
RNA Sequencing of Contaminated Seeds Reveals the State of the Seed Permissive for Pre-Harvest Aflatoxin Contamination and Points to a Potential Susceptibility Factor


Abstract
:1. Introduction
2. Results
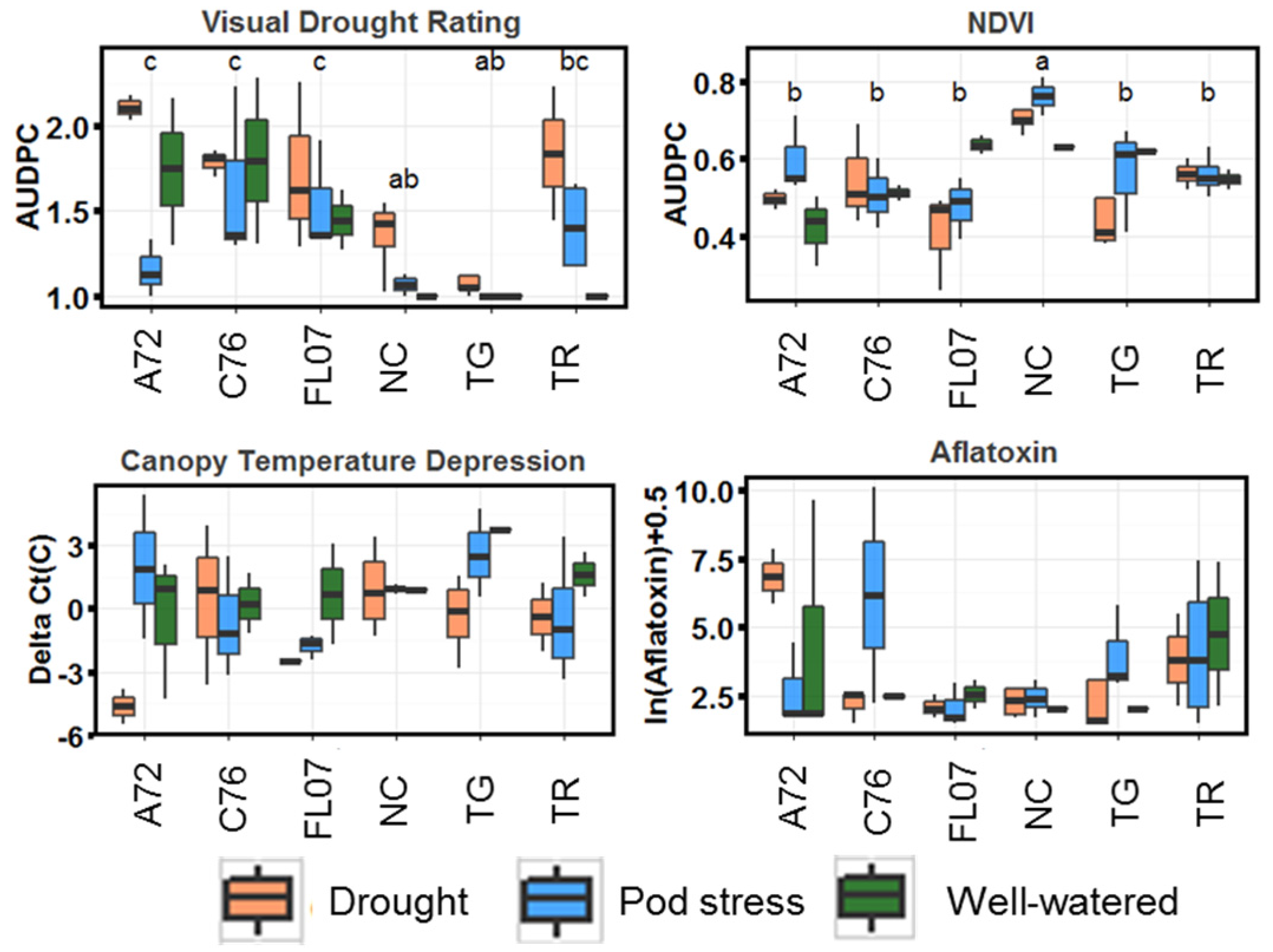
2.1. Prediction of Aflatoxin by Drought Tolerance-Related Traits
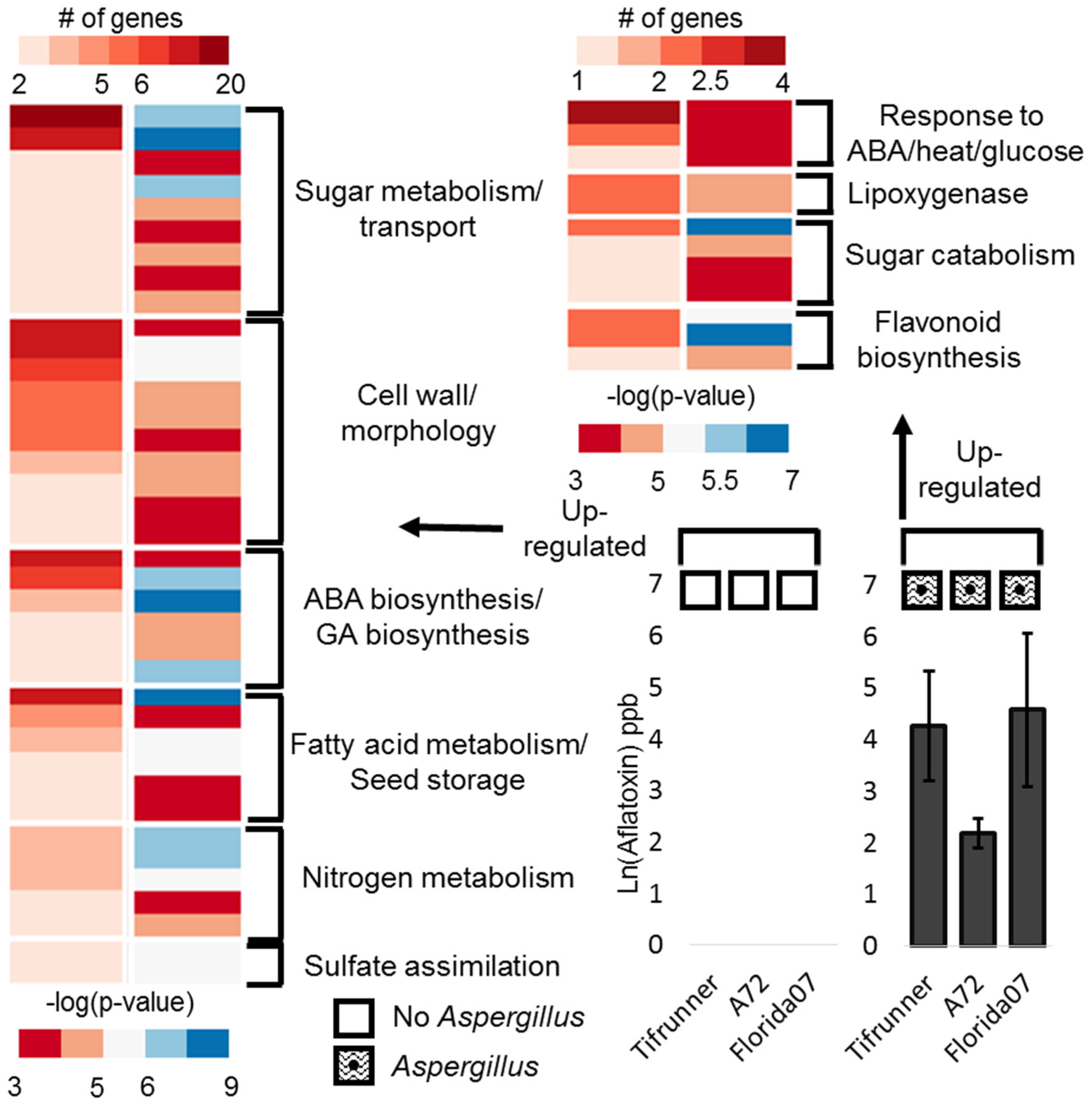
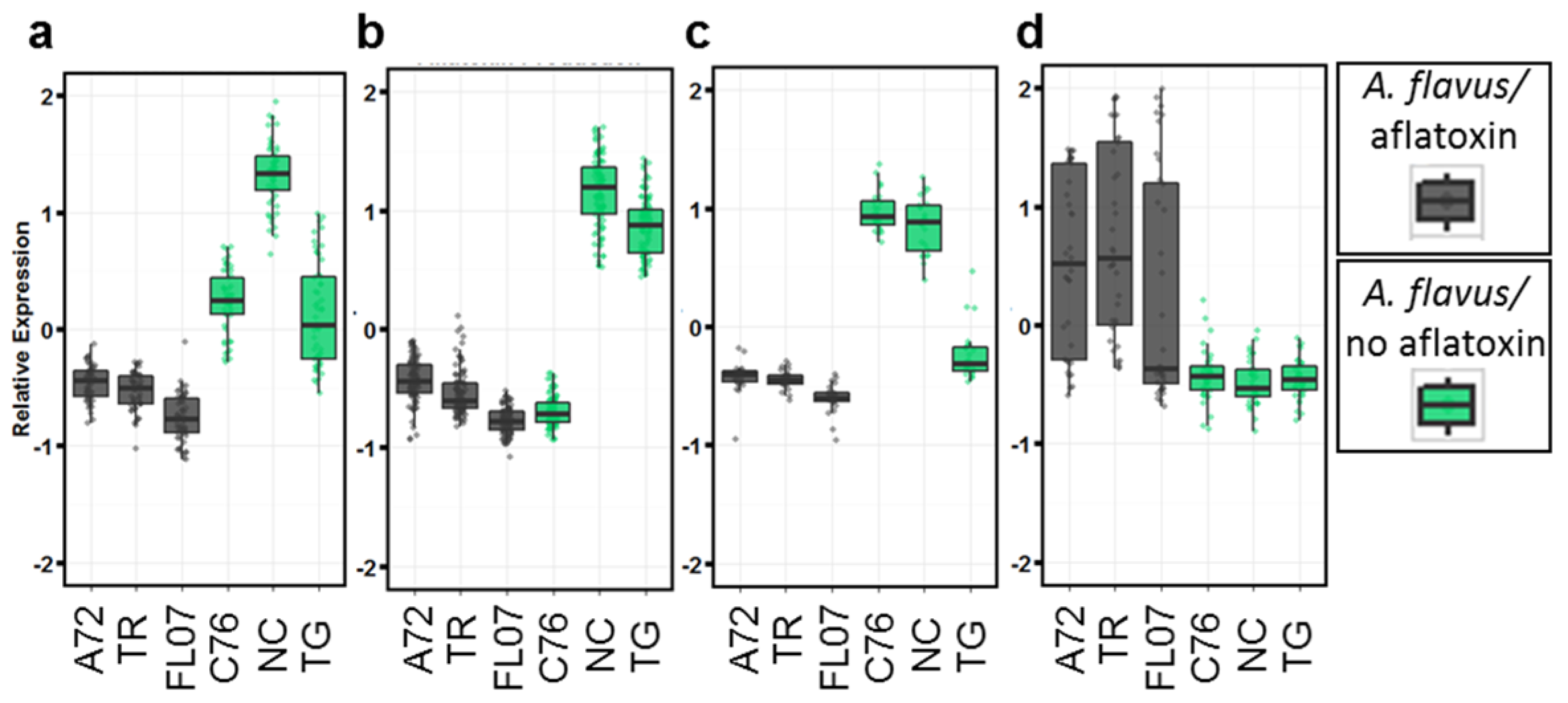
2.2. Differentially Expressed Genes in Seeds Contaminated with Aflatoxin
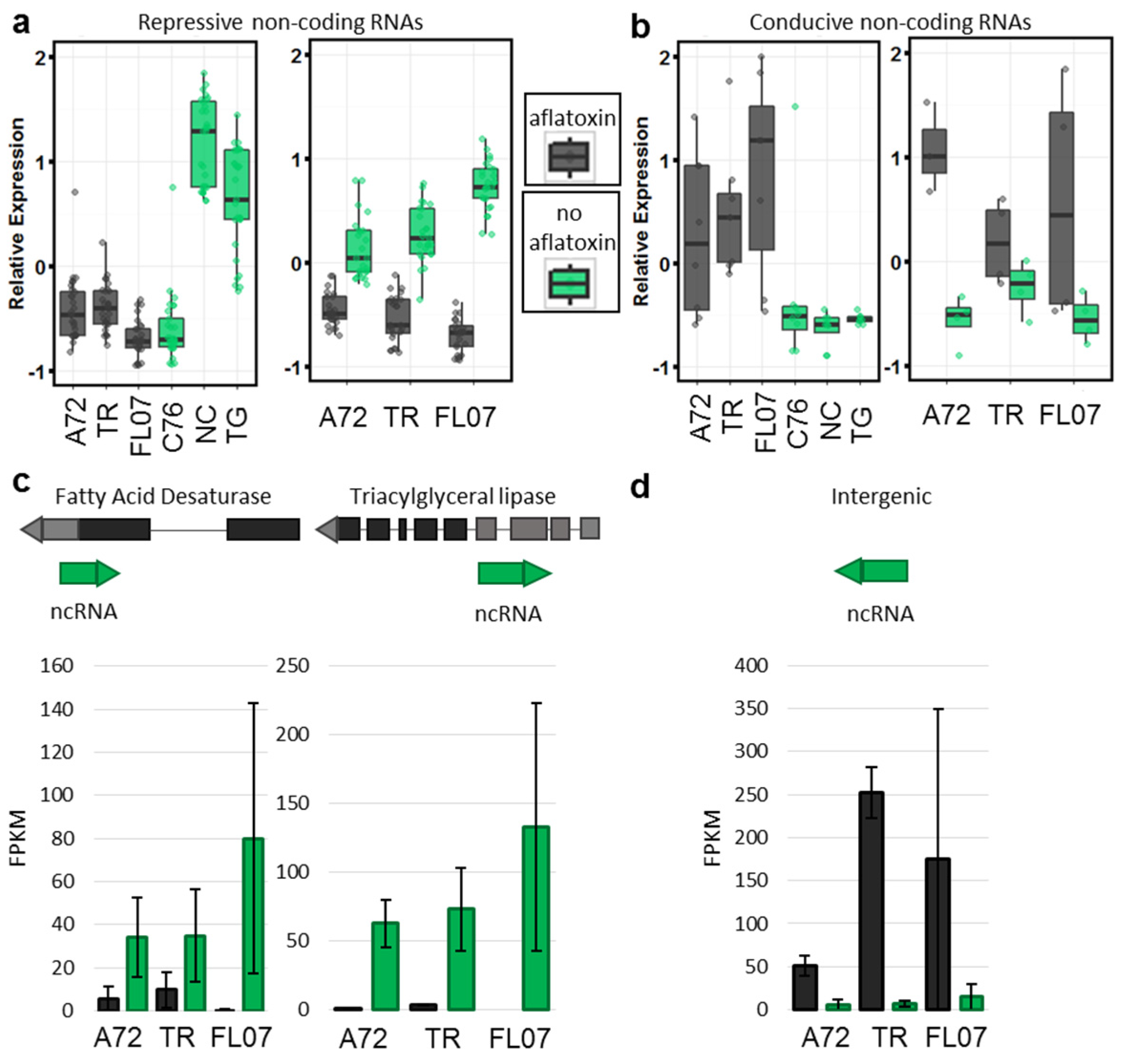
2.3. Aflatoxin Contamination Reveals Differential Expression of Non-Coding RNAs
2.4. SNPs Associated with Differentially Expressed Genes as Possible Aflatoxin-Associated eQTL
3. Discussion
4. Materials and Methods
4.1. Automated Rainout Shelter
4.2. Drought Tolerance Phenotyping
4.3. Aflatoxin Quantification
4.4. Seed Processing for RNA Sequencing
4.5. RNA Sequencing
4.6. Expression Analysis
4.7. Grouping of Differentially Expressed Genes and Enriched GO Analysis
4.8. Non-Coding RNAs
4.9. Statistical Analysis
4.10. SNP Identification
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yu, J.; Cleveland, T.E.; Nierman, W.C.; Bennett, J.W. Aspergillus flavus genomics: Gateway to human and animal health, food safety, and crop resistance to diseases. Rev. Iberoam. Micol. 2005, 22, 194–202. [Google Scholar] [CrossRef]
- Yard, E.; Daniel, J.H.; Lewis, L.S.; Rybak, M.E.; Paliakov, E.M.; Kim, A.A.; Montgomery, J.M.; Bunnell, R.; Abudo, M.U.; Akhwale, W.; et al. Human aflatoxin exposure in Kenya, 2007: A cross-sectional study. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2013, 30, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Groopman, J.; Egner, P.A.; Schulze, K.J.; Wu, L.S.; Merrill, R.; Mehra, S.; Shamim, A.A.; Ali, H.; Shaikh, S.; Gernand, A.; et al. Aflatoxin exposure during the first 1000 days of life in rural South Asia assessed by aflatoxin B1-lysine albumin biomarkers. Food Chem. Toxicol. 2014, 74, 184–189. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group. Mycotoxin Control in Low- and Middle Income Countries; International Agency for Research on Cancer and World Health Organization: Lyon, France, 2015. [Google Scholar]
- Gong, Y.; Wilson, S.; Mwatha, J.K.; Routledge, M.N.; Castelino, J.M.; Zhao, B.; Kimani, G.; Kariuki, H.C.; Vennervald, B.J.; Dunne, D.W.; et al. Aflatoxin exposure may contribute to chronic hepatomegaly in Kenyan school children. Environ. Health Perspect. 2012, 120, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Lamb, M.; Sternitzk, D.A. Cost of aflatoxin to the farmer, buying point, and sheller segments of the southeast United States peanut industry. Peanut Sci. 2001, 28, 59–63. [Google Scholar] [CrossRef]
- Burow, G.; Nesbitt, T.C.; Dunlap, J.; Keller, N.P. Seed lipoxygenase products modulate Aspergillus mycotoxin biosynthesis. Mol. Plant Microbe Interact. 1997, 10, 380–387. [Google Scholar] [CrossRef]
- Xue, H.; Isleib, T.G.; Payne, G.A.; Wilson, R.F.; Novitzky, W.P.; O’Brian, G. Comparison of aflatoxin production in normal- and high-oleic backcross-derived peanut lines. Plant Dis. 2003, 87, 1360–1365. [Google Scholar] [CrossRef]
- Burow, G.; Gardner, H.W.; Keller, N.P. A peanut seed lipoxygenase responsive to Aspergillus colonization. Plant Mol. Biol. 2000, 42, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Tsitsigiannis, D.; Kunze, S.; Willis, D.K.; Feussner, I.; Keller, N.P. Aspergillus infection inhibits the expression of peanut 13S-hpode-forming seed lipoxygenases. Mol. Plant Microbe Interact. 2005, 18, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Goodrich-Tanrikulu, M.; Mahoney, N.E.; Rodriguez, S.B. The plant growth regulator methyl jasmonate inhibits aflatoxin production by Aspergillus flavus. Microbiology 1995, 141, 2831–2837. [Google Scholar] [CrossRef] [PubMed]
- Meimaroglou, D.; Galanopoulou, D.; Markaki, P. Study of the effect of methyl jasmonate concentration on aflatoxin B1 biosynthesis by Aspergillus parasiticus in yeast extract sucrose medium. Int. J. Microbiol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Brodhagen, M.; Isakeit, T.; Brown, S.H.; Göbel, C.; Betran, J.; Feussner, I.; Keller, N.P.; Kolomiets, M.V. Inactivation of the lipoxygenase ZMLOX3 increases susceptibility of maize to Aspergillus spp. Mol. Plant Microbe Interact. 2009, 22, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, C.; Wilson, D.M.; Matheron, M.E.; Hunter, J.E.; Knauft, D.A.; Gorbet, D.W. Aspergillus colonization and aflatoxin contamination in peanut genotypes with reduced linoleic acid composition. Plant Dis. 2000, 84, 148–150. [Google Scholar] [CrossRef]
- Wotton, H.; Strange, R.N. Increased susceptibility and reduced phytoalexin accumulation in drought-stressed peanut kernels challenged with Aspergillus flavus. Appl. Environ. Microbiol. 1987, 53, 270–273. [Google Scholar] [PubMed]
- Dorner, J.; Cole, R.J.; Sanders, T.H.; Blankenship, P.D. Interrelationship of kernel water activity, soil temperature, maturity, and phytoalexin production in preharvest aflatoxin contamination of drought-stressed peanuts. Mycopathologia 1989, 105, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.; Holbrook, C.C.; Wilson, D.M. Development of greenhouse screening for resistance to Aspergillus parasiticus infection and preharvest aflatoxin contamination in peanut. Mycopathologia 1996, 115, 115–118. [Google Scholar] [CrossRef]
- Holbrook, C.; Kvien, C.K.; Rucker, K.S.; Wilson, D.W.; Hook, S.J. Preharvest aflatoxin contamination in drought tolerant and intolerant peanut genotypes. Peanut Sci. 2000, 27, 45–48. [Google Scholar] [CrossRef]
- Arunyanark, A.; Jogloy, S.; Wongkaew, S.; Akkasaeng, C.; Vorasoot, N.; Wright, G.C.; Rachaputi Rao, C.N.; Patanothai, A. Association between aflatoxin contamination and drought tolerance traits in peanut. Field Crops Res. 2009, 114, 14–22. [Google Scholar] [CrossRef]
- Girdthai, T.; Jogloy, S.; Vorasoot, N.; Akksaeng, C.; Wongkaew, S.; Holbrook, C.C.; Patanothal, A. Associations between physiological traits for drought tolerance and aflatoxin contamination in peanut genotypes under terminal drought. Plant Breed. 2010, 129, 693–699. [Google Scholar] [CrossRef]
- Kebede, H.; Abbas, H.K.; Fisher, D.K.; Bellaloui, N. Relationship between aflatoxin contamination and physiological responses of corn plants under drought and heat stress. Toxins (Basel) 2012, 4, 1385–1403. [Google Scholar] [CrossRef] [PubMed]
- Hamidoua, F.; Rathored, A.; Waliyarc, F.; Vadezd, V. Although drought intensity increases aflatoxin contamination, drought tolerance does not lead to less aflatoxin contamination. Field Crops Res. 2014, 156, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Chen, X.; Dang, P.; Scully, B.T.; Liang, X.; Holbrook, C.C.; Yu, J.; Culbreath, A.K. Peanut gene expression profiling in developing seeds at different reproduction stages during Aspergillus parasiticus infection. BMC Dev. Biol. 2008, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Fedorova, N.D.; Chen, X.; Wan, C.H.; Wang, W.; Nierman, W.C.; Bhatnagar, D.; Yu, J. Gene expression profiling and identification of resistance genes to Aspergillus flavus infection in peanut through est and microarray strategies. Toxins (Basel) 2011, 3, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Bedre, R.; Rajasekaran, K.; Mangu, V.R.; Sanchez Timm, L.E.; Bhatnagar, D.; Baisakh, N. Genome-wide transcriptome analysis of cotton (Gossypium hirsutum L.) identifies candidate gene signatures in response to aflatoxin producing fungus Aspergillus flavus. PLoS ONE 2015, 10, e0138025. [Google Scholar] [CrossRef] [PubMed]
- Dolezal, A.; Shu, X.; OBrian, G.R.; Nielsen, D.M.; Woloshuk, C.P.; Boston, R.S.; Payne, G.A. Aspergillus flavus infection induces transcriptional and physical changes in developing maize kernels. Front. Microbiol. 2014, 5, 384. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Perkins, A.; Williams, W.P.; Warburton, M.L. Using genome-wide associations to identify metabolic pathways involved in maize aflatoxin accumulation resistance. BMC Genom. 2015, 16, 673. [Google Scholar] [CrossRef] [PubMed]
- Pelechano, V.; Steinmetz, L.M. Gene regulation by antisense transcription. Nat. Rev. Genet. 2013, 14, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Clevenger, J.; Ozias-Akins, P. SWEEP: A tool for filtering high-quality SNPs in polyploid crops. G3 (Bethesda) 2015, 5, 1797–1803. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, T.; Dorner, I.W.; Giesbrecht, E.G.; Slate, A.B. Variability among aflatoxin test results on runner peanuts harvested from small field plots. Peanut Sci. 2004, 31, 59–63. [Google Scholar] [CrossRef]
- Baud, S.; Mendoza, M.S.; To, A.; Harscoët, E.; Lepiniec, L.; Dubreucq, B. WRINKLED1 specifies the regulatory action of LEAFY COTYLEDON2 towards fatty acid metabolism during seed maturation in Arabidopsis. Plant J. 2007, 50, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Maeo, K.; Tokuda, T.; Ayame, A.; Mitsui, N.; Kawai, T.; Tsukagoshi, H.; Ishiguro, S.; Nakamura, K. An AP2-type transcription factor, WRINKLED1, of Arabidopsis thaliana binds to the AW-box sequence conserved among proximal upstream regions of genes involved in fatty acid synthesis. Plant J. 2009, 60, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, R.; Gampala, S.S.; Rock, C.D. Abscisic acid signaling in seeds and seedlings. Plant Cell 2002, 14, S15–S45. [Google Scholar] [PubMed]
- Brocard, I.; Lynch, T.J.; Finkelstein, R.R. Regulation and role of the Arabidopsis abscisic acid-insensitive 5 gene in abscisic acid, sugar, and stress response. Plant Physiol. 2002, 129, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.; Gazzarrini, S. AKIN10 and FUSCA3 interact to control lateral organ development and phase transitions in Arabidopsis. Plant J. 2012, 69, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, J.; Lambert, K.N.; Lin, Y. Developmental control of Arabidopsis seed oil biosynthesis. Planta 2007, 226, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Elahi, N.; Duncan, R.W.; Stasolla, C. Decreased seed oil production in FUSCA3 brassica napus mutant plants. Plant Physiol. Biochem. 2015, 96, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Xiong, W.; Ye, T.; Wu, Y. Overexpression of the aspartic protease ASPG1 gene confers drought avoidance in Arabidopsis. J. Exp. Bot. 2012, 63, 2579–2593. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Sun, L.; Song, Y.; Wang, L.; Liu, L.; Zhang, L.; Liu, B.; Li, N.; Miao, C.; Hao, F. AtrbohD and AtrbohF positively regulate abscisic acid-inhibited primary root growth by affecting Ca2+ signalling and auxin response of roots in Arabidopsis. J. Exp. Bot. 2013, 64, 4183–4192. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Molina, L.; Mongrand, S.; McLachlin, D.T.; Chait, B.T.; Chua, N.H. ABI5 acts downstream of ABI3 to execute an ABA-dependent growth arrest during germination. Plant J. 2002, 32, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Olszewski, N.; Sun, T.P.; Gubler, F. Gibberellin signaling: Biosynthesis, catabolism, and response pathways. Plant Cell 2002, 14 (Suppl. 1), S61–S80. [Google Scholar] [PubMed]
- Bueso, E.; Muñoz-Bertomeu, J.; Campos, F.; Brunaud, V.; Martínez, L.; Sayas, E.; Ballester, P.; Yenush, L.; Serrano, R. ARABIDOPSIS THALIANA HOMEOBOX25 uncovers a role for gibberellins in seed longevity. Plant Physiol. 2014, 164, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.; Grant, J.J.; Cheong, Y.H.; Kim, B.G.; Li, L.; Luan, S. ABR1, an APETALA2-domain transcription factor that functions as a repressor of ABA response in Arabidopsis. Plant Physiol. 2005, 139, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, C.; Culbreath, A.K. Registration of ‘Tifrunner’ peanut. J. Plant Regist. 2007, 1, 124. [Google Scholar] [CrossRef]
- Holbrook, C.; Timper, P.; Culbreath, A.K.; Kvien, C.K. Registration of ‘Tifguard’ peanut. J. Plant Regist. 2008, 2, 92–94. [Google Scholar] [CrossRef]
- Gorbet, D.; Tillman, B.L. Registration of ‘Florida-07’ peanut. J. Plant Regist. 2009, 3, 14–18. [Google Scholar] [CrossRef]
- Rucker, K.; Kvien, C.K.; Holbrook, C.C.; Hook, J.E. Identification of peanut genotypes with improved drought avoidance traits. Peanut Sci. 1995, 22, 14–18. [Google Scholar] [CrossRef]
- Hadley, B.; Beute, M.K.; Wynne, J.C. Heritability of Cylindrocladium black rot resistance in peanut. Peanut Sci. 1979, 6, 51–54. [Google Scholar] [CrossRef]
- Bertioli, D.; Cannon, S.B.; Froenicke, L.; Huang, G.; Farmer, A.D.; Cannon, E.K.; Liu, X.; Gao, D.; Clevenger, J.; Dash, S.; et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 2016, 48, 438–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Haas, B.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 36, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Pohlert, T. The pairwise multiple comparison of mean ranks package (PMCMR). R Package. 2014. Available online: http://CRAN.R-project.org/package=PMCMR (accessed on 1 January 2016).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | Effect Testing | Genotypes | DE Genes |
|---|---|---|---|
| A. flavus invasion | |||
| Aspergillus present vs. Control | A. flavus | All | 8 |
| Aspergillus present vs. Control | A. flavus | C76-16 | 0 |
| Aspergillus present vs. Control | A. flavus | NC 3033 | 0 |
| Aspergillus present vs. Control | A. flavus | Tifguard | 45 |
| Aflatoxin production | |||
| Aflatoxin present vs. Control | Aflatoxin/A. flavus | A72, Tifrunner, Florida-07 | 543 |
| Aflatoxin present vs. not present | Aflatoxin | mixed | 506 |
| Aflatoxin present vs. Control | Aflatoxin/A. flavus | A72 | 28 |
| Aflatoxin present vs. Control | Aflatoxin/A. flavus | Tifrunner | 520 |
| Aflatoxin present vs. Control | Aflatoxin/A. flavus | Florida07 | 1347 |
| Fold Change (FC) | Other Genotype’s FC | Other Genotypes’ Base | Florida 07 Base | Annotation |
|---|---|---|---|---|
| 832.23 | 69.46 ± 147.28 | G | A | Bidirectional sugar transporter |
| 399.89 | 4.85 ± 6.81 | C | T | Beta-galactosidase 3 |
| 147.30 | 4.37 ± 6.99 | C | T | Cannabidiolic acid synthase-like 1 |
| 103.33 | 1.84 ± 0.67 | G | C | Kinesin-like calmodulin-binding protein |
| 26.68 | 1.76 ± 0.83 | A | T | (+)-neomenthol dehydrogenase |
| 23.95 | 1.34 ± 0.70 | C | A | Plasma membrane ATPase 4 |
| 23.41 | 1.59 ± 0.69 | C | G | ATP synthase subunit a, chloroplastic |
| 18.49 | 1.77 ± 1.61 | G | A | Protein argonaute 5 |
| 18.48 | 1.85 ± 0.87 | G | C | Receptor-like protein kinase HSL1 |
| - | - | - | Tifrunner base | - |
| 0.29 | 0.92 ± 1.01 | A | T | Alpha-L-fucosidase 2 |
| 0.37 | 0.84 ± 0.42 | T | C | Cytochrome P450 82C2 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clevenger, J.; Marasigan, K.; Liakos, V.; Sobolev, V.; Vellidis, G.; Holbrook, C.; Ozias-Akins, P. RNA Sequencing of Contaminated Seeds Reveals the State of the Seed Permissive for Pre-Harvest Aflatoxin Contamination and Points to a Potential Susceptibility Factor. Toxins 2016, 8, 317. https://doi.org/10.3390/toxins8110317
Clevenger J, Marasigan K, Liakos V, Sobolev V, Vellidis G, Holbrook C, Ozias-Akins P. RNA Sequencing of Contaminated Seeds Reveals the State of the Seed Permissive for Pre-Harvest Aflatoxin Contamination and Points to a Potential Susceptibility Factor. Toxins. 2016; 8(11):317. https://doi.org/10.3390/toxins8110317
Chicago/Turabian StyleClevenger, Josh, Kathleen Marasigan, Vasileios Liakos, Victor Sobolev, George Vellidis, Corley Holbrook, and Peggy Ozias-Akins. 2016. "RNA Sequencing of Contaminated Seeds Reveals the State of the Seed Permissive for Pre-Harvest Aflatoxin Contamination and Points to a Potential Susceptibility Factor" Toxins 8, no. 11: 317. https://doi.org/10.3390/toxins8110317
APA StyleClevenger, J., Marasigan, K., Liakos, V., Sobolev, V., Vellidis, G., Holbrook, C., & Ozias-Akins, P. (2016). RNA Sequencing of Contaminated Seeds Reveals the State of the Seed Permissive for Pre-Harvest Aflatoxin Contamination and Points to a Potential Susceptibility Factor. Toxins, 8(11), 317. https://doi.org/10.3390/toxins8110317