Staphylococcal enterotoxins in the Etiopathogenesis of Mucosal Autoimmunity within the Gastrointestinal Tract

Abstract
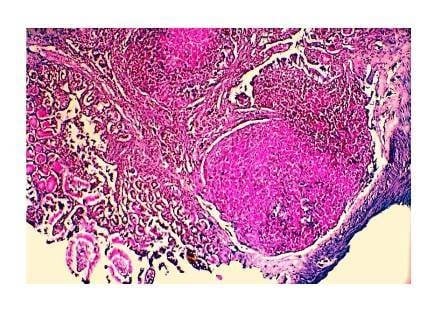
:
{kind=link}
{kind=link}
{kind=link}
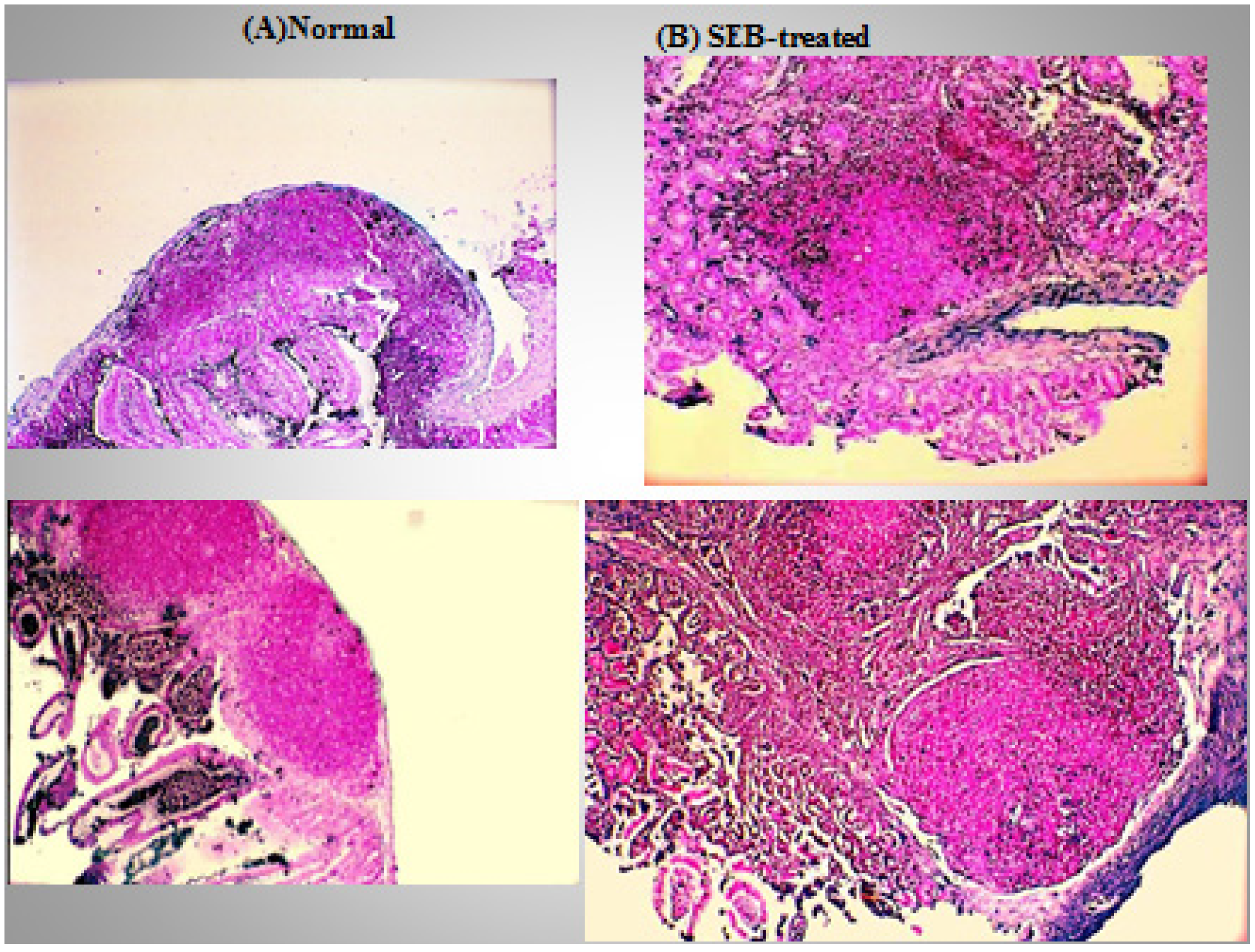{kind=link}
1. The Staphylococcal enterotoxins and Disease
2. Immunity within the Gut Associated Lymphoid Tissue
Superantigenic Response in the Gastrointestinal Mucosa
3. Immunopathology of the Staphylococcal enterotoxins within the Intestinal Mucosa


4. Staphylococcal enterotoxins and Immune Tolerance
5. Staphylococcal enterotoxins in Inflammatory Bowel Diseases
6. Conclusions
Acknowledgments
Conflicts of Interest
Declare
References
- Friedman, S.M.; Tumang, J.R.; Crow, M.K. Microbial superantigens as etiopathogenic agents in autoimmunity. Rheum. Dis. Clin. N. Am. 1993, 19, 207–222. [Google Scholar]
- Friedman, S.M.; Posnett, D.N.; Tumang, J.R.; Cole, B.C.; Crow, M.K. A potential role for microbial superantigens in the pathogenesis of systemic autoimmune disease. Arthr. Rheum. 1991, 34, 468–480. [Google Scholar] [CrossRef]
- Lee, J.H.; Lin, Y.T.; Yang, Y.H.; Wang, L.C.; Chiang, B.L. Increased levels of serum-specific immunoglobulin E to staphylococcal enterotoxin A and B in patients with allergic rhinitis and bronchial asthma. Int. Arch. Allergy Immunol. 2005, 138, 305–311. [Google Scholar] [CrossRef]
- Bachert, C.; Gevaert, P.; van Cauwenberge, P. Staphylococcus aureus enterotoxins: A key in airway disease? Allergy 2002, 57, 480–487. [Google Scholar] [CrossRef]
- Orfali, R.L.; Sato, M.N.; Takaoka, R.; Azor, M.H.; Rivitti, E.A.; Hanifin, J.M.; Aoki, V. Atopic dermatitis in adults: Evaluation of peripheral blood mononuclear cells proliferation response to Staphylococcus aureus enterotoxins A and B and analysis of interleukin-18 secretion. Exp. Dermatol. 2009, 18, 628–633. [Google Scholar] [CrossRef]
- Yudate, T.; Yamada, H.; Tezuka, T. Role of staphylococcal enterotoxins in pathogenesis of atopic dermatitis: Growth and expression of T cell receptro Vbeta of peripheral blood mononuclear cells stimulated by enterotoxins A and B. J. Dermatol. Sci. 1996, 13, 63–70. [Google Scholar] [CrossRef]
- Yang, P.-C.; Liu, T.; Wang, B.-Q.; Zhang, T.-Y.; An, Z.-Y.; Zheng, P.-Y.; Tian, D.-F. Rhinosinusitis derived Staphylococcal enterotoxin B possibly associates with pathogenesis of ulcerative colitis. BMC Gastroenterol. 2005, 5. [Google Scholar] [CrossRef] [Green Version]
- Rossi, R.E.; Monasterolo, G. Prevalence of serum IgE antibodies to the Staphylococcus aureus enterotoxins (SAE, SEB, SEC, SED, TSST-1) in patients with persistent allergic rhinitis. Int. Arch. Allergy Immunol. 2004, 133, 261–266. [Google Scholar] [CrossRef]
- Forbes-Blom, E.; Camberis, M.; Prout, M.; Tang, S.C.; le Gros, G. Staphylococcal-derived superantigen enhances peanut induced Th2 responses in the skin. J. Br. Soc. Allergy Clin. Immunol. 2012, 42, 305–314. [Google Scholar] [CrossRef]
- Bergdoll, M.S. Importance of Staphylococci that produce nanogram quantities of enterotoxin. Zbl. Bakt. 1995, 282, 1–6. [Google Scholar] [CrossRef]
- Breckinridge, J.C.; Bergdoll, M.S. Outbreak of food-borne gastroenteritis due to a coagulase-negative enterotoxin-producing staphylococcus. N. Engl. J. Med. 1971, 284, 541–543. [Google Scholar] [CrossRef]
- Marrack, P.; Kappler, J. The staphylococcal enterotoxins and their relatives. Science 1990, 248, 705–711. [Google Scholar]
- Krakauer, T. Update on staphylococcal superantigen-induced signaling pathways and therapeutic interventions. Toxins 2013, 5, 1629–1654. [Google Scholar] [CrossRef]
- Jarraud, S.; Peyrat, M.A.; Lim, A.; Tristan, A.; Bes, M.; Mougel, C.; Etienne, J.; Vandenesch, F.; Bonneville, M.; Lina, G. EGC, a highly prevalent operon of enterotoxin gene, forms a putative nursery of superantigens in Staphylococcus aureus. J. Immunol. 2001, 166, 669–677. [Google Scholar] [CrossRef]
- Su, Y.C.; Wong, A.C. Identification and purification of a new staphylococcal enterotoxin, H. Appl. Environ. Microbiol. 1995, 61, 1438–1443. [Google Scholar]
- Chiang, Y.C.; Chang, L.T.; Lin, C.W.; Yang, C.Y.; Tsen, H.Y. PCR primers for the detection of staphylococcal enterotoxins K, L, and M and survey of staphylococcal enterotoxin types in Staphylococcus aureus isolates from food poisoning cases in Taiwan. J. Food Prot. 2006, 69, 1072–1079. [Google Scholar]
- Orwin, P.M.; Leung, D.Y.; Donahue, H.L.; Novick, R.P.; Schlievert, P.M. Biochemical and biological properties of Staphylococcal enterotoxin K. Infect. Immun. 2001, 69, 360–366. [Google Scholar] [CrossRef]
- Chiang, Y.-C.; Liao, W.-W.; Fan, C.-M.; Pai, W.-Y.; Chiou, C.-S.; Tsen, H.-Y. PCR detection of Staphylococcal enterotoxins (SEs) N, O, P, Q, R, U, and survey of SE types in Staphylococcus aureus isolates from food-poisoning cases in Taiwan. Int. J. Food Microbiol. 2008, 121, 66–73. [Google Scholar] [CrossRef]
- Omoe, K.; Imanishi, K.; Hu, D.-L.; Kato, H.; Takahashi-Omoe, H.; Nakane, A.; Uchiyama, T.; Shinagawa, K. Biological properties of staphylococcal enterotoxin-like toxin type R. Infect. Immun. 2004, 72, 3664–3667. [Google Scholar] [CrossRef]
- Letertre, C.; Perelle, S.; Dilasser, F.; Fach, P. Identification of a new putative enterotoxin SEU encoded by the egc cluster of Staphylococcus aureus. J. Appl. Microbiol. 2003, 95, 38–43. [Google Scholar] [CrossRef]
- Asao, T.; Kumeda, Y.; Kawai, T.; Shibata, T.; Oda, H.; Haruki, K.; Nakazawa, H.; Kozaki, S. An extensive outbreak of staphylococcal food poisoning due to low-fat milk in Japan: Estimation of enterotoxin A in the incriminated milk and powdered skim milk. Epidemiol. Infect. 2003, 130, 33–40. [Google Scholar] [CrossRef]
- Becker, K.; Friedrich, A.W.; Lubritz, G.; Weilert, M.; Peters, G.; von Eiff, C. Prevalence of genes encoding pyrogenic toxin superantigens and exfoliative toxins among strains of Staphylococcus aureus isolated from blood and nasal specimens. J. Clin. Microbiol. 2003, 41, 1434–1439. [Google Scholar] [CrossRef]
- Do Carmo, L.S.; Cummings, C.; Linardi, V.R.; Dias, R.S.; De Souza, J.M.; De Sena, M.J.; Dos Santos, D.A.; Shupp, J.W.; Pereira, R.K.P.; Jett, M. A case study of a massive staphylococcal food poisoning incident. Foodborne Pathog. Dis. 2004, 1, 241–246. [Google Scholar] [CrossRef]
- Evenson, M.L.; Hinds, M.W.; Bernstein, R.S.; Bergdoll, M.S. Estimation of human dose of staphylococcal enterotoxin A from a large outbreak of staphylococcal food poisoning involving chocolate milk. Int. J. Food Microbiol. 1988, 7, 311–316. [Google Scholar] [CrossRef]
- Levine, W.C.; Bennett, R.W.; Choi, Y.; Henning, K.J.; Rager, J.R.; Hendricks, K.A.; Hopkins, D.P.; Gunn, R.A.; Griffin, P.M. Staphylococcal food poisoning caused by imported canned mushrooms. J. Infect. Dis. 1996, 173, 1263–1267. [Google Scholar] [CrossRef]
- Hennekinne, J.-A.; Ostyn, A.; Guillier, F.; Herbin, S.; Prufer, A.-L.; Dragacci, S. How should staphylococcal food poisoning outbreaks be characterized? Toxins 2010, 2, 2106–2116. [Google Scholar] [CrossRef]
- Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Staphylococcal enterotoxins. Toxins 2010, 2, 2177–2197. [Google Scholar] [CrossRef]
- Omoe, K.; Hu, D.-L.; Takahashi-Omoe, H.; Nakane, A.; Shinagawa, K. Comprehensive analysis of classical and newly described staphlyococcal superantigenic toxin genes in Staphylococcus aureus isolates. FEMS Microbiol. Lett. 2005, 246, 191–198. [Google Scholar] [CrossRef]
- Yarwood, J.M.; McCormick, J.K.; Paustian, M.L.; Orwin, P.M.; Kapur, V.; Schlievert, P.M. Characterization and expression analysis of Staphylococcus aureus pathogenicity island 3. Implications for the evoluation of staphylococcal pathogenicity islands. Biol. Chem. 2002, 277, 13138–13147. [Google Scholar]
- Fleischer, B.; Schrezenmeier, H. T cell stimulation by staphylococcal enterotoxins. Clonally variable response and requirement for major histocompatibility complex class II molecules on accessory or target cells. J. Exp. Med. 1988, 167, 1697–1707. [Google Scholar] [CrossRef]
- Kappler, J.; Kotzin, B.; Herron, L.; Gelfand, E.W.; Bigler, R.D.; Boylston, A.; Carrel, S.; Posnett, D.N.; Choi, Y.; Marrack, P. V beta-specific stimulation of human T cells by staphylococcal toxins. Science 1989, 244, 811–813. [Google Scholar]
- Mead, P.S.; Slutsker, L.; Dietz, V.; McCaig, L.F.; Bresee, J.S.; Shapiro, C.; Griffin, P.M.; Tauxe, R.V. Food-related illness and death in the United States. Emerg. Infect. Dis. 1999, 5, 607–625. [Google Scholar] [CrossRef]
- Zorgani, A.; Essery, S.D.; Madani, O.A.; Bentley, A.J.; James, V.S.; MacKenzie, D.A.C.; Keeling, J.W.; Rambaud, C.; Hilton, J.; Blackwell, C.C. Detection of pyrogenic toxins of Staphylococcus aureus in sudden infant death syndrome. FEMS Immunol. Med. Microbiol. 1999, 25, 103–108. [Google Scholar]
- Principato, M. Infant Formulas and Feeding:Risks Associated with Staphylococcus aureus and Its Enterotoxins. In Dietary and Nutritional Aspects of Bottle Feeding; Preedy, V., Ed.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2014. [Google Scholar]
- Lawrynowicz-Paciorek, M.; Kochman, M.; Piekarska, K.; Grochowska, A.; Windyga, B. The distribution of enterotoxin and enterotoxin-like genes in Staphylococcus aureus strains isolated from nasal carriers and food samples. Int. J. Food Microbiol. 2007, 117, 319–323. [Google Scholar] [CrossRef]
- Holmberg, S.D.; Blake, P.A. Staphylococcal food poisoning in the United States. New facts and old misconceptions. JAMA 1984, 251, 487–489. [Google Scholar] [CrossRef]
- Udo, E.E.; Al-Bustan, M.A.; Jacob, L.E.; Chugh, T.D. Enterotoxin production by coagulase-negativestaphylococci in restaurant workers from Kuwait City may be a potential cause of food poisoning. J. Med. Microbiol. 1999, 48, 819–823. [Google Scholar] [CrossRef]
- Le Loir, Y.; Baron, F.; Gautier, M. Staphylococcus aureus and food poisoning. Genet. Mol. Res. 2003, 2, 63–76. [Google Scholar]
- Read, R., Jr.; Bradshaw, J. Staphylococcal enterotoxin B thermal inactivation in milk. J. Dairy Sci. 1966, 49, 202–203. [Google Scholar] [CrossRef]
- Principato, M.; Boyle, T.; Njoroge, J.; Jones, R.L.; O’Donnell, M. Effect of thermal processing during yogurt production upon the detection of staphylococcal enterotoxin B. J. Food Protect. 2009, 72, 2212–2216. [Google Scholar]
- Raj, H.D.; Bergdoll, M.S. Effect of enterotoxin B on human volunteers. J. Bacteriol. 1969, 98, 833–834. [Google Scholar]
- Ikeda, T.; Tamate, N.; Yamaguchi, K.; Makino, S. Mass outbreak of food poisoning disease caused by small amounts of staphylococcal enterotoxins A and H. Appl. Environ. Microbiol. 2005, 71, 2793–2795. [Google Scholar] [CrossRef]
- Balaban, N.; Rasooly, A. Staphylococcal enterotoxins. Int. J. Food Microbiol. 2000, 61, 1–10. [Google Scholar] [CrossRef]
- Dinges, M.M.; Orwin, P.M.; Schlievert, P.M. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 2000, 13, 16–34. [Google Scholar] [CrossRef]
- Danielsen, E.M.; Hansen, G.H.; Karlsdottir, E. Staphylococcus aureus enterotoxins A and B: Binding to the enterocyte brush border and uptake by perturbation of the apical endocytic membrane traffic. Histochem. Cell Biol. 2013, 139, 513–524. [Google Scholar] [CrossRef]
- Edwards, L.A.; O’Neill, C.; Furman, M.A.; Hicks, S.; Torrente, F.; Pérez-Machado, M.; Wellington, E.M.; Phillips, A.D.; Murch, S.H. Enterotoxin-producing staphylococci cause intestinal inflammation by a combination of direct epithelial cytopathy and superantigen-mediated T cell activation. Inflamm. Bowel Dis. 2012, 18, 624–640. [Google Scholar] [CrossRef]
- Hu, D.-L.; Zhu, G.; Mori, F.; Omoe, K.; Okada, M.; Wakabayashi, K.; Kaneko, S.; Shinagawa, K.; Nakane, A. Staphylococcal enterotoxin induces emesis through increasing serotonin release in intestine and it is downregulated by cannabinoid receptor 1. Cell Microbiol. 2007, 9, 2267–2277. [Google Scholar] [CrossRef]
- Luongo, D.; D’Arienzo, R.; Bergamo, P.; Maurano, F.; Rossi, M. Immunomodulation of gut-associated lymphoid tissue: Current perspectives. Int. Rev. Immunol. 2009, 28, 446–464. [Google Scholar] [CrossRef]
- van Wijk, F.; Cheroutre, H. Intestinal T cells: Facing the mucosal immune dilemma with synergy and diversity. Semin. Immunol. 2009, 21, 130–138. [Google Scholar] [CrossRef]
- Strober, W.; Kelsall, B.; Marth, T. Oral tolerance. J. Clin. Immunol. 1998, 18, 1–30. [Google Scholar] [CrossRef]
- LeFrancois, L.; Lynn, P. Basic Aspects of Intraepithelial Lymphocytes Immunobiology; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Coombes, J.L.; Siddiqui, K.R.; Arancibia-Carcamo, C.V.; Hall, J.; Sun, C.M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef]
- Sun, C.M.; Hall, J.A.; Blank, R.B.; Bouladoux, N.; Oukka, M.; Mora, J.R.; Belkaid, Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 2007, 204, 1775–1785. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Li, M.O.; Flavell, R.A. TGF-beta: A master of all T cell trades. Cell 2008, 134, 392–404. [Google Scholar] [CrossRef]
- Marie, J.C.; Liggitt, D.; Rudensky, A.Y. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity 2006, 25, 441–454. [Google Scholar] [CrossRef]
- Aggeler, J.; Werb, Z. Initial events during phagocytosis by macrophages viewed from the outside and inside the cell: Membrane-particle interactions and clathrin. J. Cell Biol. 1982, 94, 613–623. [Google Scholar] [CrossRef]
- Unanue, E.R.; Allen, P.M. The basis for the immunoregulatory role of macrophages and other accessory cells. Science 1987, 236, 551–557. [Google Scholar]
- Unanue, E.R.; Ungewickell, E.; Branton, D. The binding of clathrin triskelions to membranes from coated vesicles. Cell 1981, 26, 439–446. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recogntion receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Gordon, S.; Hamann, J.; Lin, H.-H.; Stacey, M. F4/80 and the related adhesion-GPCRs. Eur. J. Immunol. 2011, 41, 2472–2476. [Google Scholar] [CrossRef]
- Neiss, J.H.; Brand, S.; Giu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.; Boes, M.; Plooegh, H.; Fox, J. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005, 307, 254–258. [Google Scholar] [CrossRef]
- Zeigler, K.; Unanue, E.R. Identification of a macrophage antigen-processing event required for I-region-restricted antigen presentation to T lymphocytes. J. Immunol. 1981, 127, 1869–1875. [Google Scholar]
- Hadis, U.; Wahl, B.; Schulz, O.; Hardtke-Wolenski, M.; Schippers, A.; Wagner, N.; Muller, W.; Sparwasser, T.; Forster, R.; Pabst, O. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 2011, 34, 237–246. [Google Scholar] [CrossRef]
- McGhee, J.R.; Kunisawa, J.; Kiyono, H. Gut lymphocyte migration: We are halfway ‘home’. Trends Immunol. 2007, 28, 150–153. [Google Scholar]
- Mayer, L.; Sperber, K.; Chan, L.; Child, J.; Toy, L. Oral tolerance to protein antigens. Allergy 2001, 56, 12–15. [Google Scholar]
- Dellabona, P.; Peccoud, J.; Kappler, J.; Marrack, P.; Benoist, C.; Mathis, D. Superantigens interact with MHC class II molecules outside of the antigen groove. Cell 1990, 62, 1115–1121. [Google Scholar] [CrossRef]
- White, J.; Herman, A.; Pullen, A.M.; Kubo, R.; Kappler, J.W.; Marrack, P. The V beta specific superantigen staphylococcal enterotoxin B: Stimulation of mature T cells and clonaldeletion in neonatal mice. Cell 1989, 56, 27. [Google Scholar] [CrossRef]
- Acharya, K.R.; Passalacqua, E.F.; Jones, E.Y.; Harlos, K.; Stuart, D.I.; Brehm, R.D.; Tranter, H.S. Structural basis of superantigen action inferred from crystal structure of toxic-shock syndrome toxin-1. Nature 1994, 367, 94–97. [Google Scholar] [CrossRef]
- MacDonald, H.R.; Schnieder, R.; Lees, R.K.; Howe, R.C.; Acha-Orbea, H.; Festenstein, F.; Zinkernagel, R.M. T cell receptor V beta use predicts reactivity andtolerance to Mls-encoded antigens. Nature 1988, 332, 40–45. [Google Scholar] [CrossRef]
- Atkins, C.L.; Cole, B.C.; Sullivan, G.J.; Washburn, L.R.; Wiley, B.B. Stimulation of mouse lymphocytes by a mitogen derived from M. arthriditis. J. Immunol. 1986, 137, 1581. [Google Scholar]
- Jardetzky, T.S.; Brown, J.H.; Gorga, J.C.; Stern, L.J.; Urban, R.G.; Chi, Y.I.; Stauffacher, C.; Strominger, J.L.; Wiley, D.C. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature 1994, 368, 711–718. [Google Scholar] [CrossRef]
- Hudson, K.R.; Tiedemann, R.E.; Urban, R.G.; Lowe, S.C.; Strominger, J.L.; Fraser, J.D. Staphylococcal enterotoxin A has two cooperative binding sites on major histocompatibility complex class II. J. Exp. Med. 1995, 182, 711–720. [Google Scholar] [CrossRef]
- Sundström, M.; Abrahmsén, L.; Antonsson, P.; Mehindate, K.; Mourad, W.; Dohlsten, M. The crystal structure of staphylococcal enterotoxin type D reveals Zn2+-mediated homodimerization. EMBO J. 1996, 15, 6832–6840. [Google Scholar]
- Proft, T.; Fraser, J.D. Bacterial superantigens. Clin. Exp. Immunol. 2003, 133, 299–306. [Google Scholar] [CrossRef]
- Tiedemann, R.E.; Fraser, J.D. Cross-linking of MHC class II molecules by staphylococcal enterotoxin A is essential for antigen-presenting cell and T cell activation. J. Immunol. 1996, 157, 3958–3966. [Google Scholar]
- Krakauer, T. Chemotherapeutics targeting immune activation by staphylococcal superantigens. Med. Sci. Monit. 2005, 11, 290–295. [Google Scholar]
- Li, H.; Llera, A.; Tsuchiya, D.; Leder, L.; Ysern, X.; Schlievert, P.M.; Karjalainen, K.; Mariuzza, R.A. Three-dimensional structure of the complex between a T cell receptor β chain and the superantigen staphylococcal enterotoxin B. Immunity 1998, 9, 807–816. [Google Scholar] [CrossRef]
- Marrack, P.; Blackman, M.; Kushnir, E.; Kappler, J. The toxicity of staphylococcal enterotoxin B in mice is mediated by T cells. J. Exp. Med. 1990, 171, 455–464. [Google Scholar] [CrossRef]
- Benjamin, M.A.; Lu, J.; Donnelly, G.; Dureja, P.; McKay, D.M. Changes in murine jejunal morphology evoked by the bacterial superantigen Staphylococcus aureus enterotoxin B are mediated by CD4+ T cells. Infect. Immun. 1998, 66, 2193–2199. [Google Scholar]
- McKay, D.M.; Benjamin, M.A.; Lu, J. CD4+ T cells mediate superantigen-induced abnormalities in murine jejunal ion transport. Am. J. Physiol. 1998, 275, G29–G38. [Google Scholar]
- Hamad, A.R.; Marrack, P.; Kappler, J.W. Transcytosis of staphylococcal superantigen toxins. J. Exp. Med. 1997, 185, 1447–1454. [Google Scholar] [CrossRef]
- Kappler, J.; Herman, A.; Clements, J.; Marrack, P. Mutations defining functional regions of hte superantigen staphylcoccal enterotoxin B. J. Exp. Med. 1992, 175, 387–396. [Google Scholar] [CrossRef]
- Moreto, M.; Perez-Bosque, A. Dietary plasma proteins, the intestinal immune system, and the barrier functions of the intestinal mucosa. J. Anim. Sci. 2009, 87, E92–E100. [Google Scholar] [CrossRef]
- Perez-Bosque, A.; Moreto, M. A rat model of mild intestinal inflammation induced by Staphylococcus aureus enterotoxin B. Proc. Nutr. Soc. 2010, 69, 447–453. [Google Scholar] [CrossRef]
- Kamaras, J.; Murrell, W.G. The effect of bacterial enterotoxins implicated in SIDS on the rabbit intestine. Pathology 2001, 33, 187–196. [Google Scholar] [CrossRef]
- Beery, J.T.; Taylor, S.L.; Schlunz, L.R.; Freed, R.C.; Bergdoll, M.S. Effects of staphylococcal enterotoxin A on the rat gastrointestinal tract. Infect. Immun. 1984, 44, 234–240. [Google Scholar]
- Spiekermann, G.M.; Nagler-Anderson, C. Oral administration of the bacterial superantigen staphylococcal enterotoxin B induces activation and cytokine production by T cells in murine gut-associated lymphoid tissue. J. Immunol. 1998, 161, 5825–5831. [Google Scholar]
- Principato, M.A. Tissue-specific immune responsiveness to Staphylococcal enterotoxin in the aged gut lymphatics. FASEB J. 1999, 13, A291. [Google Scholar]
- Principato, M. Gastrointestinal Immunoregulation and the Challenges of Nanotechnology in Foods. In Food Industry; Muzzalupo, I., Ed.; InTech: Rijeka, Croatia, 2013. [Google Scholar]
- Perez-Bosque, A.; Miro, L.; Polo, J.; Russell, L.; Campbell, J.; Weaver, E.; Crenshaw, J.; Moreto, M. Dietary plasma proteins modulate the immune response of diffuse gut-associated lymphoid tissue in rats challenged with Staphylococcus aureus enterotoxin B. J. Nutr. 2008, 138, 533–537. [Google Scholar]
- Perez-Bosque, A.; Pelegri, C.; Vicario, M.; Castell, M.; Russell, L.; Campbell, J.M.; Quigley, J.D., 3rd; Polo, J.; Amat, C.; Moreto, M. Dietary plasma protein affects the immune response of weaned rats challenged with S. aureus Superantigen B. J. Nutr. 2004, 134, 2667–2672. [Google Scholar]
- Roberts, A.I.; Blumberg, R.S.; Christ, A.D.; Brolin, R.E.; Ebert, E.C. Staphylococcal enterotoxin B induces potent cytotoxic activity by intraepithelial lymphocytes. Immunology 2000, 101, 185–190. [Google Scholar] [CrossRef]
- Faria, A.M.; Weiner, H.L. Oral tolerance. Immunol. Rev. 2005, 206, 232–259. [Google Scholar]
- Friedman, A.; Weiner, H.L. Induction of anergy or active suppression following oral tolerance is determined by antigen dosage. Proc. Natl. Acad. Sci. USA 1994, 91, 6688–6692. [Google Scholar] [CrossRef]
- Chen, Y.; Inobe, J.; Marks, R.; Gonnella, P.; Kuchroo, V.K.; Weiner, H.L. Peripheral deletion of antigen-reactive T cells in oral tolerance. Nature 1995, 376, 177–180. [Google Scholar] [CrossRef]
- Weiner, H.L.; da Cunha, A.P.; Quintana, F.; Wu, H. Oral tolerance. Immunol. Rev. 2011, 241, 241–259. [Google Scholar] [CrossRef]
- Whitacre, C.C.; Gienapp, I.E.; Orosz, C.G.; Bitar, D.M. Oral tolerance in experimental autoimmune encephalomyelitis. III. Evidence for clonal anergy. J. Immunol. 1991, 147, 2155–2163. [Google Scholar]
- Lonnqvist, A.; Ostman, S.; Almqvist, N.; Hultkrantz, S.; Telemo, E.; Wold, A.E.; Rask, C. Neonatal exposure to staphylococcal superantigen improves induction of oral tolerance in a mouse model of airway allergy. Eur. J. Immunol. 2009, 39, 447–456. [Google Scholar] [CrossRef]
- Miron, N.; Feldrihan, V.; Berindan-Neagoe, I.; Cristea, V. The role of staphylococcal enterotoxin A in acheiving oral tolerance to myelin basic protein in adult mice. Immunol. Invest. 2014, 43, 267–277. [Google Scholar] [CrossRef]
- Sundstedt, A.; Hoiden, I.; Rosendahl, A.; Kalland, T.; van Rooijen, N.; Dohlsten, M. Immunoregulatory role of IL-10 during superantigen-induced hyporesponsiveness in vivo. J. Immunol. 1997, 158, 180–186. [Google Scholar]
- Miller, C.; Ragheb, J.A.; Schwartz, R.H. Anergy and cytokine-mediated suppression as distinct superantigen-induced tolerance mechanisms in vivo. J. Exp. Med. 1999, 190, 53–64. [Google Scholar] [CrossRef]
- Grundstrom, S.; Cederbom, L.; Sundstedt, A.; Scheipers, P.; Ivars, F. Superantigen-induced regulatory T cells display different suppressive functions in the presence or absence of natural CD4+CD25+ regulatory T cells in vivo. J. Immunol. 2003, 170, 5008–5017. [Google Scholar] [CrossRef]
- Schartner, J.M.; Singh, A.M.; Dahlberg, P.E.; Nettenstrom, L.; Seroogy, C.M. Recurrent superantigen exposure in vivo leads to highly suppressive CD4+CD25+ and CD4+CD25− T cells with anergic and suppressive genetic signatures. Clin. Exp. Immunol. 2009, 155, 348–356. [Google Scholar] [CrossRef]
- Mahic, M.; Henjum, K.; Yaqub, S.; Bjornbeth, B.A.; Torgersen, K.M.; Tasken, K.; Aandahl, E.M. Generation of highly suppressive adaptive CD8(+)CD25(+)FOXP3(+) regulatory T cells by continuous antigen stimulation. Eur. J. Immunol. 2008, 38, 640–646. [Google Scholar] [CrossRef]
- Mahic, M.; Yaqub, S.; Bryn, T.; Henjum, K.; Eide, D.M.; Torgersen, K.M.; Aandahl, E.M.; Tasken, K. Differentiation of naive CD4+ T cells into CD4+CD25+FOXP3+ regulatory T cells by continuous antigen stimulation. J. Leukoc. Biol. 2008, 38, 1111–1117. [Google Scholar]
- Noel, C.; Florquin, S.; Goldman, M.; Braun, M.Y. Chronic exposure to superantigen induces regulatory CD4(+) T cells with IL-10-mediated suppressive activity. Int. Immunol. 2001, 13, 431–439. [Google Scholar] [CrossRef]
- Feunou, P.; Vanwetswinkel, S.; Gaudray, F.; Goldman, M.; Matthys, P.; Braun, M.Y. Foxp3+CD25+ T regulatory cells stimulate IFN-gamma-independent CD152-mediated activation of tryptophan catabolism that provides dendritic cells with immune regulatory activity in mice unresponsive to staphylococcal enterotoxin B. J. Immunol. 2007, 179, 910–917. [Google Scholar] [CrossRef]
- Feunou, P.; Poulin, L.; Habran, C.; Le Moine, A.; Goldman, M.; Braun, M.Y. CD4+CD25+ and CD4+CD25− T cells act respectively as inducer and effector T suppressor cells in superantigen-induced tolerance. J. Immunol. 2003, 171, 3475–3484. [Google Scholar] [CrossRef]
- Eroukhmanoff, L.; Oderup, C.; Ivars, F. T-cell tolerance induced by repeated antigen stimulation: Selective loss of Foxp3− conventional CD4 T cells and induction of CD4 T-cell anergy. Eur. J. Immunol. 2009, 39, 1078–1087. [Google Scholar] [CrossRef]
- Sartor, R.B. Microbial influences in inflammatory bowel diseases. Gastroenterology 2008, 134, 577–594. [Google Scholar] [CrossRef]
- Posnett, D.N.; Schmelkin, I.; Burton, D.A.; August, A.; McGrath, H.; Mayer, L.F. T cell antigen receptor V gene usage. Increases in V beta 8+ T cells in Crohn’s disease. J. Clin. Invest. 1990, 85, 1770–1776. [Google Scholar] [CrossRef]
- Kelsen, J.; Agnholt, J.; Hoffmann, H.J.; Kaltoft, K.; Dahlerup, J.F. Increased expression of TCR vbeta5.1 and 8 in mucosal T-cell lines cultured from patients with Crohn disease. Scand. J. Gastroenterol. 2004, 39, 238–245. [Google Scholar]
- Baca-Estrada, M.E.; Wong, D.K.; Croitoru, K. Cytotoxic activity of V beta 8+ T cells in Crohn’s disease: The role of bacterial superantigens. Clin. Exp. Immunol. 1995, 99, 398–403. [Google Scholar] [CrossRef]
- Dionne, S.; Laberge, S.; Deslandres, C.; Seidman, E.G. Modulation of cytokine release from colonic explants by bacterial antigens in inflammatory bowel disease. Clin. Exp. Immunol. 2003, 133, 108–114. [Google Scholar] [CrossRef]
- Shiobara, N.; Suzuki, Y.; Aoki, H.; Gotoh, A.; Fujii, Y.; Hamada, Y.; Suzuki, S.; Fukui, N.; Kurane, I.; Itoh, T.; et al. Bacterial superantigens and T cell receptor beta-chain-bearing T cells in the immunopathogenesis of ulcerative colitis. Clin. Exp. Immunol. 2007, 150, 13–21. [Google Scholar] [CrossRef]
- Gittelman, P.D.; Jacobs, J.B.; Lebowitz, A.S.; Tierno, P.M., Jr. Staphylococcus aureus nasal carriage in patients with rhinosinusitis. Laryngoscope 1991, 101, 733–737. [Google Scholar]
- Yang, P.C.; Wang, C.S.; An, Z.Y. A murine model of ulcerative colitis: Induced with sinusitis-derived superantigen and food allergen. BMC Gastroenterol. 2005, 5. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Wang, A.; Ansari, S.; Hershberg, R.M.; McKay, D.M. Colonic bacterial superantigens evoke an inflammatory response and exaggerate disease in mice recovering from colitis. Gastroenterology 2003, 125, 1785–1795. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Principato, M.; Qian, B.-F. Staphylococcal enterotoxins in the Etiopathogenesis of Mucosal Autoimmunity within the Gastrointestinal Tract. Toxins 2014, 6, 1471-1489. https://doi.org/10.3390/toxins6051471
Principato M, Qian B-F. Staphylococcal enterotoxins in the Etiopathogenesis of Mucosal Autoimmunity within the Gastrointestinal Tract. Toxins. 2014; 6(5):1471-1489. https://doi.org/10.3390/toxins6051471
Chicago/Turabian StylePrincipato, MaryAnn, and Bi-Feng Qian. 2014. "Staphylococcal enterotoxins in the Etiopathogenesis of Mucosal Autoimmunity within the Gastrointestinal Tract" Toxins 6, no. 5: 1471-1489. https://doi.org/10.3390/toxins6051471