
Bioactive Mimetics of Conotoxins and other Venom Peptides

Abstract
:1. Introduction
2. Peptidomimetics
3. Pain Blocking Conotoxins and Their Mimetics
3.1. Conotoxins


3.2. Commonly Used Assays
3.2.1. Rat Vas Deferens
3.2.2. Radioligand Displacement Assay
3.2.3. FLIPR assay with SH-SY5Y Neuroblastoma Cells
3.2.4. IMR-32 Human Neuroblastoma Cell Assay
3.2.5. Electrophysiological Methods—Patch Clamp Technique
3.3. ω-Conotoxin Mimetics
3.3.1. ω-Conotoxin MVIIA


3.3.2. ω-Conotoxin CVID

3.3.3. ω-Conotoxin GVIA






| Compd. No. | R1 | R2 | Radioligand Displacement Assay IC50 (μM) | SH-SY5Y Neuroblastoma Cells IC50 (μM) | HEK293 hCav2.2 + β3 + α2δ1 IC50 (μM) |
|---|---|---|---|---|---|
| 25 | H | NH2 | 214 | 160 | 232 |
| 26 | H | N=C(NH2)2 | 12 | 206 | 299 |
| 27 | F | NH2 | 65 | 286 | 288 |
| 28 | F | N=C(NH2)2 | 16 | 156 | 156 |

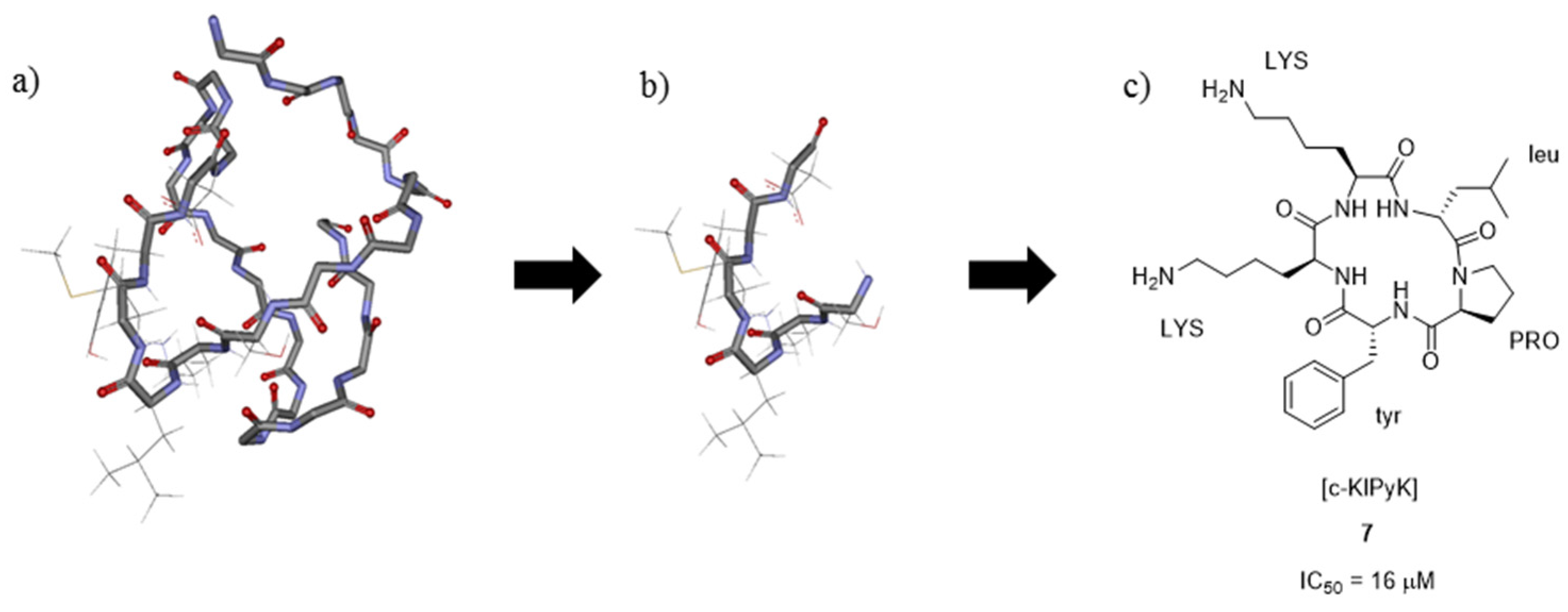
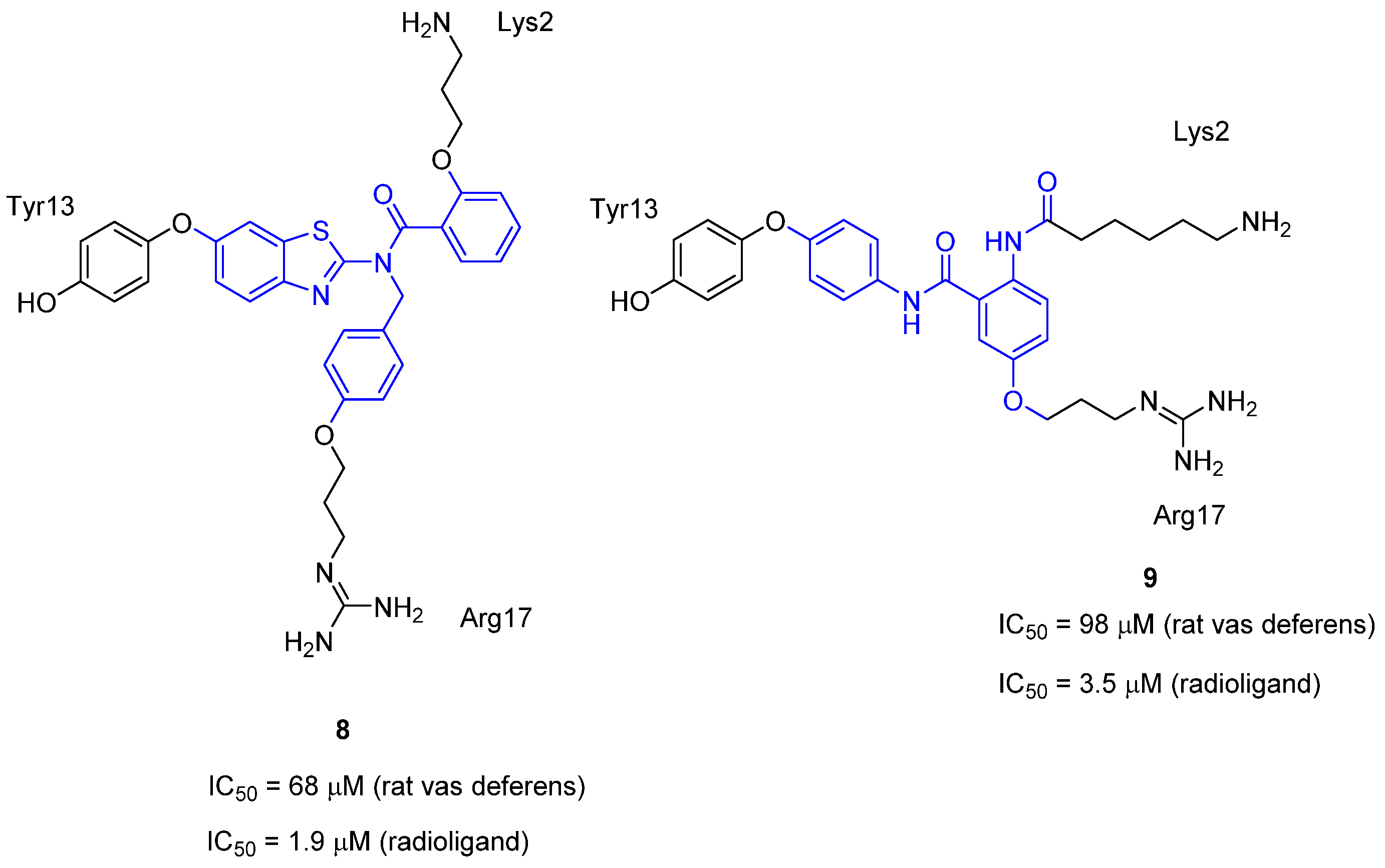
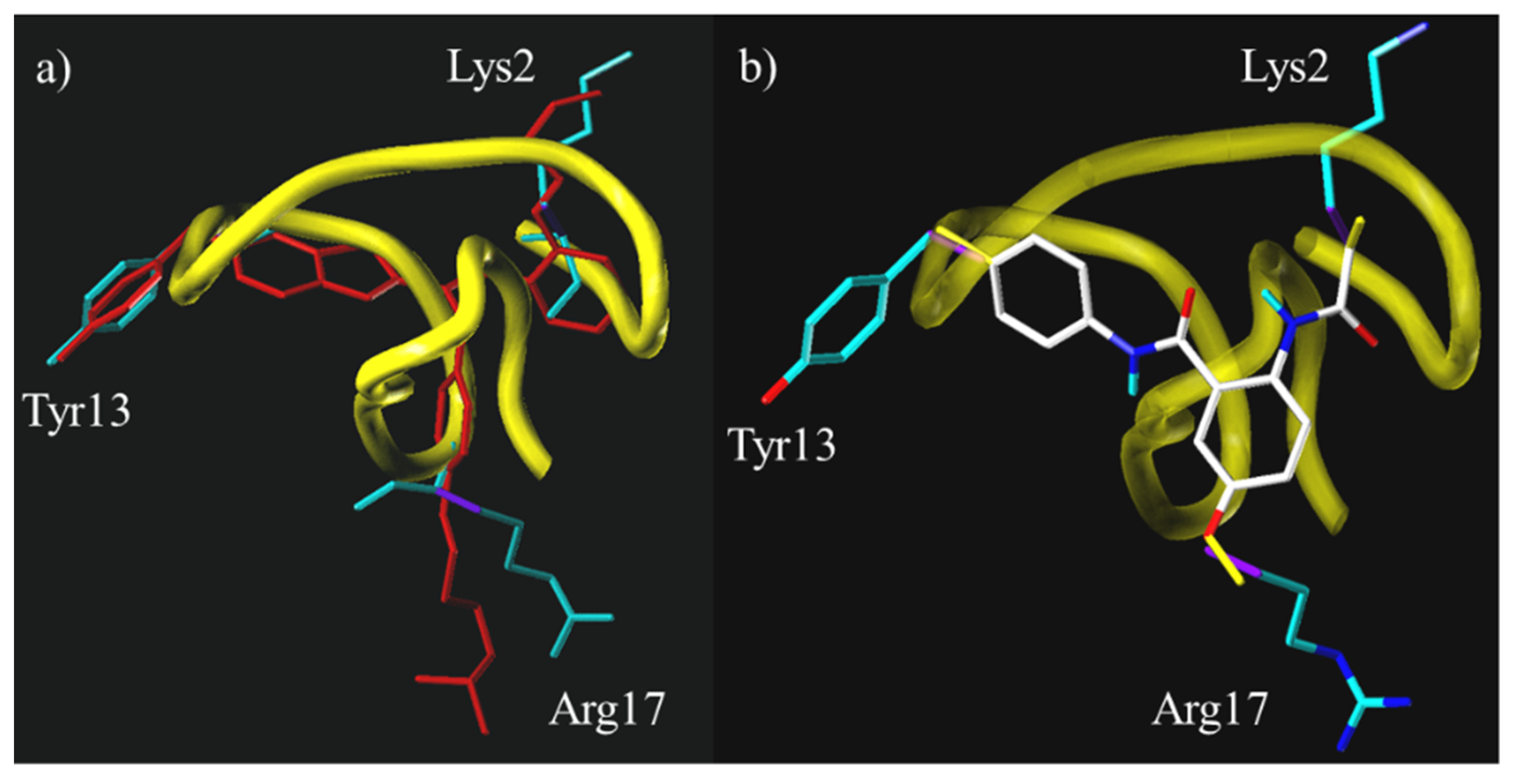
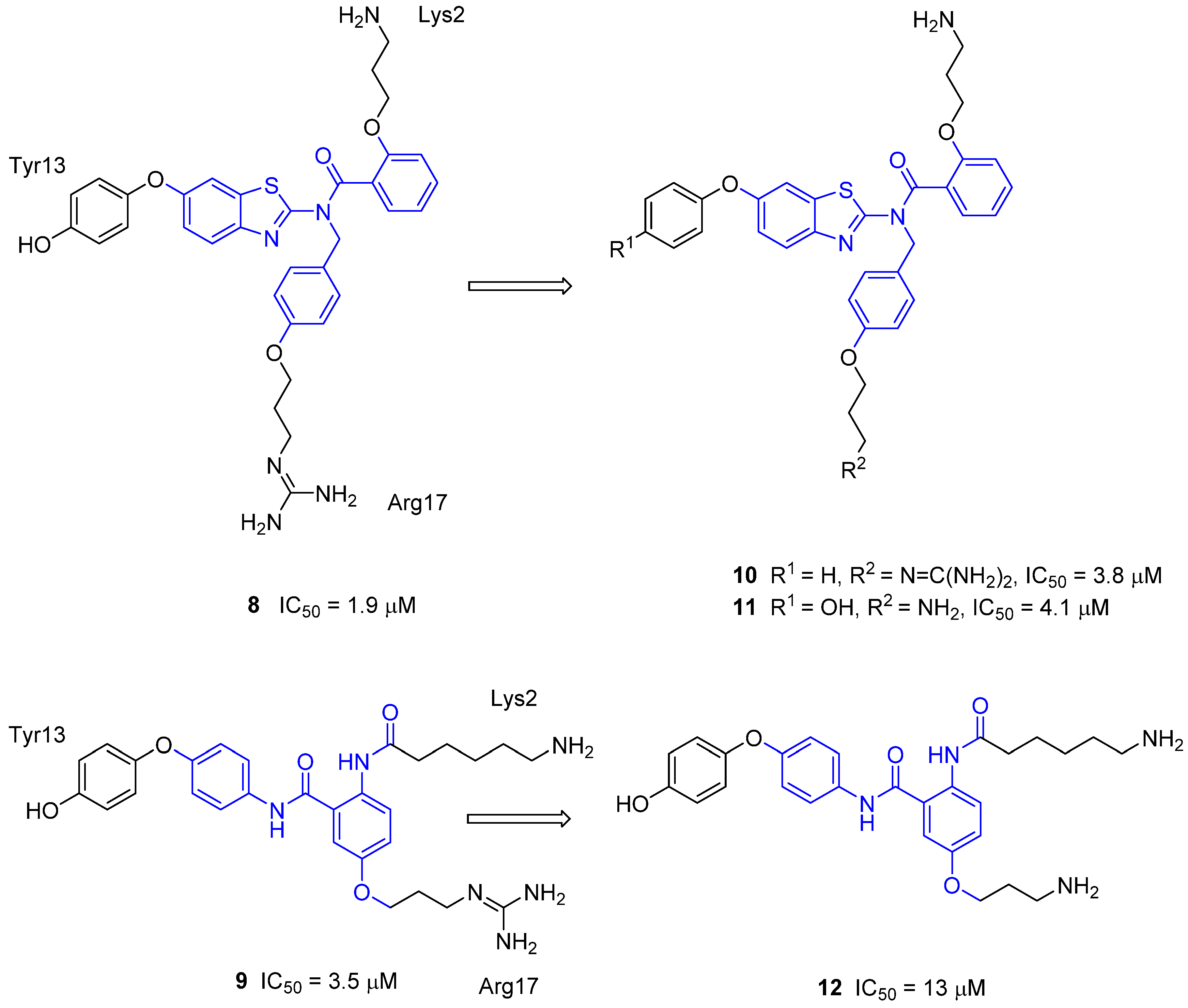
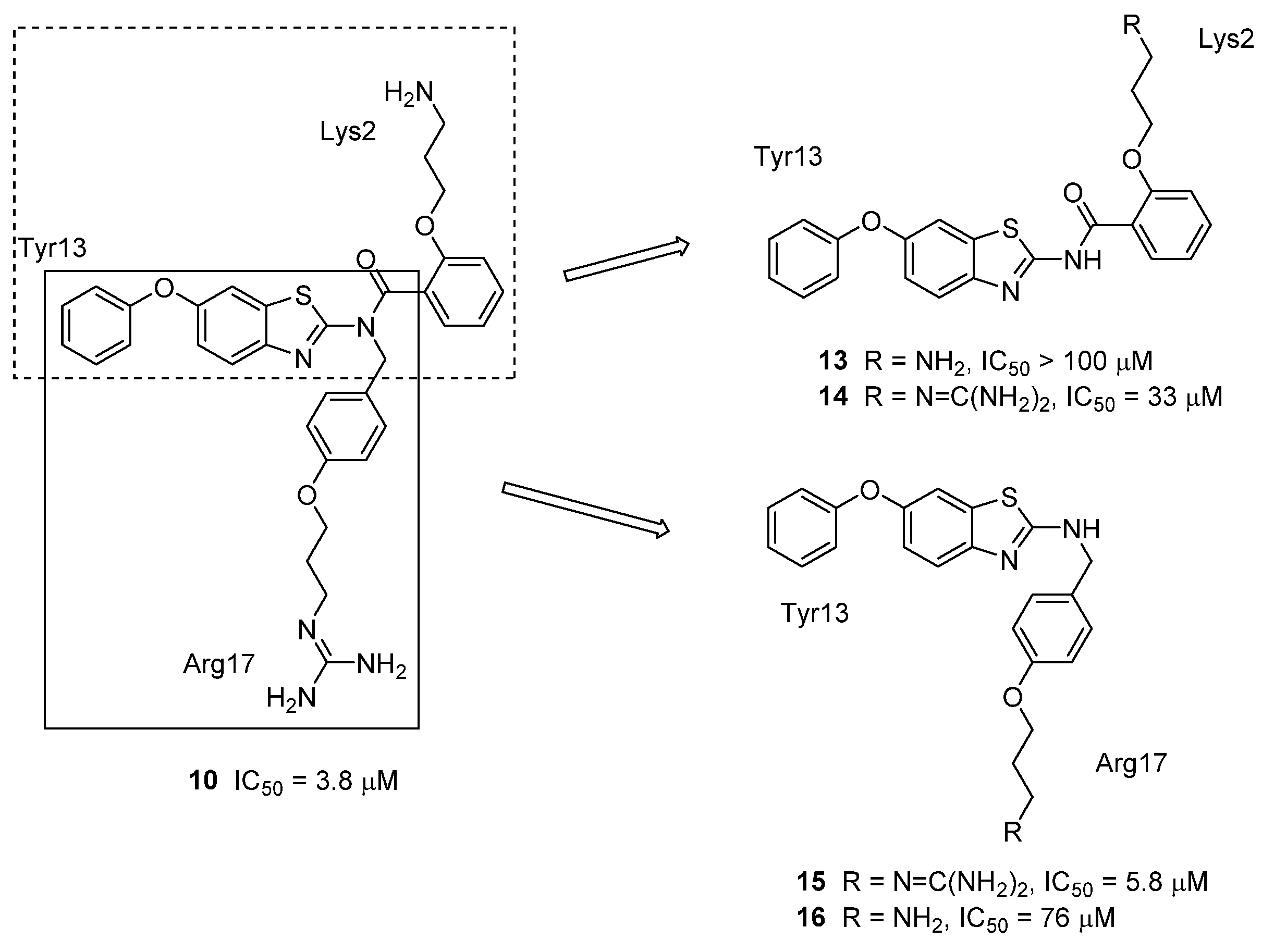
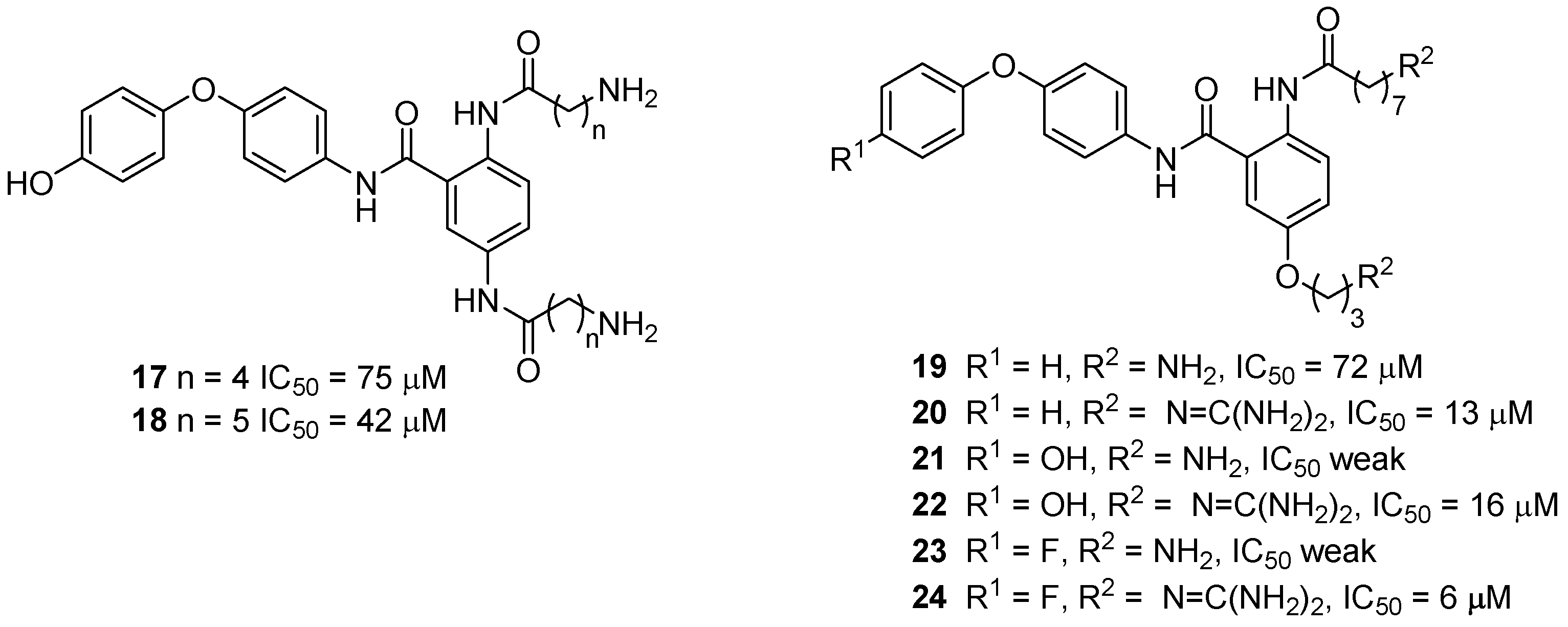
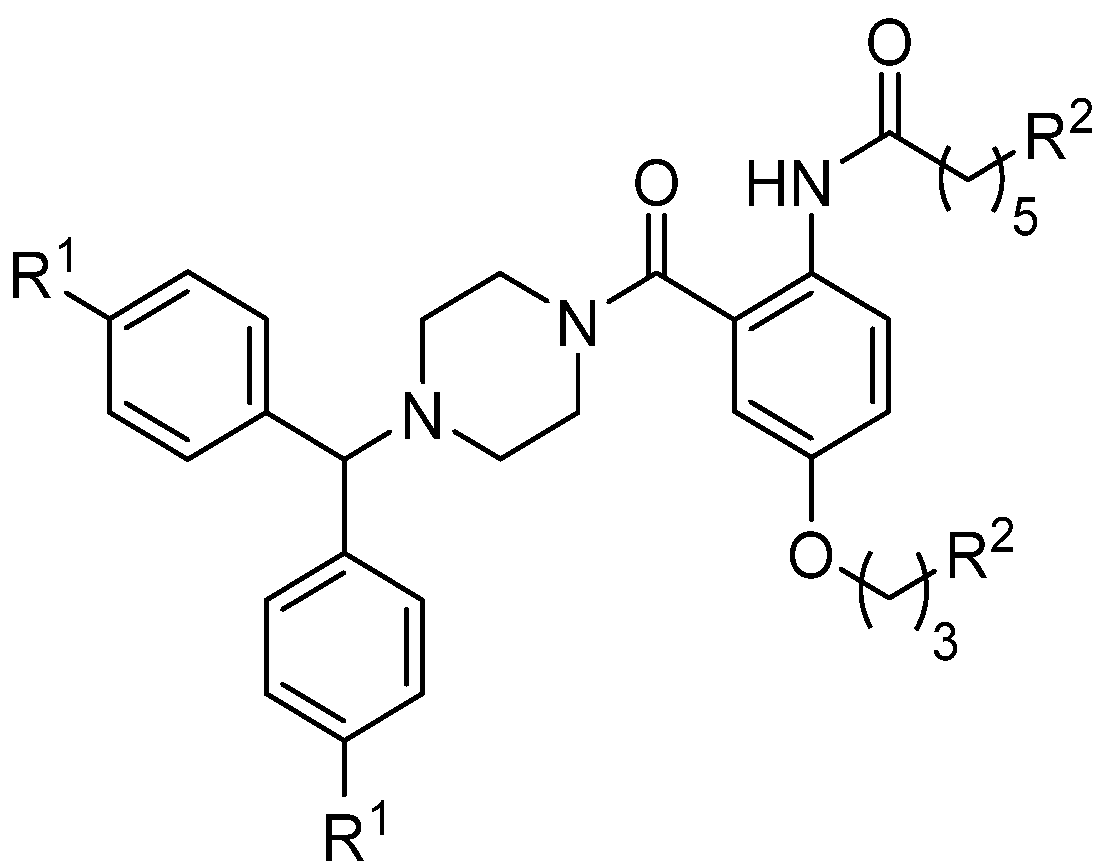
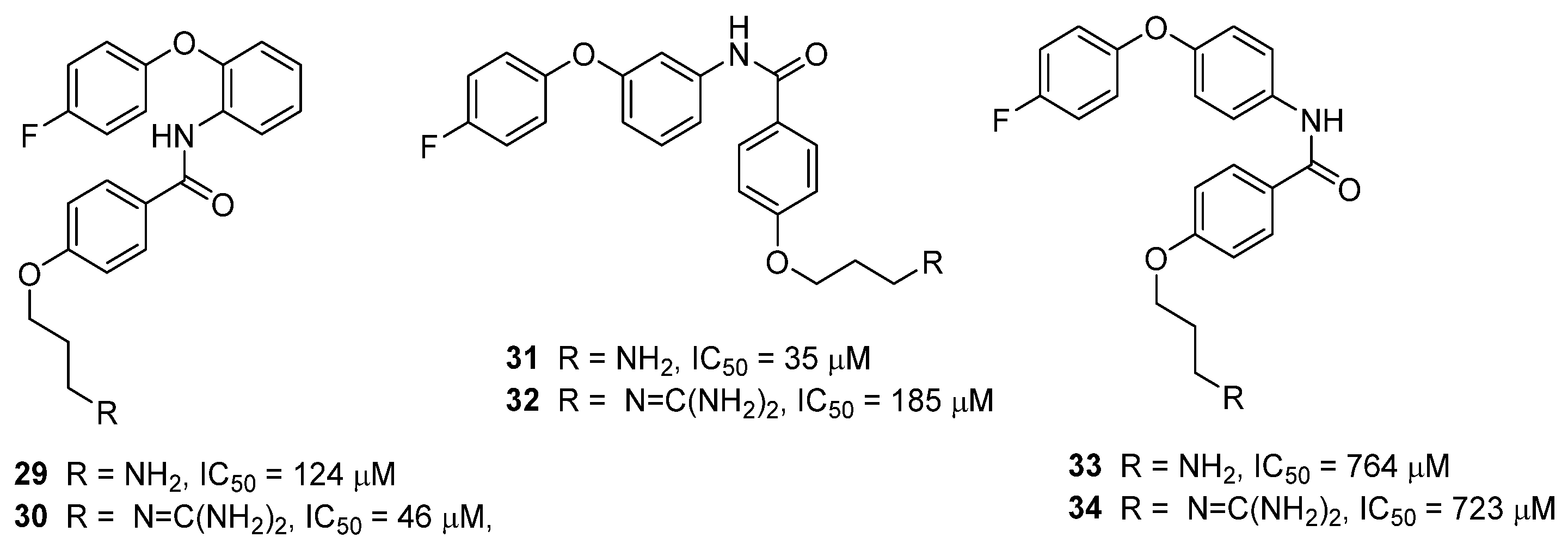
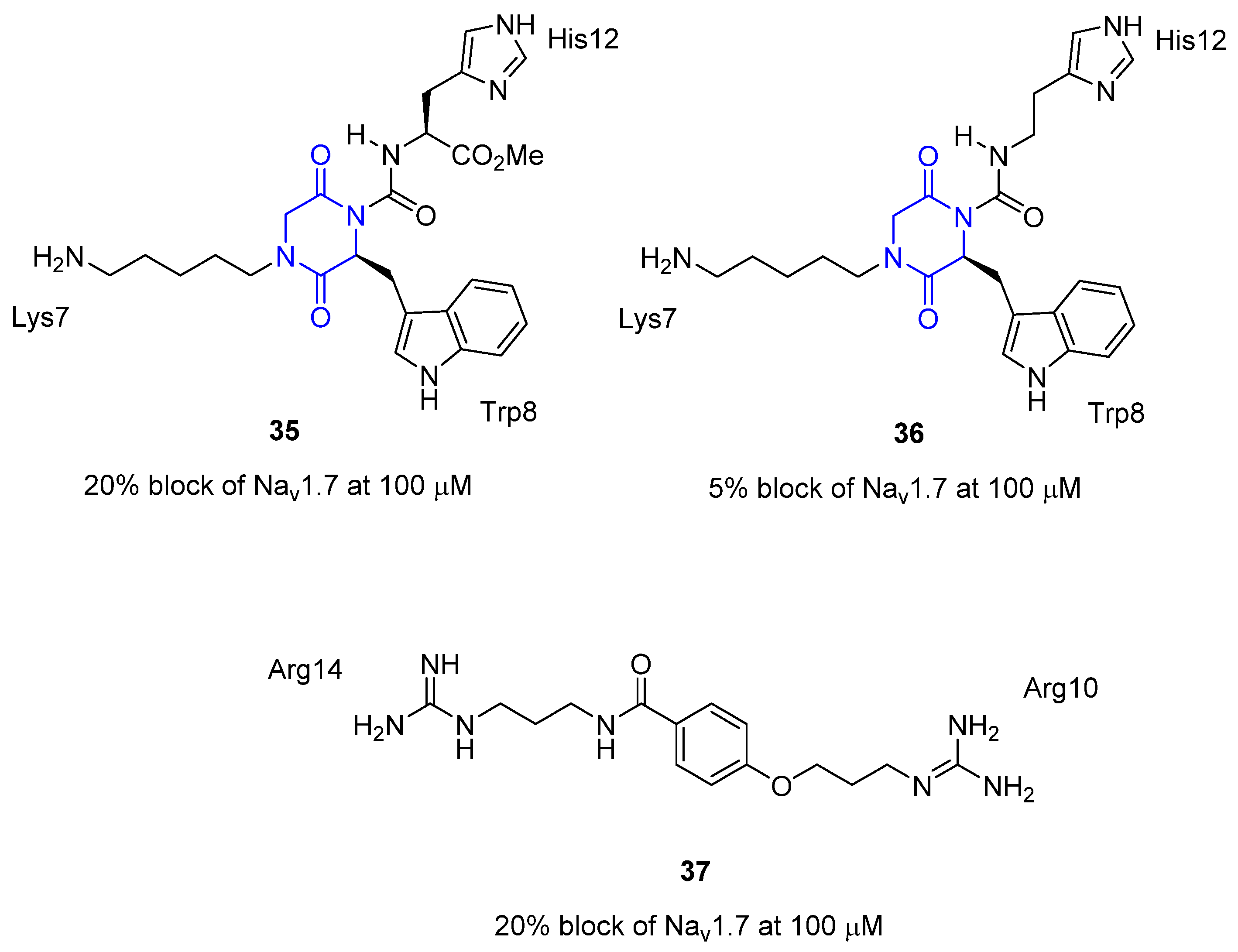
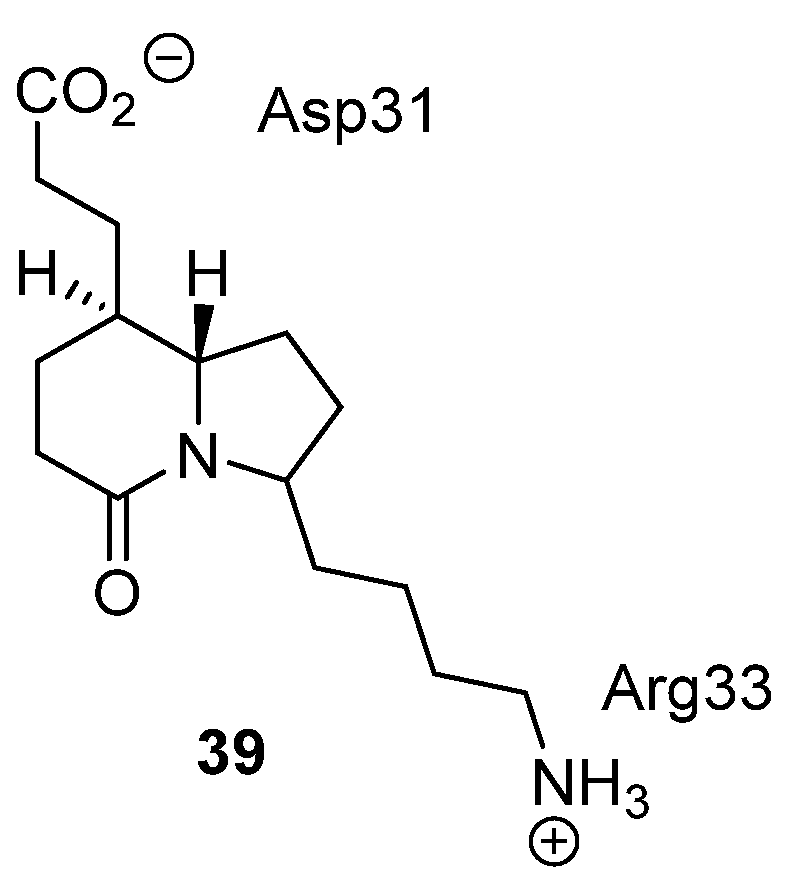
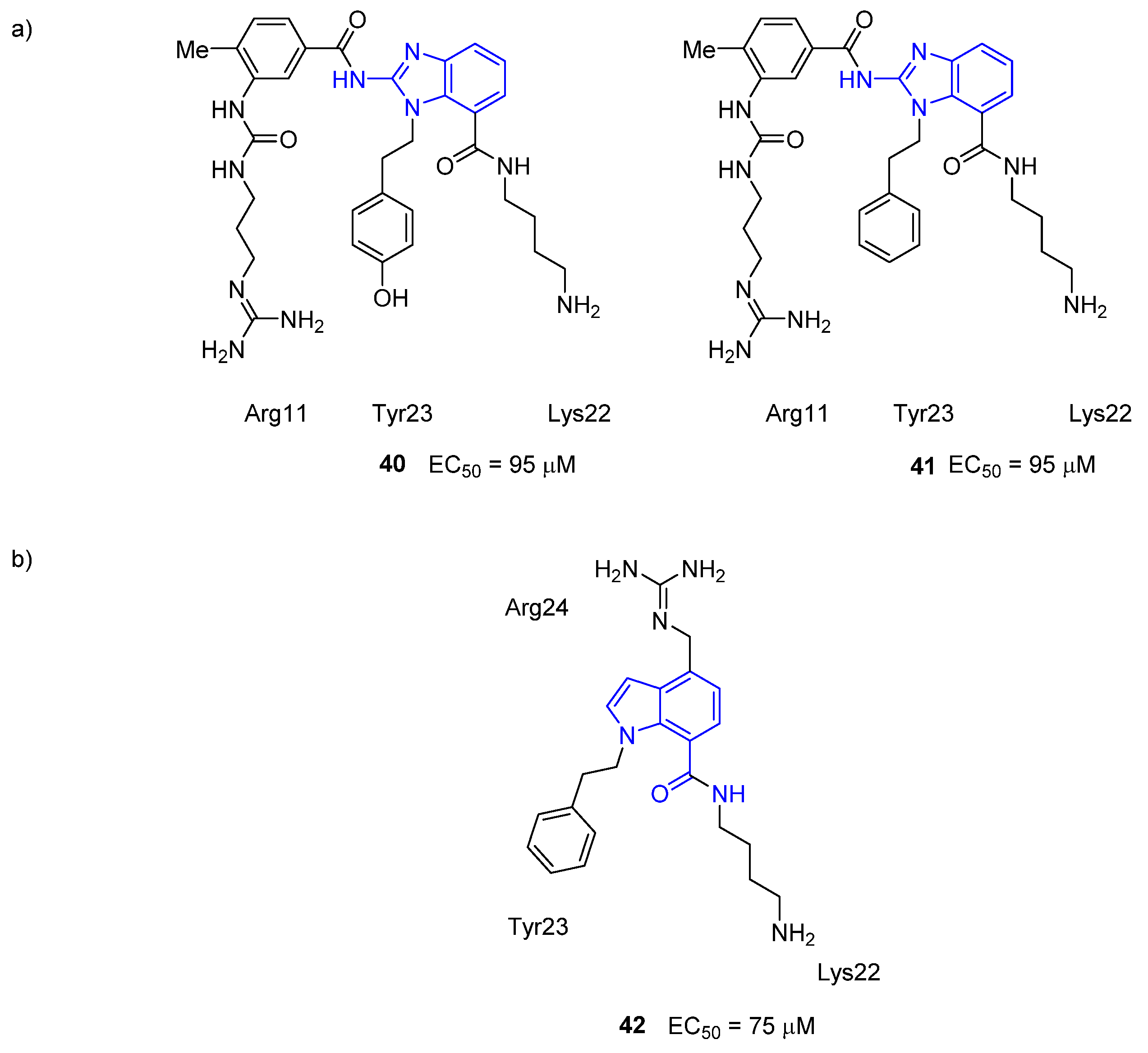
3.4. μ-Conotoxin Mimetics

{kind=link}
{kind=link}
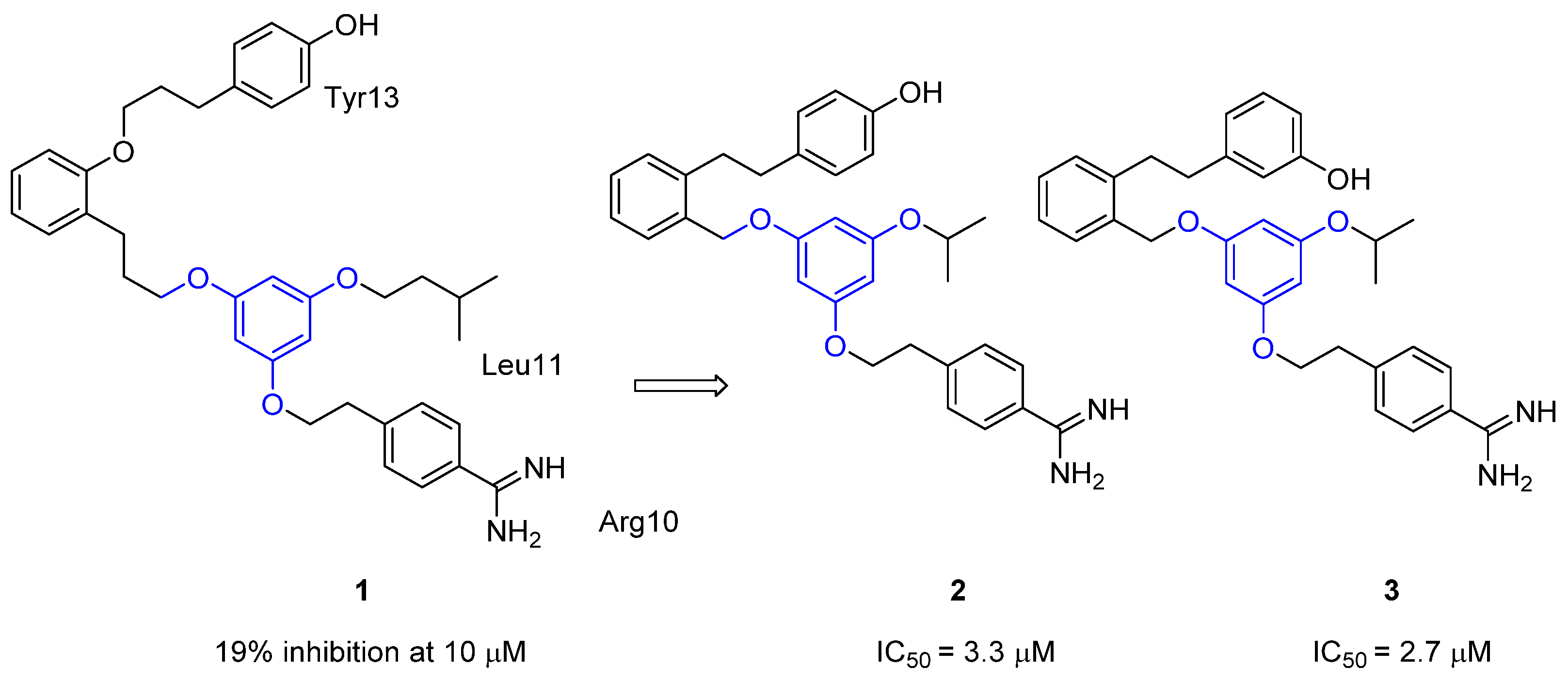{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4. Snake Venom Peptides and Their Mimetics
4.1. Echistatin

4.2. Erabutoxin B

5. Sun Anemone Toxin

6. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kaas, Q.; Craik, D. Bioinformatics-aided venomics. Toxins 2015, 7, 2159–2187. [Google Scholar] [CrossRef] [PubMed]
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.B.; Holladay, M.W. Lead Discovery and Lead Modification. In The Organic Chemistry of Drug Design and Drug Action, 3rd ed.; Holladay, R.B., Silverman, M.W., Eds.; Academic Press: Boston, MA, USA, 2014; pp. 19–122. [Google Scholar]
- Trabocchi, A.; Guarna, A. The basics of peptidomimetics. In Peptidomimetics in Organic and Medicinal Chemistry; John Wiley & Sons, Ltd.: New York, NY, USA, 2014; pp. 1–17. [Google Scholar]
- Ripka, A.S.; Rich, D.H. Peptidomimetic design. Curr. Opin. Chem. Biol. 1998, 2, 441–452. [Google Scholar] [CrossRef]
- Lauri, G.; Bartlett, P. CAVEAT: A program to facilitate the design of organic molecules. J. Comput.-Aided Mol. Des. 1994, 8, 51–66. [Google Scholar] [CrossRef] [PubMed]
- CAVEAT. Available online: http://www.cchem.berkeley.edu/pabgrp/Data/caveat.html (accessed on 1 September 2015).
- Olivera, B.M. E.E. Just Lecture, 1996: Conus venom peptides, receptor and ion channel targets, and drug design: 50 million years of neuropharmacology. Mol. Biol. Cell 1997, 8, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Livett, B.G.; Gayler, K.R.; Khalil, Z. Drugs from the sea: Conopeptides as potential therapeutics. Curr. Med. Chem. 2004, 11, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M. Peptide neurotoxins from fish-hunting cone snails. Science 1985, 230, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; White, J. Clinical toxicology of Conus snail stings. In Handbook of Clinical Toxicology of Animal Venoms and Poisons; Julian, W., Jurg, M., Eds.; CRC Press: Boca Raton, FL, USA, 1995; pp. 117–128. [Google Scholar]
- Olivera, B.M.; Cruz, L.J. Conotoxins, in retrospect. Toxicon 2001, 39, 7–14. [Google Scholar] [CrossRef]
- Olivera, B.M.; Miljanich, G.P.; Ramachandran, J.; Adams, M.E. Calcium channel diversity and neurotransmitter release: The ω-Conotoxins and ω-Agatoxins. Annu. Rev. Biochem. 1994, 63, 823–867. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.R.; Olivera, B.M.; Cruz, L.J. Peptide toxins from venomous Conus snails. Annu. Rev. Biochem. 1988, 57, 665–700. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.S.; Olivera, B.M. Conotoxins down under. Toxicon 2006, 48, 780–798. [Google Scholar] [CrossRef] [PubMed]
- Han, T.S.; Teichert, R.W.; Olivera, B.M.; Bulaj, G. Conus venoms—A rich source of peptide-based therapeutics. Curr. Pharm. Des. 2008, 14, 2462–2479. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Albillos, A.; Fernández, F.; Medrano, J.; Jurkiewicz, A.; Garcı́a, A.G. ω-Conotoxins block neurotransmission in the rat vas deferens by binding to different presynaptic sites on the N-type Ca2+ channel. Eur. J. Pharmacol. 1997, 321, 217–223. [Google Scholar] [CrossRef]
- Lewis, R.J. Conotoxins as selective inhibitors of neuronal ion channels, receptors and transporters. IUBMB Life 2004, 56, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Halai, R.; Craik, D.J. Conotoxins: Natural product drug leads. Nat. Prod. Rep. 2009, 26, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, C.; Lewis, R. ω-Conotoxins GVIA, MVIIA and CVID: SAR and clinical potential. Mar. Drugs 2006, 4, 193–214. [Google Scholar] [CrossRef]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, Synthesis, and Structure–Activity Relationships of Conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Gray, W.R.; Yoshikami, D.; Olivera, B.M. Conus venoms: A rich source of neuroactive peptides. Toxin Rev. 1985, 4, 107–132. [Google Scholar] [CrossRef]
- Lin, H.; Li, Q.-Z. Predicting conotoxin superfamily and family by using pseudo amino acid composition and modified Mahalanobis discriminant. Biochem. Biophys. Res. Commun. 2007, 354, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.-C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Norton, R.S. Conotoxin gene superfamilies. Mar. Drugs 2014, 12, 6058–6101. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.J.; Schroeder, T.; Lewis, R. Structure-activity relationships of ω-conotoxins at N-type voltage-sensitive calcium channels. J. Mol. Recognit. 2000, 13, 55–70. [Google Scholar] [CrossRef]
- Baell, J.B.; Duggan, P.J.; Lok, Y.P. ω-Conotoxins and Approaches to Their Non-Peptide Mimetics. Aust. J. Chem. 2004, 57, 179–185. [Google Scholar] [CrossRef]
- ConoServer. Available online: http://www.conoserver.org/?page=card&table=structure&id=138 (accessed on 1 September 2015).
- Winquist, R.J.; Pan, J.Q.; Gribkoff, V.K. Use-dependent blockade of Cav2.2 voltage-gated calcium channels for neuropathic pain. Biochem. Pharmacol. 2005, 70, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Vink, S.; Alewood, P.F. Targeting voltage-gated calcium channels: Developments in peptide and small-molecule inhibitors for the treatment of neuropathic pain. Br. J. Pharmacol. 2012, 167, 970–989. [Google Scholar] [CrossRef] [PubMed]
- Pexton, T.; Moeller-Bertram, T.; Schilling, J.M.; Wallace, M.S. Targeting voltage-gated calcium channels for the treatment of neuropathic pain: A review of drug development. Expert Opin. Investig. Drugs 2011, 20, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Belardetti, F.; Zamponi, G.W. Calcium channels as therapeutic targets. WIREs Membr. Transp. Signal. 2012, 1, 433–451. [Google Scholar] [CrossRef]
- Perret, D.; Luo, Z.D. Targeting voltage-gated calcium channels for neuropathic pain management. Neurotherapeutics 2009, 6, 679–692. [Google Scholar] [CrossRef] [PubMed]
- McGivern, J.G. Targeting N-type and T-type calcium channels for the treatment of pain. Drug Discov. Today 2006, 11, 245–253. [Google Scholar] [CrossRef]
- Rahman, W.; Dickenson, A.H. Voltage gated sodium and calcium channel blockers for the treatment of chronic inflammatory pain. Neurosci. Lett. 2013, 557 Pt A, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S. Recent progress in the discovery and development of N-type calcium channel modulators for the treatment of pain. Prog. Med. Chem. 2014, 53, 147–186. [Google Scholar] [PubMed]
- Green, B.R.; Bulaj, G.; Norton, R.S. Structure and function of μ-conotoxins, peptide-based sodium channel blockers with analgesic activity. Future Med. Chem. 2014, 6, 1677–1698. [Google Scholar] [CrossRef] [PubMed]
- Maggi, C.; Patacchini, R.; Santicioli, P.; Lippe, I.; Giuliani, S.; Geppetti, P.; del Bianco, E.; Selleri, S.; Meli, A. The effect of omega conotoxin GVIA, a peptide modulator of the N-type voltage sensitive calcium channels, on motor responses produced by activation of efferent and sensory nerves in mammalian smooth muscle. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1988, 338, 107–113. [Google Scholar] [CrossRef]
- Lew, M.J.; Flinn, J.P.; Pallaghy, P.K.; Murphy, R.; Whorlow, S.L.; Wright, C.E.; Norton, R.S.; Angus, J.A. Structure-function relationships of ω-conotoxin GVIA: Synthesis, structure, calcium channel binding, and functional assay of alanine substituted analogues. J. Biol. Chem. 1997, 272, 12014–12023. [Google Scholar] [CrossRef] [PubMed]
- Flinn, J.P.; Pallaghy, P.K.; Lew, M.J.; Murphy, R.; Angus, J.A.; Norton, R.S. Roles of key functional groups in ω-conotoxin GVIA. Eur. J. Biochem. 1999, 262, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Nielsen, K.J.; Craik, D.J.; Loughnan, M.L.; Adams, D.A.; Sharpe, I.A.; Luchian, T.; Adams, D.J.; Bond, T.; Thomas, L.; et al. Novel ω-conotoxins from Conus catus discriminate among neuronal calcium channel subtypes. J. Biol. Chem. 2000, 275, 35335–35344. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.; Forsyth, S.; Gable, R.; Norton, R.; Mulder, R. Design and synthesis of type-III mimetics of ω-conotoxin GVIA. J. Comput.-Aided Mol. Des. 2001, 15, 1119–1136. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Snowman, A.; Biswas, A.; Olivera, B.; Snyder, S. Omega-conotoxin GVIA binding to a high-affinity receptor in brain: Characterization, calcium sensitivity, and solubilization. J. Neurosci. 1988, 8, 3354–3359. [Google Scholar] [PubMed]
- Nielsen, K.J.; Adams, D.A.; Alewood, P.F.; Lewis, R.J.; Thomas, L.; Schroeder, T.; Craik, D.J. Effects of chirality at Tyr13 on the structure-activity relationships of ω-conotoxins from Conus magus. Biochemistry 1999, 38, 6741–6751. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Duggan, P.J.; Forsyth, S.A.; Lewis, R.J.; Lok, Y.P.; Schroeder, C.I. Synthesis and biological evaluation of nonpeptide mimetics of ω-conotoxin GVIA. Bioorg. Med. Chem. 2004, 12, 4025–4037. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Duggan, P.J.; Forsyth, S.A.; Lewis, R.J.; Lok, Y.P.; Schroeder, C.I.; Shepherd, N.E. Synthesis and biological evaluation of anthranilamide-based non-peptide mimetics of ω-conotoxin GVIA. Tetrahedron 2006, 62, 7284–7292. [Google Scholar] [CrossRef]
- Duggan, P.J.; Faber, J.M.; Graham, J.E.; Lewis, R.J.; Lumsden, N.G.; Tuck, K.L. Synthesis and Cav2.2 binding data for non-peptide mimetics of ω-conotoxin GVIA based on a 5-amino-anthranilamide core. Aust. J. Chem. 2008, 61, 11–15. [Google Scholar] [CrossRef]
- Andersson, A.; Baell, J.B.; Duggan, P.J.; Graham, J.E.; Lewis, R.J.; Lumsden, N.G.; Tranberg, C.E.; Tuck, K.L.; Yang, A. ω-Conotoxin GVIA mimetics based on an anthranilamide core: Effect of variation in ammonium side chain lengths and incorporation of fluorine. Bioorg. Med. Chem. 2009, 17, 6659–6670. [Google Scholar] [CrossRef] [PubMed]
- Duggan, P.J.; Lewis, R.J.; Lok, Y.P.; Lumsden, N.G.; Tuck, K.L.; Yang, A. Low molecular weight non-peptide mimics of ω-conotoxin GVIA. Bioorg. Med. Chem. Lett. 2009, 19, 2763–2765. [Google Scholar] [CrossRef] [PubMed]
- Tranberg, C.E.; Yang, A.; Vetter, I.; McArthur, J.R.; Baell, J.B.; Lewis, R.J.; Tuck, K.L.; Duggan, P.J. ω-Conotoxin GVIA mimetics that bind and inhibit neuronal Cav2.2 ion channels. Mar. Drugs 2012, 10, 2349–2368. [Google Scholar] [CrossRef] [PubMed]
- Passafaro, M.; Clementi, F.; Sher, E. Metabolism of omega-conotoxin-sensitive voltage-operated calcium channels in human neuroblastoma cells: Modulation by cell differentiation and anti-channel antibodies. J. Neurosci. 1992, 12, 3372–3379. [Google Scholar] [PubMed]
- Sousa, S.R.; Vetter, I.; Ragnarsson, L.; Lewis, R.J. Expression and pharmacology of endogenous Cav channels in SH-SY5Y human neuroblastoma cells. PLoS ONE 2013, 8, e59293. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, E.; Graham, J.; Spiller, S.; Vetter, I.; Lewis, R.; Duggan, P.; Tuck, K. Inhibition of N-Type Calcium Channels by Fluorophenoxyanilide Derivatives. Mar. Drugs 2015, 13, 2030–2045. [Google Scholar] [CrossRef] [PubMed]
- Sher, E.; Pandiella, A.; Clementi, F. ω-Conotoxin binding and effects on calcium channel function in human neuroblastoma and rat pheochromocytoma cell lines. FEBS Lett. 1988, 235, 178–182. [Google Scholar] [CrossRef]
- Carbone, E.; Sher, E.; Clementi, F. Ca currents in human neuroblastoma IMR32 cells: Kinetics, permeability and pharmacology. Pflügers Arch. 1990, 416, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Menzler, S.; Bikker, J.A.; Suman-Chauhan, N.; Horwell, D.C. Design and biological evaluation of non-peptide analogues of omega-conotoxin MVIIA. Bioorg. Med. Chem. Lett. 2000, 10, 345–347. [Google Scholar] [CrossRef]
- Menzler, S.; Bikker, J.A.; Horwell, D.C. Synthesis of a non-peptide analogue of omega-conotoxin MVIIA. Tetrahedron Lett. 1998, 39, 7619–7622. [Google Scholar] [CrossRef]
- Kohno, T.; Kim, J.I.; Kobayashi, K.; Kodera, Y.; Maeda, T.; Sato, K. Three-dimensional structure in solution of the calcium channel blocker omega-conotoxin MVIIA. Biochemistry 1995, 34, 10256–10265. [Google Scholar] [CrossRef] [PubMed]
- Nadasdi, L.; Yamashiro, D.; Chung, D.; Tarczy-Hornoch, K.; Adriaenssens, P.; Ramachandran, J. Structure-activity analysis of a Conus peptide blocker of N-type neuronal calcium channels. Biochemistry 1995, 34, 8076–8081. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.-X.; Cammidge, A.N.; Horwell, D.C. Dendroid peptide structural mimetics of ω-conotoxin MVIIA based on a 2(1H)-quinolinone core. Tetrahedron 2000, 56, 5169–5175. [Google Scholar] [CrossRef]
- Smith, M.T.; Cabot, P.J.; Ross, F.B.; Robertson, A.D.; Lewis, R.J. The novel N-type calcium channel blocker, AM336, produces potent dose-dependent antinociception after intrathecal dosing in rats and inhibits substance P release in rat spinal cord slices. Pain 2002, 96, 119–127. [Google Scholar] [CrossRef]
- Wright, C.E.; Robertson, A.D.; Whorlow, S.L.; Angus, J.A. Cardiovascular and autonomic effects of ω-conotoxins MVIIA and CVID in conscious rabbits and isolated tissue assays. Br. J. Pharmacol. 2000, 131, 1325–1336. [Google Scholar] [PubMed]
- Schroeder, C.; Smythe, M.; Lewis, R. Development of small molecules that mimic the binding of ω-conotoxins at the N-type voltage-gated calcium channel. Mol. Divers. 2004, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Beedle, A.M.; Zamponi, G.W. Block of voltage-dependent calcium channels by aliphatic monoamines. Biophys. J. 2000, 79, 260–270. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Feng, Z.-P.; Zhang, L.; Pajouhesh, H.; Ding, Y.; Belardetti, F.; Pajouhesh, H.; Dolphin, D.; Mitscher, L.A.; Snutch, T.P. Scaffold-based design and synthesis of potent N-type calcium channel blockers. Bioorg. Med. Chem. Lett. 2009, 19, 6467–6472. [Google Scholar] [CrossRef] [PubMed]
- Scott, V.E.; Vortherms, T.A.; Niforatos, W.; Swensen, A.M.; Neelands, T.; Milicic, I.; Banfor, P.N.; King, A.; Zhong, C.; Simler, G.; et al. A-1048400 is a novel, orally active, state-dependent neuronal calcium channel blocker that produces dose-dependent antinociception without altering hemodynamic function in rats. Biochem. Pharmacol. 2012, 83, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Bulaj, G.; West, P.J.; Garrett, J.E.; Watkins, M.; Zhang, M.-M.; Norton, R.S.; Smith, B.J.; Yoshikami, D.; Olivera, B.M. Novel conotoxins from Conus striatus and Conus kinoshitai selectively block TTX-resistant sodium channels. Biochemistry 2005, 44, 7259–7265. [Google Scholar] [PubMed]
- Zhang, M.-M.; Green, B.R.; Catlin, P.; Fiedler, B.; Azam, L.; Chadwick, A.; Terlau, H.; McArthur, J.R.; French, R.J.; Gulyas, J.; et al. Structure/function characterization of μ-conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. J. Biol. Chem. 2007, 282, 30699–30706. [Google Scholar] [CrossRef] [PubMed]
- Khoo, K.K.; Feng, Z.-P.; Smith, B.J.; Zhang, M.-M.; Yoshikami, D.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structure of the analgesic μ-conotoxin KIIIA and effects on the structure and function of disulfide deletion. Biochemistry 2009, 48, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Khoo, K.K.; Gupta, K.; Green, B.R.; Zhang, M.-M.; Watkins, M.; Olivera, B.M.; Balaram, P.; Yoshikami, D.; Bulaj, G.; Norton, R.S. Distinct disulfide isomers of μ-conotoxins KIIIA and KIIIB block voltage-gated sodium channels. Biochemistry 2012, 51, 9826–9835. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.M.; Zhang, M.; Gable, R.; Norton, R.S.; Baell, J.B. De novo design and synthesis of a μ-conotoxin KIIIA peptidomimetic. Bioorg. Med. Chem. Lett. 2013, 23, 4892–4895. [Google Scholar] [PubMed]
- Brady, R.; Baell, J.; Norton, R. Strategies for the development of conotoxins as new therapeutic leads. Mar. Drugs 2013, 11, 2293–2313. [Google Scholar] [PubMed]
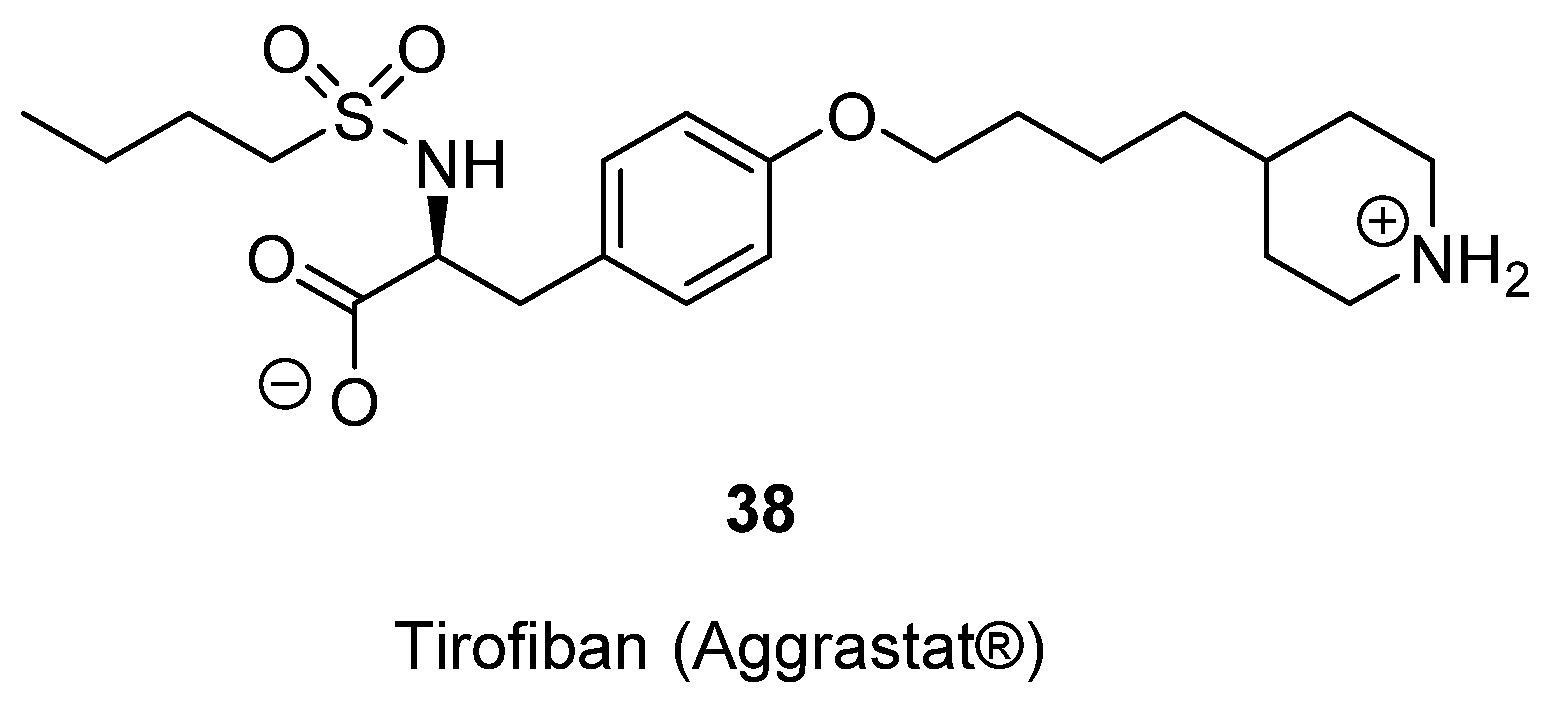
- Hartman, G.D.; Egbertson, M.S.; Halczenko, W.; Laswell, W.L.; Duggan, M.E.; Smith, R.L.; Naylor, A.M.; Manno, P.D.; Lynch, R.J. Non-peptide fibrinogen receptor antagonists. 1. Discovery and design of exosite inhibitors. J. Med. Chem. 1992, 35, 4640–4642. [Google Scholar] [CrossRef] [PubMed]
- Egbertson, M.S.; Chang, C.T.C.; Duggan, M.E.; Gould, R.J.; Halczenko, W.; Hartman, G.D.; Laswell, W.L.; Lynch, J.J.; Lynch, R.J. Non-peptide fibrinogen receptor antagonists. 2. Optimization of a tyrosine template as a mimic for Arg-Gly-Asp. J. Med. Chem. 1994, 37, 2537–2551. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.J.; Cook, J.J.; Sitko, G.R.; Holahan, M.A.; Ramjit, D.R.; Mellott, M.J.; Stranieri, M.T.; Stabilito, I.I.; Zhang, G.; Lynch, R.J. Nonpeptide glycoprotein IIb/IIIa inhibitors. 5. Antithrombotic effects of MK-0383. J. Pharmacol. Exp. Ther. 1995, 272, 20–32. [Google Scholar] [PubMed]
- Peerlinck, K.; de Lepeleire, I.; Goldberg, M.; Farrell, D.; Barrett, J.; Hand, E.; Panebianco, D.; Deckmyn, H.; Vermylen, J.; Arnout, J. MK-383 (L-700,462), a selective nonpeptide platelet glycoprotein IIb/IIIa antagonist, is active in man. Circulation 1993, 88, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.S.; Murphy, G.; Peerlinck, K.; Lepeleire, I.D.; Gould, R.J.; Panebianco, D.; Hand, E.; Deckmyn, H.; Vermylen, J.; Arnout, J. Pharmacokinetics and pharmacodynamics of MK-383, a selective non-peptide platelet glycoprotein-IIb/IIIa receptor antagonist, in healthy men. Clin. Pharmacol. Ther. 1994, 56, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.J.; Bednar, B.; Lynch, J.J.; Gould, R.J.; Egbertson, M.S.; Halczenko, W.; Duggan, M.E.; Hartman, G.D.; Lo, M.-W.; Murphy, G.M.; et al. Tirofiban (Aggrastat®). Cardiovasc. Drug Rev. 1999, 17, 199–224. [Google Scholar] [CrossRef]
- Lee, C.Y. Chemistry and pharmacology of polypeptide toxins in snake venoms. Annu. Rev. Pharmacol. 1972, 12, 265–286. [Google Scholar] [CrossRef] [PubMed]
- Tu, A.T. Neurotoxins of animal venoms: Snakes. Annu. Rev. Biochem. 1973, 42, 235–258. [Google Scholar] [CrossRef] [PubMed]
- Tsernoglou, D.; Petsko, G.A. The crystal structure of a post-synaptic neurotoxin from sea snake at 2.2 Å resolution. FEBS Lett. 1976, 68, 1–4. [Google Scholar] [CrossRef]
- Low, B.W.; Potter, R.; Jackson, R.B.; Tamiya, N.; Sato, S. X-ray crystallographic study of the erabutoxins and of a diiodo derivative. J. Biol. Chem. 1971, 246, 4366–4368. [Google Scholar] [PubMed]
- Kahn, M.; Chen, B.; Zieske, P. The design and synthesis of a nonpeptide mimic of erabutoxin. Heterocycles 1987, 25, 29–31. [Google Scholar] [CrossRef]
- Castañeda, O.; Sotolongo, V.; Amor, A.M.; Stöcklin, R.; Anderson, A.J.; Harvey, A.L.; Engström, Å.; Wernstedt, C.; Karlsson, E. Characterization of a potassium channel toxin from the Caribbean sea anemone Stichodactyla helianthus. Toxicon 1995, 33, 603–613. [Google Scholar]
- Pennington, M.W.; Mahnir, V.M.; Krafte, D.S.; Zaydenberg, I.; Byrnes, M.E.; Khaytin, I.; Crowley, K.; Kem, W.R. Identification of three separate binding sites on SHK toxin, a potent inhibitor of voltage-dependent potassium channels in human T-lymphocytes and rat brain. Biochem. Biophys. Res. Commun. 1996, 219, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Rauer, H.; Pennington, M.; Cahalan, M.; Chandy, K.G. Structural conservation of the pores of calcium-activated and voltage-gated potassium channels determined by a sea anemone toxin. J. Biol. Chem. 1999, 274, 21885–21892. [Google Scholar] [CrossRef] [PubMed]
- Tudor, J.E.; Pallaghy, P.K.; Pennington, M.W.; Norton, R.S. Solution structure of ShK toxin, a novel potassium channel inhibitor from a sea anemone. Nat. Struct. Mol. Biol. 1996, 3, 317–320. [Google Scholar] [CrossRef]
- Baell, J.; Harvey, A.; Norton, R. Design and synthesis of type-III mimetics of ShK toxin. J. Comput.-Aided Mol. Des. 2002, 16, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.J.; Gable, R.W.; Baell, J.B. A three-residue, continuous binding epitope peptidomimetic of ShK toxin as a Kv1.3 inhibitor. Bioorg. Med. Chem. Lett. 2005, 15, 3193–3196. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duggan, P.J.; Tuck, K.L. Bioactive Mimetics of Conotoxins and other Venom Peptides. Toxins 2015, 7, 4175-4198. https://doi.org/10.3390/toxins7104175
Duggan PJ, Tuck KL. Bioactive Mimetics of Conotoxins and other Venom Peptides. Toxins. 2015; 7(10):4175-4198. https://doi.org/10.3390/toxins7104175
Chicago/Turabian StyleDuggan, Peter J., and Kellie L. Tuck. 2015. "Bioactive Mimetics of Conotoxins and other Venom Peptides" Toxins 7, no. 10: 4175-4198. https://doi.org/10.3390/toxins7104175
APA StyleDuggan, P. J., & Tuck, K. L. (2015). Bioactive Mimetics of Conotoxins and other Venom Peptides. Toxins, 7(10), 4175-4198. https://doi.org/10.3390/toxins7104175