
Bioinformatics-Aided Venomics

{kind=link}
Abstract
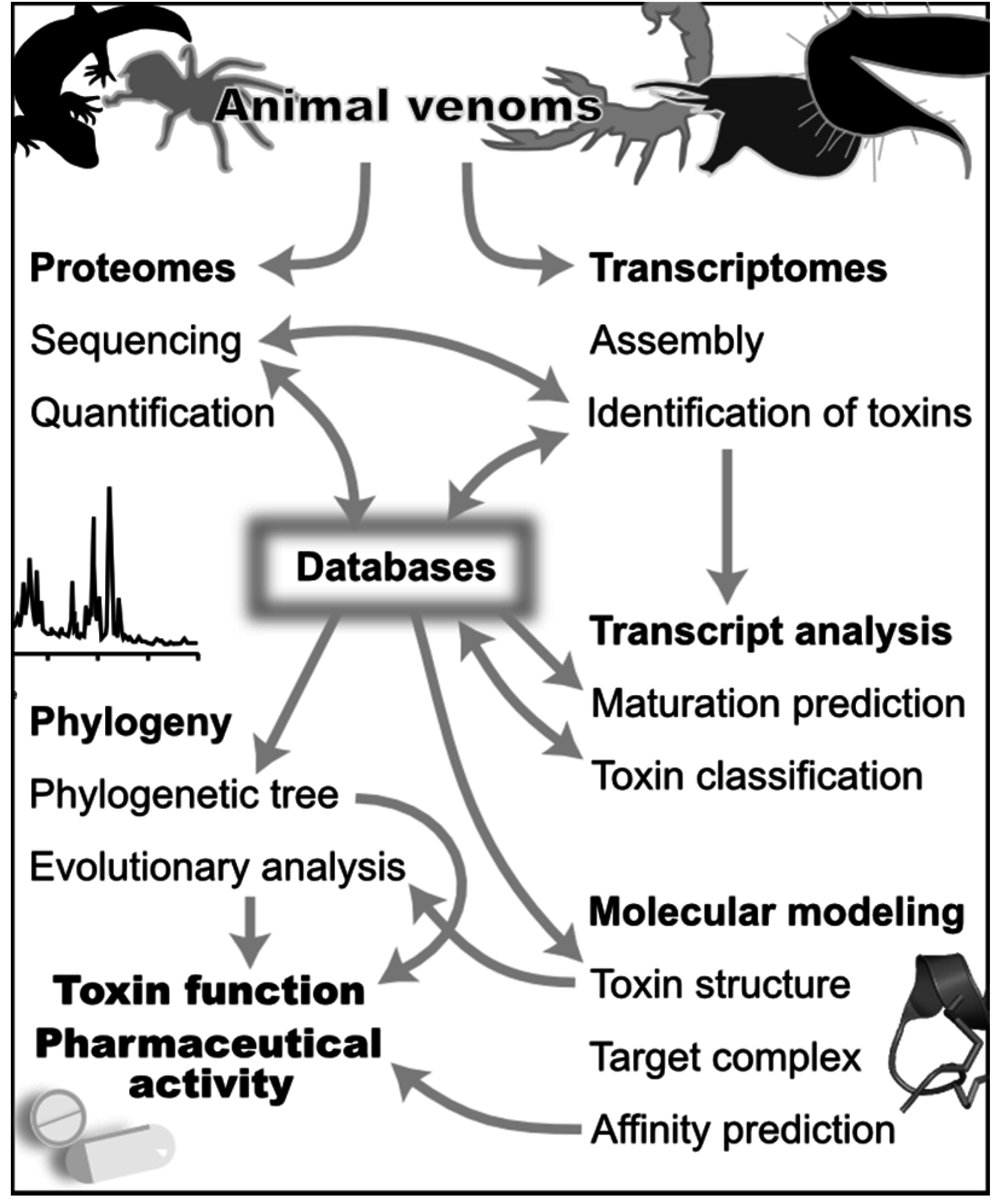
:1. Introduction

2. Databases
3. Discovery of Toxins from Venom Gland Transcriptomes
4. Discovery of Toxins from Proteomes
5. Bioinformatics Analysis of Toxin Transcripts and Classifications
6. Phylogenies and Evolutionary Analysis
6.1. Building Phylogenetic Trees
6.2. Evolutionary Analysis
7. Prediction of Toxin Structures and Activities
7.1. Molecular Modeling of Toxin Structures
7.2. Molecular Modeling of Complexes with Molecular Targets
7.3. Prediction of Toxin Binding Affinities and Specificities
8. Future Perspective: Integrating the Biology of Venoms and Prediction of Toxin Activity
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Georgieva, D.; Arni, R.K.; Betzel, C. Proteome analysis of snake venom toxins: Pharmacological insights. Expert Rev. Proteomics 2008, 5, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Escoubas, P.; Quinton, L.; Nicholson, G.M. Venomics: Unravelling the complexity of animal venoms with mass spectrometry. J. Mass Spectrom. 2008, 43, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Jones, A.; Lewis, R.J. Remarkable inter- and intra-species complexity of conotoxins revealed by LC/MS. Peptides 2009, 30, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Duda, T.F., Jr.; Kohn, A.J.; Matheny, A.M. Cryptic species differentiated in Conus ebraeus, a widespread tropical marine gastropod. Biol. Bull. 2009, 217, 292–305. [Google Scholar] [PubMed]
- Puillandre, N.; Bouchet, P.; Duda, T.F.; Kauferstein, S.; Kohn, A.J.; Olivera, B.M.; Watkins, M.; Meyer, C. Molecular phylogeny and evolution of the cone snails (Gastropoda, Conoidea). Mol. Phylogenet. Evol. 2014, 78, 290–303. [Google Scholar] [CrossRef] [PubMed]
- World Spider Catalog. Available online: http://www.wsc.nmbe.ch/ (accessed on 5 June 2015).
- Vidal, N.; Delmas, A.-S.; David, P.; Cruaud, C.; Couloux, A.; Hedges, S.B. The phylogeny and classification of caenophidian snakes inferred from seven nuclear protein-coding genes. C. R. Biol. 2007, 330, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Uetz, P. The Reptile Database. Available online: http://www.reptile-database.org (accessed on 5 June 2015).
- Cao, Z.; Yu, Y.; Wu, Y.; Hao, P.; Di, Z.; He, Y.; Chen, Z.; Yang, W.; Shen, Z.; He, X.; et al. The genome of Mesobuthus martensii reveals a unique adaptation model of arthropods. Nat. Commun. 2013, 4, 2602. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J. Venomics, what else? Toxicon 2012, 60, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Miljanich, G.P. Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef] [PubMed]
- Beraud, E.; Chandy, K.G. Therapeutic potential of peptide toxins that target ion channels. Inflamm. Allergy Drug Targets 2011, 10, 322–342. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Mebs, D. Toxicity in animals. Trends in evolution? Toxicon 2001, 39, 87–96. [Google Scholar] [CrossRef]
- Kaas, Q.; Westermann, J.-C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G.; Gilad, Y.; Avidan, N.; Ben-Asher, E.; Levy, Z.; Fainzilber, M. Mechanisms for evolving hypervariability: The case of conopeptides. Mol. Biol. Evol. 2001, 18, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Buczek, O.; Bulaj, G.; Olivera, B.M. Conotoxins and the posttranslational modification of secreted gene products. Cell. Mol. Life Sci. 2005, 62, 3067–3079. [Google Scholar] [CrossRef] [PubMed]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, synthesis, and structure-activity relationships of conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Duda, T.F., Jr. Extensive and continuous duplication facilitates rapid evolution and diversification of gene families. Mol. Biol. Evol. 2012, 29, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Prashanth, J.R.; Lewis, R.J.; Dutertre, S. Towards an integrated venomics approach for accelerated conopeptide discovery. Toxicon 2012, 60, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Juárez, P.; Sanz, L.; Calvete, J.J. Snake venomics: Characterization of protein families in Sistrurus barbouri venom by cysteine mapping, N-terminal sequencing, and tandem mass spectrometry analysis. Proteomics 2004, 4, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.T.J.; Khan, A.M.; Brusic, V. Bioinformatics for venom and toxin sciences. Brief. Bioinform. 2003, 4, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2014, 42, D32–D37. [Google Scholar] [CrossRef] [PubMed]
- Jungo, F.; Estreicher, A.; Bairoch, A.; Bougueleret, L.; Xenarios, I. Animal toxins: How is complexity represented in databases? Toxins 2010, 2, 262–282. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Yu, R.; Jin, A.-H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef] [PubMed]
- Herzig, V.; Wood, D.L.A.; Newell, F.; Chaumeil, P.-A.; Kaas, Q.; Binford, G.J.; Nicholson, G.M.; Gorse, D.; King, G.F. ArachnoServer 2.0, an updated online resource for spider toxin sequences and structures. Nucleic Acids Res. 2011, 39, D653–D657. [Google Scholar] [CrossRef] [PubMed]
- Roly, Z.Y.; Hakim, M.A.; Zahan, A.S.; Hossain, M.M.; Reza, M.A. ISOB: A Database of Indigenous Snake Species of Bangladesh with respective known venom composition. Bioinformation 2015, 11, 107–114. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. Activities at the universal protein resource (UniProt). Nucleic Acids Res. 2014, 42, D191–D198. [Google Scholar]
- Gutmanas, A.; Alhroub, Y.; Battle, G.M.; Berrisford, J.M.; Bochet, E.; Conroy, M.J.; Dana, J.M.; Fernandez Montecelo, M.A.; van Ginkel, G.; Gore, S.P.; et al. PDBe: Protein Data Bank in Europe. Nucleic Acids Res. 2014, 42, D285–D291. [Google Scholar] [CrossRef] [PubMed]
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.-C.; Halai, R.; Wang, C.K.L.; Craik, D.J. ConoServer, a database for conopeptide sequences and structures. Bioinform. Oxf. Engl. 2008, 24, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.L.A.; Miljenović, T.; Cai, S.; Raven, R.J.; Kaas, Q.; Escoubas, P.; Herzig, V.; Wilson, D.; King, G.F. ArachnoServer: A database of protein toxins from spiders. BMC Genomics 2009, 10, 375. [Google Scholar] [CrossRef] [PubMed]
- Jungo, F.; Bairoch, A. Tox-Prot, the toxin protein annotation program of the Swiss-Prot protein knowledgebase. Toxicon 2005, 45, 293–301. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Han, W.; He, Q.; Huo, L.; Zhang, J.; Lin, Y.; Chen, P.; Liang, S. ATDB 2.0: A database integrated toxin-ion channel interaction data. Toxicon 2010, 56, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Pawson, A.J.; Sharman, J.L.; Benson, H.E.; Faccenda, E.; Alexander, S.P.H.; Buneman, O.P.; Davenport, A.P.; McGrath, J.C.; Peters, J.A.; Southan, C.; et al. The IUPHAR/BPS Guide to PHARMACOLOGY: An expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2014, 42, D1098–D1106. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Ulrich, E.L.; Berman, H.M.; Henrick, K.; Nakamura, H.; Akutsu, H. BioMagResBank (BMRB) as a partner in the Worldwide Protein Data Bank (wwPDB): New policies affecting biomolecular NMR depositions. J. Biomol. NMR 2008, 40, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, D503–D509. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.T.J.; Veeramani, A.; Srinivasan, K.N.; Ranganathan, S.; Brusic, V. SCORPION2: A database for structure-function analysis of scorpion toxins. Toxicon 2006, 47, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, K.; Undheim, E.A.B.; Chan, A.H.C.; Koludarov, I.; Muñoz-Gómez, S.A.; Antunes, A.; Fry, B.G. Evolution stings: The origin and diversification of scorpion toxin peptide scaffolds. Toxins 2013, 5, 2456–2487. [Google Scholar] [CrossRef] [PubMed]
- Duda, T.F.; Chang, D.; Lewis, B.D.; Lee, T. Geographic variation in venom allelic composition and diets of the widespread predatory marine gastropod Conus ebraeus. PLoS ONE 2009. [Google Scholar] [CrossRef] [PubMed]
- Castoe, T.A.; de Koning, A.P.J.; Hall, K.T.; Card, D.C.; Schield, D.R.; Fujita, M.K.; Ruggiero, R.P.; Degner, J.F.; Daza, J.M.; Gu, W.; et al. The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc. Natl. Acad. Sci. USA 2013, 110, 20645–20650. [Google Scholar] [CrossRef] [PubMed]
- Vonk, F.J.; Casewell, N.R.; Henkel, C.V.; Heimberg, A.M.; Jansen, H.J.; McCleary, R.J.R.; Kerkkamp, H.M.E.; Vos, R.A.; Guerreiro, I.; Calvete, J.J.; et al. The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc. Natl. Acad. Sci. USA 2013, 110, 20651–20656. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Bandyopadhyay, P.K.; Olivera, B.M.; Yandell, M. Characterization of the Conus bullatus genome and its venom-duct transcriptome. BMC Genomics 2011, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Terrat, Y.; Biass, D.; Dutertre, S.; Favreau, P.; Remm, M.; Stöcklin, R.; Piquemal, D.; Ducancel, F. High-resolution picture of a venom gland transcriptome: Case study with the marine snail Conus consors. Toxicon 2012, 59, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Sanggaard, K.W.; Bechsgaard, J.S.; Fang, X.; Duan, J.; Dyrlund, T.F.; Gupta, V.; Jiang, X.; Cheng, L.; Fan, D.; Feng, Y.; et al. Spider genomes provide insight into composition and evolution of venom and silk. Nat. Commun. 2014, 5, 3765. [Google Scholar] [PubMed]
- Honeybee Genome Sequencing Consortium. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 2006, 443, 931–949. [Google Scholar]
- Werren, J.H.; Richards, S.; Desjardins, C.A.; Niehuis, O.; Gadau, J.; Colbourne, J.K.; Nasonia Genome Working Group; Werren, J.H.; Richards, S.; Desjardins, C.A.; et al. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science 2010, 327, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.H.; Gasser, R.B.; Ranganathan, S. A hitchhiker’s guide to expressed sequence tag (EST) analysis. Brief. Bioinform. 2007, 8, 6–21. [Google Scholar] [CrossRef] [PubMed]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology information. Nucleic Acids Res. 2014, 42, D7–D17. [Google Scholar]
- Jin, A.; Dutertre, S.; Kaas, Q.; Lavergne, V.; Kubala, P.; Lewis, R.J.; Alewood, P.F. Transcriptomic messiness in the venom duct of Conus miles contributes to conotoxin diversity. Mol. Cell. Proteomics 2013, 12, 3824–3833. [Google Scholar] [CrossRef] [PubMed]
- Gilles, A.; Meglécz, E.; Pech, N.; Ferreira, S.; Malausa, T.; Martin, J.-F. Accuracy and quality assessment of 454 GS-FLX Titanium pyrosequencing. BMC Genomics 2011, 12, 245. [Google Scholar] [CrossRef] [PubMed]
- Lavergne, V.; Dutertre, S.; Jin, A.; Lewis, R.J.; Taft, R.J.; Alewood, P.F. Systematic interrogation of the Conus marmoreus venom duct transcriptome with ConoSorter reveals 158 novel conotoxins and 13 new gene superfamilies. BMC Genomics 2013, 14, 708. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Green, P. Consed: A graphical editor for next-generation sequencing. Bioinformatics 2013, 29, 2936–2937. [Google Scholar] [CrossRef] [PubMed]
- Swindell, S.R.; Plasterer, T.N. SEQMAN. Contig assembly. Methods Mol. Biol. 1997, 70, 75–89. [Google Scholar] [PubMed]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.J.; Müller, W.E.G.; Wetter, T.; Suhai, S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Gorson, J.; Ramrattan, G.; Verdes, A.; Wright, M.E.; Kantor, Y.; Srinivasan, R.; Musunuri, R.; Packer, D.; Albano, G.; Qiu, W.G.; et al. Molecular diversity and gene evolution of the venom arsenal of terebridae predatory marine snails. Genome Biol. Evol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Jin, A.; Kaas, Q.; Jones, A.; Alewood, P.; Lewis, R. Deep venomics reveals the mechanism for expanded peptide diversity in cone snail venom. Mol. Cell. Proteomics 2013, 12, 312–329. [Google Scholar] [CrossRef] [PubMed]
- Archer, J.; Whiteley, G.; Casewell, N.R.; Harrison, R.A.; Wagstaff, S.C. VTBuilder: A tool for the assembly of multi isoform transcriptomes. BMC Bioinform. 2014, 15, 389. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Letunic, I.; Okuda, S.; Kanehisa, M.; Bork, P. iPath2.0: Interactive pathway explorer. Nucleic Acids Res. 2011, 39, W412–W415. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Bandyopadhyay, P.K.; Olivera, B.M.; Yandell, M. Elucidation of the molecular envenomation strategy of the cone snail conus geographus through transcriptome sequencing of its venom duct. BMC Genomics 2012, 13, 284. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Safavi-Hemami, H.; McIntosh, L.D.; Purcell, A.W.; Norton, R.S.; Papenfuss, A.T. Diversity of conotoxin gene superfamilies in the venomous snail, Conus victoriae. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.F.; Diego-Garcia, E.; de la Vega, R.C.R.; Possani, L.D. Transcriptome analysis of the venom gland of the Mexican scorpion Hadrurus gertschi (Arachnida: Scorpiones). BMC Genomics 2007, 8, 119. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Koua, D.; Brauer, A.; Laht, S.; Kaplinski, L.; Favreau, P.; Remm, M.; Lisacek, F.; Stöcklin, R. ConoDictor: A tool for prediction of conopeptide superfamilies. Nucleic Acids Res. 2012, 40, W238–W241. [Google Scholar] [CrossRef] [PubMed]
- Koua, D.; Laht, S.; Kaplinski, L.; Stöcklin, R.; Remm, M.; Favreau, P.; Lisacek, F. Position-specific scoring matrix and hidden Markov model complement each other for the prediction of conopeptide superfamilies. Biochim. Biophys. Acta 2013, 1834, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Petrel, C.; Hocking, H.G.; Reynaud, M.; Upert, G.; Favreau, P.; Biass, D.; Paolini-Bertrand, M.; Peigneur, S.; Tytgat, J.; Gilles, N.; et al. Identification, structural and pharmacological characterization of τ-CnVA, a conopeptide that selectively interacts with somatostatin sst3 receptor. Biochem. Pharmacol. 2013, 85, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. A probabilistic model of local sequence alignment that simplifies statistical significance estimation. PLoS Comput. Biol. 2008, 4, e1000069. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.; Malyavka, A.; McCutchen, B.; Lu, A.; Schepers, E.; Herrmann, R.; Grishin, E. A novel strategy for the identification of toxinlike structures in spider venom. Proteins 2005, 59, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Gracy, J.; Le-Nguyen, D.; Gelly, J.-C.; Kaas, Q.; Heitz, A.; Chiche, L. KNOTTIN: The knottin or inhibitor cystine knot scaffold in 2007. Nucleic Acids Res. 2008, 36, D314–D319. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.F.C.; Huang, C.; Chong, C.-M.; Leung, S.W.; Prieto-da-Silva, Á.R.B.; Havt, A.; Quinet, Y.P.; Martins, A.M.C.; Lee, S.M.Y.; Rádis-Baptista, G. Transcriptome analysis in venom gland of the predatory giant ant Dinoponera quadriceps: Insights into the polypeptide toxin arsenal of hymenopterans. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Naamati, G.; Askenazi, M.; Linial, M. ClanTox: A classifier of short animal toxins. Nucleic Acids Res. 2009, 37, W363–W368. [Google Scholar] [CrossRef] [PubMed]
- Freund, Y.; Schapire, R.E. A decision-theoretic generalization of on-line learning and an application to boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef]
- Tirosh, Y.; Ofer, D.; Eliyahu, T.; Linial, M. Short toxin-like proteins attack the defense line of innate immunity. Toxins 2013, 5, 1314–1331. [Google Scholar] [CrossRef] [PubMed]
- Lyukmanova, E.N.; Shulepko, M.A.; Buldakova, S.L.; Kasheverov, I.E.; Shenkarev, Z.O.; Reshetnikov, R.V.; Filkin, S.Y.; Kudryavtsev, D.S.; et al. Water-soluble LYNX1 residues important for interaction with muscle-type and/or neuronal nicotinic receptors. J. Biol. Chem. 2013, 288, 15888–15899. [Google Scholar] [CrossRef] [PubMed]
- Shulepko, M.A.; Lyukmanova, E.N.; Paramonov, A.S.; Lobas, A.A.; Shenkarev, Z.O.; Kasheverov, I.E.; Dolgikh, D.A.; Tsetlin, V.I.; Arseniev, A.S.; Kirpichnikov, M.P. Human neuromodulator SLURP-1: Bacterial expression, binding to muscle-type nicotinic acetylcholine receptor, secondary structure, and conformational heterogeneity in solution. Biochemistry 2013, 78, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Starcevic, A.; Moura-da-Silva, A.M.; Cullum, J.; Hranueli, D.; Long, P.F. Combinations of long peptide sequence blocks can be used to describe toxin diversification in venomous animals. Toxicon 2015, 95, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Jin, A.-H.; Vetter, I.; Hamilton, B.; Sunagar, K.; Lavergne, V.; Dutertre, V.; Fry, B.G.; Antunes, A.; Venter, D.J.; et al. Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails. Nat. Commun. 2014, 5, 3521. [Google Scholar] [CrossRef] [PubMed]
- Inceoglu, B.; Lango, J.; Jing, J.; Chen, L.; Doymaz, F.; Pessah, I.N.; Hammock, B.D. One scorpion, two venoms: Prevenom of Parabuthus transvaalicus acts as an alternative type of venom with distinct mechanism of action. Proc. Natl. Acad. Sci. USA 2003, 100, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.; Pisarewicz, K.; Moller, C.; Mora, D.; Fields, G.B.; Marì, F. Hyperhydroxylation: A new strategy for neuronal targeting by venomous marine molluscs. Prog. Mol. Subcell. Biol. 2006, 43, 83–103. [Google Scholar] [PubMed]
- Tawfik, D.S. Messy biology and the origins of evolutionary innovations. Nat. Chem. Biol. 2010, 6, 692–696. [Google Scholar] [PubMed]
- Gordon, A.J.E.; Satory, D.; Halliday, J.A.; Herman, C. Lost in transcription: Transient errors in information transfer. Curr. Opin. Microbiol. 2015, 24, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Gout, J.-F.; Thomas, W.K.; Smith, Z.; Okamoto, K.; Lynch, M. Large-scale detection of in vivo transcription errors. Proc. Natl. Acad. Sci. USA 2013, 110, 18584–18589. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.W.; Serrano, S.M.T. Exploring snake venom proteomes: Multifaceted analyses for complex toxin mixtures. Proteomics 2008, 8, 909–920. [Google Scholar] [CrossRef] [PubMed]
- De Queiroz, M.R.; Mamede, C.C.N.; de Morais, N.C.; Fonseca, K.C.; de Sousa, B.B.; Migliorini, T.M.; Pereira, D.F.; Stanziola, L.; Calderon, L.A.; Simões-Silva, R.; et al. Purification and Characterization of BmooAi: A New Toxin from Bothrops moojeni Snake Venom That Inhibits Platelet Aggregation. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Hardy, M.C.; Daly, N.L.; Mobli, M.; Morales, R.A.V.; King, G.F. Isolation of an orally active insecticidal toxin from the venom of an Australian tarantula. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J.; Ghezellou, P.; Paiva, O.; Matainaho, T.; Ghassempour, A.; Goudarzi, H.; Kraus, F.; Sanz, L.; Williams, D.J. Snake venomics of two poorly known Hydrophiinae: Comparative proteomics of the venoms of terrestrial Toxicocalamus longissimus and marine Hydrophis cyanocinctus. J. Proteomics 2012, 75, 4091–4101. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Kil, Y.J.; Ueberheide, B.; Chait, B.T.; Tayo, L.; Cruz, L.; Lu, B.; Yates, J.R., 3rd; Bern, M. Constrained de novo sequencing of conotoxins. J. Proteome Res. 2012, 11, 4191–4200. [Google Scholar] [CrossRef] [PubMed]
- Escoubas, P.; Sollod, B.; King, G.F. Venom landscapes: Mining the complexity of spider venoms via a combined cDNA and mass spectrometric approach. Toxicon 2006, 47, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Beavis, R.C. TANDEM: Matching proteins with tandem mass spectra. Bioinform. Oxf. Engl. 2004, 20, 1466–1467. [Google Scholar] [CrossRef] [PubMed]
- Hoopmann, M.R.; Moritz, R.L. Current algorithmic solutions for peptide-based proteomics data generation and identification. Curr. Opin. Biotechnol. 2013, 24, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Bunkenborg, J.; Matthiesen, R. Interpretation of tandem mass spectra of posttranslationally modified peptides. Methods Mol. Biol. 2013, 1007, 139–171. [Google Scholar] [PubMed]
- Skjaerbaek, N.; Nielsen, K.J.; Lewis, R.J.; Alewood, P.; Craik, D.J. Determination of the solution structures of conantokin-G and conantokin-T by CD and NMR spectroscopy. J. Biol. Chem. 1997, 272, 2291–2299. [Google Scholar] [PubMed]
- Dutton, J.L.; Bansal, P.S.; Hogg, R.C.; Adams, D.J.; Alewood, P.F.; Craik, D.J. A new level of conotoxin diversity, a non-native disulfide bond connectivity in α-conotoxin AuIB reduces structural definition but increases biological activity. J. Biol. Chem. 2002, 277, 48849–48857. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Gasteiger, E.; Gooley, A.A.; Herbert, B.R.; Molloy, M.P.; Binz, P.A.; Ou, K.; Sanchez, J.C.; Bairoch, A.; Williams, K.L.; et al. High-throughput mass spectrometric discovery of protein post-translational modifications. J. Mol. Biol. 1999, 289, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Pang, C.N.I.; Gasteiger, E.; Wilkins, M.R. Identification of arginine- and lysine-methylation in the proteome of Saccharomyces cerevisiae and its functional implications. BMC Genomics 2010, 11, 92. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J.; Marcinkiewicz, C.; Sanz, L. Snake venomics of Bitis gabonica gabonica. Protein family composition, subunit organization of venom toxins, and characterization of dimeric disintegrins bitisgabonin-1 and bitisgabonin-2. J. Proteome Res. 2007, 6, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Durban, J.; Pérez, A.; Sanz, L.; Gómez, A.; Bonilla, F.; Rodríguez, S.; Chacón, D.; Sasa, M.; Angulo, Y.; Gutiérrez, J.M.; et al. Integrated “omics” profiling indicates that miRNAs are modulators of the ontogenetic venom composition shift in the Central American rattlesnake, Crotalus simus simus. BMC Genomics 2013, 14, 234. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Petersen 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.A.; Grishin, E.V. The universal algorithm of maturation for secretory and excretory protein precursors. Toxicon 2007, 49, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhangsun, D.; Wu, Y.; Zhu, X.; Xie, L.; Hu, Y.; Zhang, J.; Zhao, X. Identification and molecular diversity of T-superfamily conotoxins from Conus lividus and Conus litteratus. Chem. Biol. Drug Des. 2006, 68, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Duckert, P.; Brunak, S.; Blom, N. Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel. 2004, 17, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.S.W.; Hardy, M.C.; Wood, D.; Bailey, T.; King, G.F. SVM-based prediction of propeptide cleavage sites in spider toxins identifies toxin innovation in an Australian tarantula. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Milne, T.J.; Abbenante, G.; Tyndall, J.D.A.; Halliday, J.; Lewis, R.J. Isolation and characterization of a cone snail protease with homology to CRISP proteins of the pathogenesis-related protein superfamily. J. Biol. Chem. 2003, 278, 31105–31110. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Bhavna, R.; Mohan Babu, R.; Ramakumar, S. Pseudo amino acid composition and multi-class support vector machines approach for conotoxin superfamily classification. J. Theor. Biol. 2006, 243, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Li, Q.-Z. Predicting conotoxin superfamily and family by using pseudo amino acid composition and modified Mahalanobis discriminant. Biochem. Biophys. Res. Commun. 2007, 354, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.-B.; Fan, Y.-X.; Shen, H.-B. Conotoxin superfamily prediction using diffusion maps dimensionality reduction and subspace classifier. Curr. Protein Pept. Sci. 2011, 12, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Zaki, N.; Wolfsheimer, S.; Nuel, G.; Khuri, S. Conotoxin protein classification using free scores of words and support vector machines. BMC Bioinform. 2011, 12, 217. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Peigneur, S.; Gao, B.; Luo, L.; Jin, D.; Zhao, Y.; Tytgat, J. Molecular diversity and functional evolution of scorpion potassium channel toxins. Mol. Cell. Proteomics 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, K.; Jackson, T.N.W.; Undheim, E.A.B.; Ali, S.A.; Antunes, A.; Fry, B.G. Three-fingered RAVERs: Rapid Accumulation of Variations in Exposed Residues of snake venom toxins. Toxins 2013, 5, 2172–2208. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Undheim, E.A.B.; Ali, S.A.; Jackson, T.N.W.; Debono, J.; Scheib, H.; Ruder, T.; Morgenstern, D.; Cadwallader, L.; Whitehead, D.; et al. Squeezers and leaf-cutters: Differential diversification and degeneration of the venom system in toxicoferan reptiles. Mol. Cell. Proteomics 2013, 12, 1881–1899. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B. Conus venom peptides: Reflections from the biology of clades and species. Annu. Rev. Ecol. Syst. 2002, 33, 25–47. [Google Scholar] [CrossRef]
- Biggs, J.S.; Watkins, M.; Puillandre, N.; Ownby, J.-P.; Lopez-Vera, E.; Christensen, S.; Moreno, K.J.; Bernaldez, J.; Licea-Navarro, A.; Corneli, P.S.; et al. Evolution of Conus peptide toxins: Analysis of Conus californicus Reeve, 1844. Mol. Phylogenet. Evol. 2010, 56, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Wüster, W.; Kini, R.M.; Brusic, V.; Khan, A.; Venkataraman, D.; Rooney, A.P. Molecular evolution and phylogeny of elapid snake venom three-finger toxins. J. Mol. Evol. 2003, 57, 110–129. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Deng, M.; He, Q.; Meng, E.; Jiang, L.; Liao, Z.; Rong, M.; Liang, S. Molecular diversity and evolution of cystine knot toxins of the tarantula Chilobrachys jingzhao. Cell. Mol. Life Sci. 2008, 65, 2431–2444. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Puillandre, N.; Koua, D.; Favreau, P.; Olivera, B.M.; Stöcklin, R. Molecular phylogeny, classification and evolution of conopeptides. J. Mol. Evol. 2012, 74, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Duda, T.F., Jr.; Remigio, E.A. Variation and evolution of toxin gene expression patterns of six closely related venomous marine snails. Mol. Ecol. 2008, 17, 3018–3032. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Keane, T.M.; Creevey, C.J.; Pentony, M.M.; Naughton, T.J.; Mclnerney, J.O. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol. 2006, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. MODELTEST: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, H.; Moran, Y.; Gordon, D.; Turkov, M.; Kahn, R.; Gurevitz, M. Positions under positive selection—Key for selectivity and potency of scorpion α-toxins. Mol. Biol. Evol. 2010, 27, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Rosenberg, H.F.; Nei, M. Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc. Natl. Acad. Sci. USA 1998, 95, 3708–3713. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wong, W.S.W.; Nielsen, R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Pond, S.L.K.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Woolley, S.; Johnson, J.; Smith, M.J.; Crandall, K.A.; McClellan, D.A. TreeSAAP: Selection on amino acid properties using phylogenetic trees. Bioinform. Oxf. Engl. 2003, 19, 671–672. [Google Scholar] [CrossRef]
- Diochot, S.; Drici, M.D.; Moinier, D.; Fink, M.; Lazdunski, M. Effects of phrixotoxins on the Kv4 family of potassium channels and implications for the role of Ito1 in cardiac electrogenesis. Br. J. Pharmacol. 1999, 126, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Oswald, R.E.; Suchyna, T.M.; McFeeters, R.; Gottlieb, P.; Sachs, F. Solution structure of peptide toxins that block mechanosensitive ion channels. J. Biol. Chem. 2002, 277, 34443–34450. [Google Scholar] [CrossRef] [PubMed]
- Edgerton, G.B.; Blumenthal, K.M.; Hanck, D.A. Inhibition of the activation pathway of the T-type calcium channel Ca(V)3.1 by ProTxII. Toxicon 2010, 56, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Middleton, R.E.; Warren, V.A.; Kraus, R.L.; Hwang, J.C.; Liu, C.J.; Dai, G.; Brochu, R.M.; Kohler, M.G.; Gao, Y.-D.; Garsky, V.M.; et al. Two tarantula peptides inhibit activation of multiple sodium channels. Biochemistry 2002, 41, 14734–14747. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.T.J.; Srinivasan, K.N.; Seah, S.H.; Koh, J.L.Y.; Tan, T.W.; Ranganathan, S.; Brusic, V. Accurate prediction of scorpion toxin functional properties from primary structures. J. Mol. Graph. Model. 2005, 24, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.-F.; Ding, C.; Guo, S.-H.; Ding, H.; Chen, W.; Lin, H. Prediction of the types of ion channel-targeted conotoxins based on radial basis function network. Toxicol. In Vitro 2013, 27, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Luo, L. Use of tetrapeptide signals for protein secondary-structure prediction. Amino Acids 2008, 35, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; MacKinnon, R. A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature 2004, 430, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Almeida, D.D.; Torres, T.M.; Barbosa, E.G.; Lima, J.P.M.S.; de Freitas Fernandes-Pedrosa, M. Molecular approaches for structural characterization of a new potassium channel blocker from Tityus stigmurus venom: cDNA cloning, homology modeling, dynamic simulations and docking. Biochem. Biophys. Res. Commun. 2013, 430, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Maupetit, J.; Derreumaux, P.; Tuffery, P. PEP-FOLD: An online resource for de novo peptide structure prediction. Nucleic Acids Res. 2009, 37, W498–W503. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chang, S.; Yang, L.; Shi, J.; McFarland, K.; Yang, X.; Moller, A.; Wang, C.; Zou, X.; Chi, C.; et al. Conopeptide Vt3.1 preferentially inhibits BK potassium channels containing β4 subunits via electrostatic interactions. J. Biol. Chem. 2014, 289, 4735–4742. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, D.L.; Konstantakopoulos, N.; McInerney, B.V.; Mulvenna, J.; Seymour, J.E.; Isbister, G.K.; Hodgson, W.C. Chironex fleckeri (box jellyfish) venom proteins: Expansion of a cnidarian toxin family that elicits variable cytolytic and cardiovascular effects. J. Biol. Chem. 2014, 289, 4798–4812. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.J.; Thomas, L.; Lewis, R.J.; Alewood, P.F.; Craik, D.J. A consensus structure for omega-conotoxins with different selectivities for voltage-sensitive calcium channel subtypes: Comparison of MVIIA, SVIB and SNX-202. J. Mol. Biol. 1996, 263, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, M.J.; Naranjo, D.; Thomas, L.; Alewood, P.F.; Lewis, R.J.; Craik, D.J. Solution structure and proposed binding mechanism of a novel potassium channel toxin κ-conotoxin PVIIA. Structure 1997, 5, 1585–1597. [Google Scholar] [CrossRef]
- Fletcher, J.I.; Chapman, B.E.; Mackay, J.P.; Howden, M.E.; King, G.F. The structure of versutoxin (δ-atracotoxin-Hv1) provides insights into the binding of site 3 neurotoxins to the voltage-gated sodium channel. Structure 1997, 5, 1525–1535. [Google Scholar] [CrossRef]
- Baconguis, I.; Gouaux, E. Structural plasticity and dynamic selectivity of acid-sensing ion channel-spider toxin complexes. Nature 2012, 489, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Ulens, C.; Büttner, R.; Fish, A.; van Elk, R.; Kendel, Y.; Hopping, G.; Alewood, P.F.; Schroeder, C.; Nicke, A.; et al. AChBP-targeted α-conotoxin correlates distinct binding orientations with nAChR subtype selectivity. EMBO J. 2007, 26, 3858–3867. [Google Scholar] [CrossRef] [PubMed]
- Tsetlin, V.; Utkin, Y.; Kasheverov, I. Polypeptide and peptide toxins, magnifying lenses for binding sites in nicotinic acetylcholine receptors. Biochem. Pharmacol. 2009, 78, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Long, S.B.; Campbell, E.B.; MacKinnon, R. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science 2005, 309, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Payandeh, J.; Scheuer, T.; Zheng, N.; Catterall, W.A. The crystal structure of a voltage-gated sodium channel. Nature 2011, 475, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Unwin, N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J. Mol. Biol. 2005, 346, 967–989. [Google Scholar] [CrossRef] [PubMed]
- Jasti, J.; Furukawa, H.; Gonzales, E.B.; Gouaux, E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 2007, 449, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Craik, D.J.; Kaas, Q. Blockade of neuronal α7-nAChR by α-conotoxin ImI explained by computational scanning and energy calculations. PLoS Comput. Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Kompella, S.N.; Adams, D.J.; Craik, D.J.; Kaas, Q. Determination of the α-conotoxin Vc1.1 binding site on the α9α10 nicotinic acetylcholine receptor. J. Med. Chem. 2013, 56, 3557–3567. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Kaas, Q.; Craik, D.J. Delineation of the unbinding pathway of α-conotoxin ImI from the α7 nicotinic acetylcholine receptor. J. Phys. Chem. B 2012, 116, 6097–6105. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhangsun, D.; Schroeder, C.I.; Zhu, X.; Hu, Y.; Wu, Y.; Weltzin, M.M.; Eberhard, S.; Kaas, Q.; Craik, D.J.; et al. A novel α4/7-conotoxin LvIA from Conus lividus that selectively blocks α3β2 vs. α6/α3β2β3 nicotinic acetylcholine receptors. FASEB J. 2014, 28, 1842–1853. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.A.; Cuny, H.; Hung, A.; Clark, R.J.; Brust, A.; Akondi, K.; Alewood, P.F.; Craik, D.J.; Adams, D.J. Identifying key amino acid residues that affect α-conotoxin AuIB inhibition of α3β4 nicotinic acetylcholine receptors. J. Biol. Chem. 2013, 288, 34428–34442. [Google Scholar] [CrossRef] [PubMed]
- Pucci, L.; Grazioso, G.; Dallanoce, C.; Rizzi, L.; de Micheli, C.; Clementi, F.; Bertrand, S.; Bertrand, D.; Longhi, R.; de Amici, M.; et al. Engineering of α-conotoxin MII-derived peptides with increased selectivity for native α6β2* nicotinic acetylcholine receptors. FASEB J. 2011, 25, 3775–3789. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, J.; Wang, X.; Yan, S.; Xu, Y.; San, M.; Tang, W.; Yang, F.; Cao, Z.; Li, W.; et al. Functional characterization of a new non-Kunitz serine protease inhibitor from the scorpion Lychas mucronatus. Int. J. Biol. Macromol. 2015, 72C, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Chen, R.; Chung, S.-H. Computational methods of studying the binding of toxins from venomous animals to biological ion channels: Theory and applications. Physiol. Rev. 2013, 93, 767–802. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Hourai, Y.; Weng, Z. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, P.L.; Rodrigues, J.P.G.L.M.; Bonvin, A.M.J.J. HADDOCK(2P2I): A biophysical model for predicting the binding affinity of protein-protein interaction inhibitors. J. Chem. Inf. Model. 2014, 54, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Salinas, M.; Besson, T.; Delettre, Q.; Diochot, S.; Boulakirba, S.; Douguet, D.; Lingueglia, E. Binding site and inhibitory mechanism of the mambalgin-2 pain-relieving peptide on acid-sensing ion channel 1a. J. Biol. Chem. 2014, 289, 13363–13373. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.-H. Binding modes and functional surface of anti-mammalian scorpion α-toxins to sodium channels. Biochemistry 2012, 51, 7775–7782. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.-H. Structural basis of the selective block of Kv1.2 by maurotoxin from computer simulations. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Wee, C.L.; Gavaghan, D.; Sansom, M.S.P. Interactions between a voltage sensor and a toxin via multiscale simulations. Biophys. J. 2010, 98, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Chen, R.; Ho, J.; Coote, M.L.; Chung, S.-H. Rigid body Brownian dynamics as a tool for studying ion channel blockers. J. Phys. Chem. B 2012, 116, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Fu, W.; Liu, H.; Luo, X.; Chen, K.X.; Ding, J.; Shen, J.; Jiang, H. Computational simulations of interactions of scorpion toxins with the voltage-gated potassium ion channel. Biophys. J. 2004, 86, 3542–3555. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Zhou, H.-X. Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Mahdavi, S.; Kuyucak, S. Computational studies of marine toxins targeting ion channels. Mar. Drugs 2013, 11, 848–869. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Chen, R.; Chung, S.-H. Conserved functional surface of antimammalian scorpion β-toxins. J. Phys. Chem. B 2012, 116, 4796–4800. [Google Scholar] [CrossRef] [PubMed]
- Jarzynski, C. Equalities and inequalities: Irreversibility and the second law of thermodynamics at the nanoscale. Annu. Rev. Condens. Matter Phys. 2011, 2, 329–351. [Google Scholar] [CrossRef]
- Baştuğ, T.; Chen, P.-C.; Patra, S.M.; Kuyucak, S. Potential of mean force calculations of ligand binding to ion channels from Jarzynski’s equality and umbrella sampling. J. Chem. Phys. 2008, 128, 155104. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Qiu, S.; Wu, Y.; Li, W.; Wang, B. Differential molecular information of maurotoxin peptide recognizing IK(Ca) and Kv1.2 channels explored by computational simulation. BMC Struct. Biol. 2011, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Weis, A.; Katebzadeh, K.; Söderhjelm, P.; Nilsson, I.; Ryde, U. Ligand affinities predicted with the MM/PBSA method: Dependence on the simulation method and the force field. J. Med. Chem. 2006, 49, 6596–6606. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaas, Q.; Craik, D.J. Bioinformatics-Aided Venomics. Toxins 2015, 7, 2159-2187. https://doi.org/10.3390/toxins7062159
Kaas Q, Craik DJ. Bioinformatics-Aided Venomics. Toxins. 2015; 7(6):2159-2187. https://doi.org/10.3390/toxins7062159
Chicago/Turabian StyleKaas, Quentin, and David J. Craik. 2015. "Bioinformatics-Aided Venomics" Toxins 7, no. 6: 2159-2187. https://doi.org/10.3390/toxins7062159
APA StyleKaas, Q., & Craik, D. J. (2015). Bioinformatics-Aided Venomics. Toxins, 7(6), 2159-2187. https://doi.org/10.3390/toxins7062159