Grosmaire, L.S.; Blake, J.; Schieven, G.L.; Masewicz, S.; Odum, N.; Ledbetter, J.A. Zap-70 and p72syk are signaling response elements through mhc class ii molecules. Tissue Antigens 1995, 46, 145–154. [Google Scholar] [CrossRef]
- Kanner, S.B.; Odum, N.; Grosmaire, L.; Masewicz, S.; Svejgaard, A.; Ledbetter, J.A. Superantigen and hla-dr ligation induce phospholipase-c gamma 1 activation in class ii+ T cells. J. Immunol. 1992, 149, 3482–3488. [Google Scholar]
- Odum, N.; Martin, P.J.; Schieven, G.L.; Norris, N.A.; Grosmaire, L.S.; Hansen, J.A.; Ledbetter, J.A. Signal transduction by hla-dr is mediated by tyrosine kinase(s) and regulated by cd45 in activated T cells. Hum. Immunol. 1991, 32, 85–94. [Google Scholar]
- Odum, N.; Martin, P.J.; Schieven, G.L.; Masewicz, S.; Hansen, J.A.; Ledbetter, J.A. Hla-dr molecules enhance signal transduction through the cd3/ti complex in activated T cells. Tissue Antigens 1991, 38, 72–77. [Google Scholar] [CrossRef]
- Daniel, D.; Meyer-Morse, N.; Bergsland, E.K.; Dehne, K.; Coussens, L.M.; Hanahan, D. Immune enhancement of skin carcinogenesis by cd4+ T cells. J. Exp. Med. 2003, 197, 1017–1028. [Google Scholar] [CrossRef]
- Talpur, R.; Bassett, R.; Duvic, M. Prevalence and treatment of Staphylococcus aureus colonization in patients with mycosis fungoides and sezary syndrome. Br. J. Dermatol. 2008, 159, 105–112. [Google Scholar] [CrossRef]
- Tokura, Y.; Yagi, H.; Ohshima, A.; Kurokawa, S.; Wakita, H.; Yokote, R.; Shirahama, S.; Furukawa, F.; Takigawa, M. Cutaneous colonization with Staphylococci influences the disease activity of sezary syndrome: A potential role for bacterial superantigens. Br. J. Dermatol. 1995, 133, 6–12. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Willerslev-Olsen, A.; Krejsgaard, T.; Lindahl, L.M.; Bonefeld, C.M.; Wasik, M.A.; Koralov, S.B.; Geisler, C.; Kilian, M.; Iversen, L.; Woetmann, A.; et al. Bacterial Toxins Fuel Disease Progression in Cutaneous T-Cell Lymphoma. Toxins 2013, 5, 1402-1421. https://doi.org/10.3390/toxins5081402
Willerslev-Olsen A, Krejsgaard T, Lindahl LM, Bonefeld CM, Wasik MA, Koralov SB, Geisler C, Kilian M, Iversen L, Woetmann A, et al. Bacterial Toxins Fuel Disease Progression in Cutaneous T-Cell Lymphoma. Toxins. 2013; 5(8):1402-1421. https://doi.org/10.3390/toxins5081402
Chicago/Turabian StyleWillerslev-Olsen, Andreas, Thorbjørn Krejsgaard, Lise M. Lindahl, Charlotte Menne Bonefeld, Mariusz A. Wasik, Sergei B. Koralov, Carsten Geisler, Mogens Kilian, Lars Iversen, Anders Woetmann, and et al. 2013. "Bacterial Toxins Fuel Disease Progression in Cutaneous T-Cell Lymphoma" Toxins 5, no. 8: 1402-1421. https://doi.org/10.3390/toxins5081402