
Clostridium perfringens Sialidases: Potential Contributors to Intestinal Pathogenesis and Therapeutic Targets


Abstract
:1. An Introduction to Clostridium perfringens
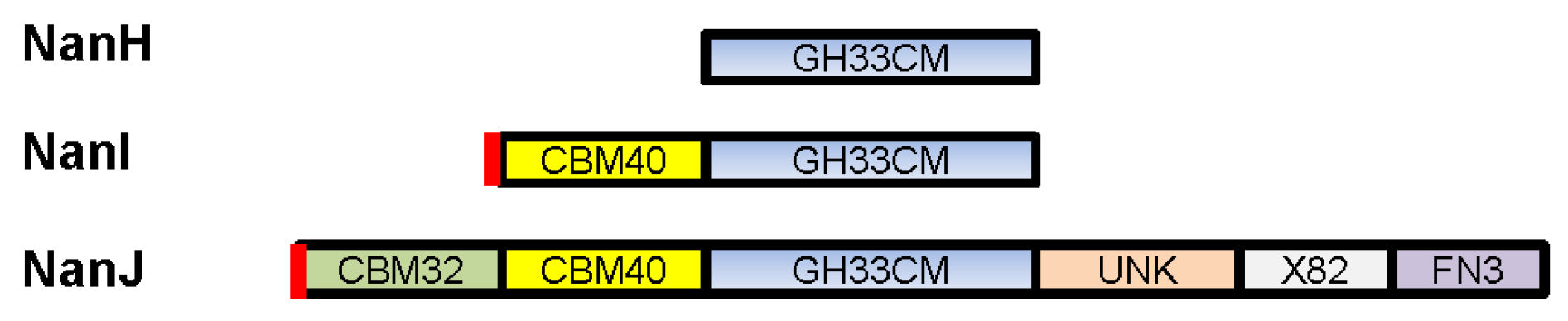
2. C. perfringens Sialidases
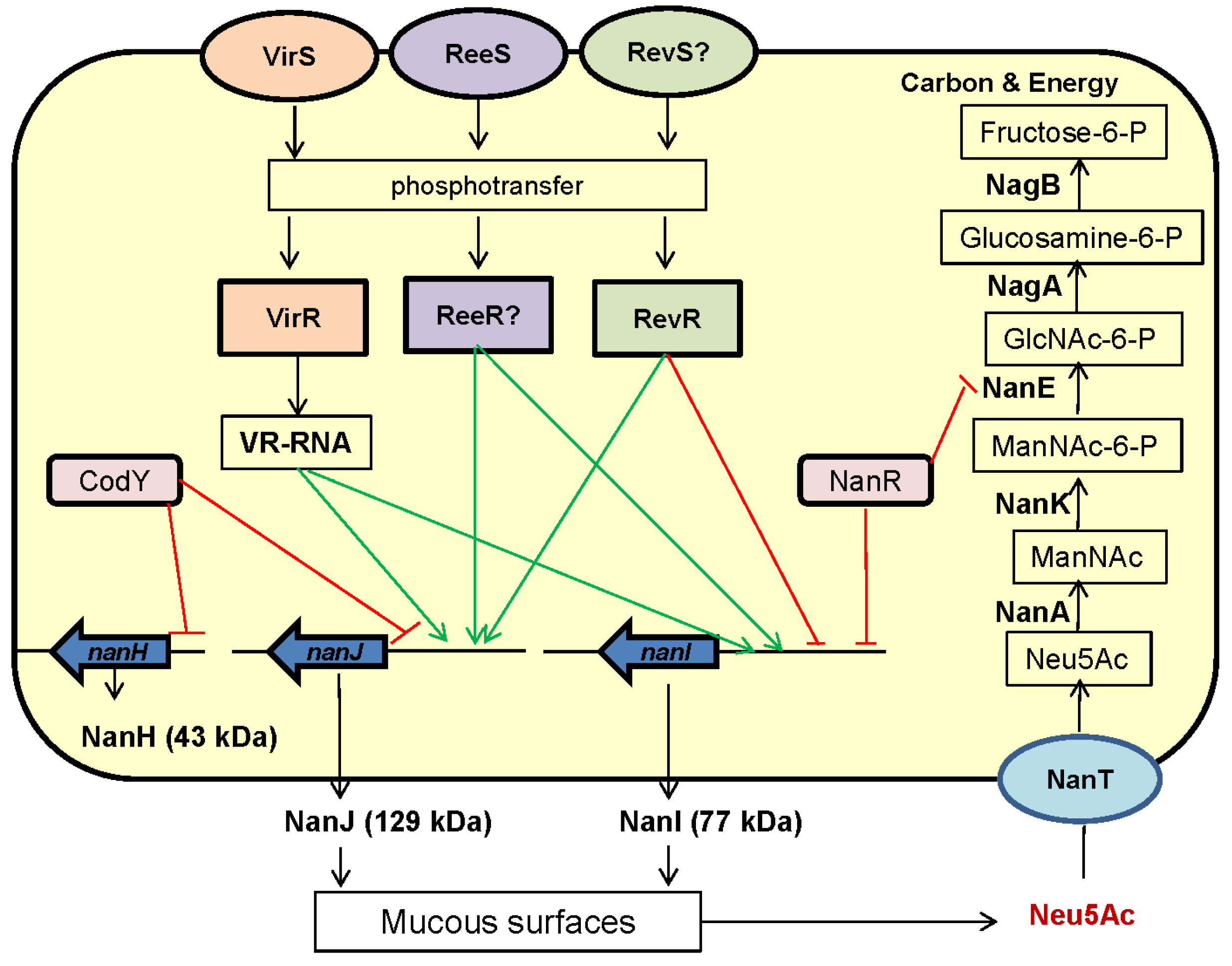
3. C. perfringens Sialidases: Genetics and Regulation of Expression
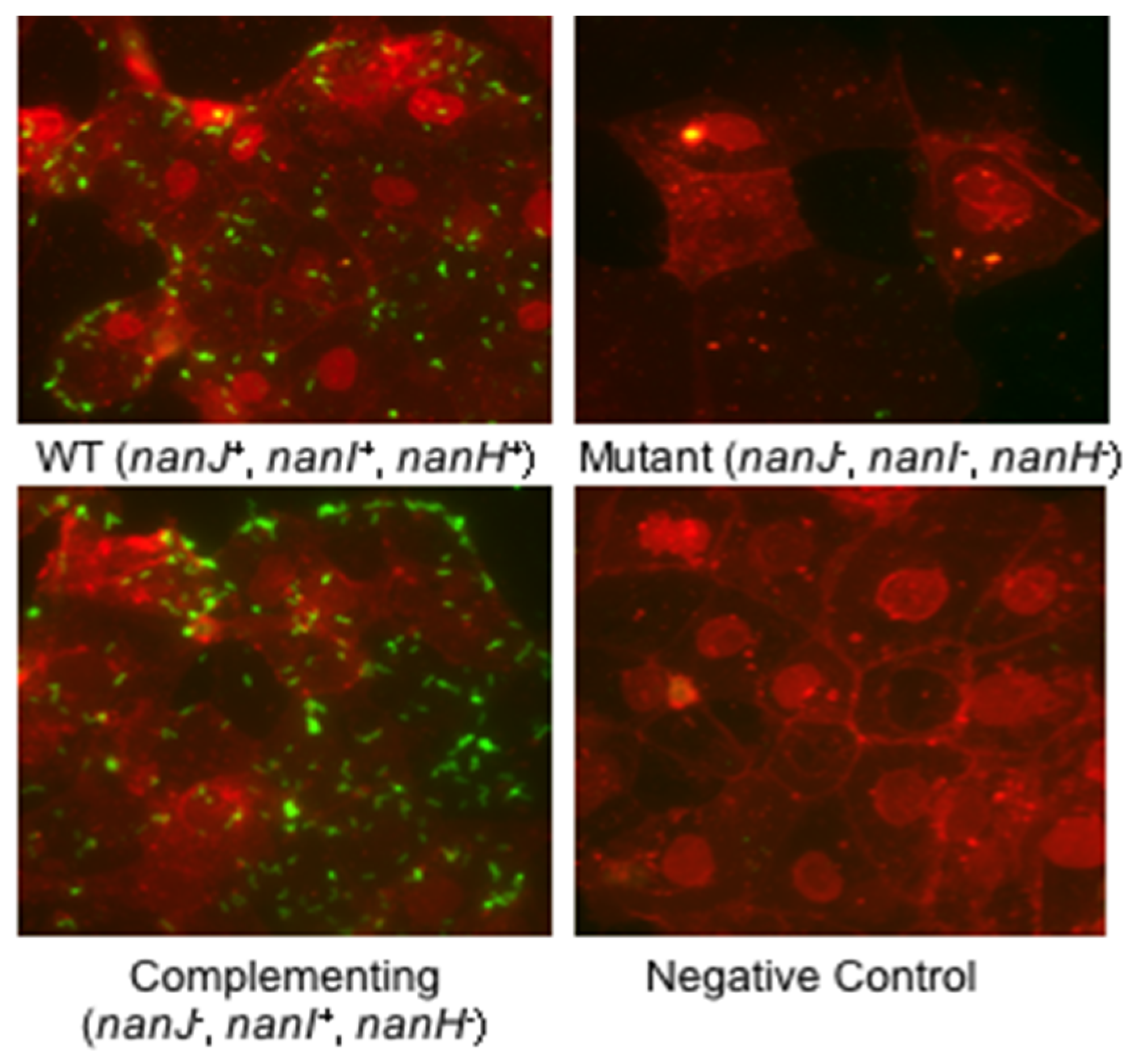
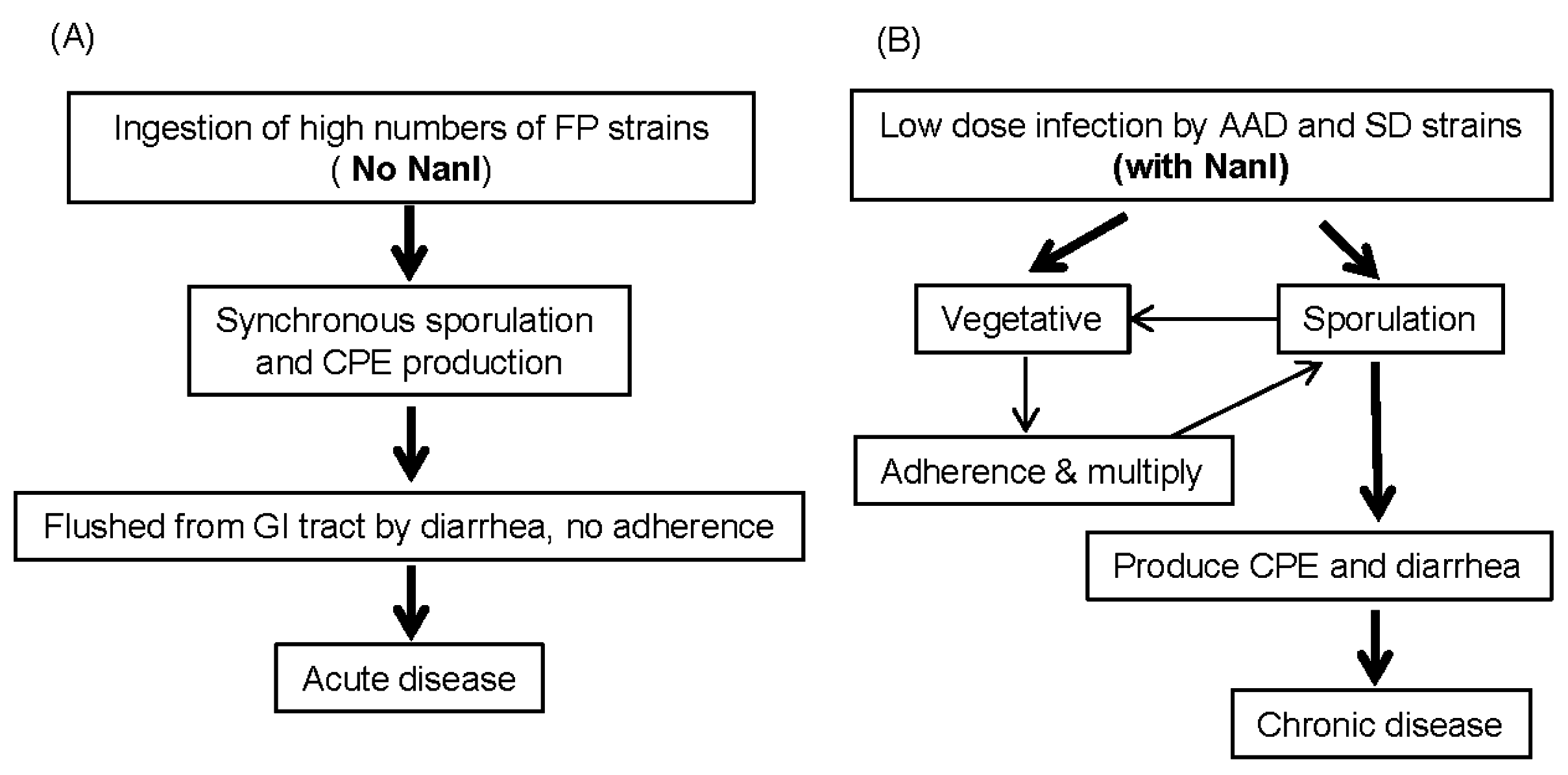
4. Possible Contributions of Sialidases to C. perfringens Diseases
5. Sialidase Inhibitors: Potential Therapeutic Agents?
6. Concluding Remarks and Future Directions
Acknowledgments
Conflicts of Interest
References
- McClane, B.A.; Robertson, S.L.; Li, J. Clostridium perfringens. In Food Microbiology: Fundamentals and Frontiers, 4th ed.; Doyle, M.P., Buchanan, R.L., Eds.; ASM Press: Washington, DC, USA, 2013; pp. 465–489. [Google Scholar]
- McClane, B.A.; Uzal, F.A.; Miyakawa, M.F.; Lyerly, D.; Wilkins, T.D. The enterotoxic clostridia. In The Prokaryotes, 3rd ed.; Dworkin, M., Falkow, S., Rosenburg, E., Schleifer, H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 688–752. [Google Scholar]
- Uzal, F.A.; Freedman, J.C.; Shrestha, A.; Theoret, J.R.; Garcia, J.; Awad, M.M.; Adams, V.; Moore, R.J.; Rood, J.I.; McClane, B.A. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 2014, 9, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Rood, J.I. Clostridium perfringens and histotoxic disease. In The prokaryotes: A Handbook on the Biology of Bacteria, 3rd ed.; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2007; Volume 4, pp. 753–770. [Google Scholar]
- Titball, R.W.; Rood, J.I. Clostridium pefringens: Wound indections. In Molecular Medical Microbiology; Sussman, M., Ed.; Academic Press: London, UK, 2002; pp. 1875–1903. [Google Scholar]
- Uzal, F.A.; Vidal, J.E.; McClane, B.A.; Gurjar, A.A. Clostridium perfringens toxins involved in mammalian veterinary diseases. Open toxinol. J. 2010, 2, 24–42. [Google Scholar] [CrossRef]
- Songer, J.G. Clostridial enteric diseases of domestic animals. Clin. Microbiol. Rev. 1996, 9, 216–234. [Google Scholar] [PubMed]
- Hatheway, C. Toxigenic clostridia. Clin. Microb. Rev. 1990, 3, 66–76. [Google Scholar] [CrossRef]
- McDonel, J.L. Toxins of Clostridium perfringens types A, B, C, D, and E. In Pharmacology of Bacterial Toxins; Dorner, F., Drews, H., Eds.; Pergamon Press: Oxford, UK, 1986; pp. 477–517. [Google Scholar]
- Petit, L.; Gilbert, M.; Popoff, M. Clostridium perfringens: Toxinotype and genotype. Trends Microbiol. 1999, 7, 104–110. [Google Scholar] [CrossRef]
- Animoto, K.; Noro, T.; Oishi, E.; Shimizu, M. A novel toxin homologous to large clostridial cytotoxins found in culture supernatant of Clostridium perfringens type C. Microbiology 2007, 153, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Keyburn, A.L.; Boyce, J.D.; Vaz, P.; Bannam, T.L.; Ford, M.E.; Parker, D.; Di Rubbo, A.; Rood, J.I.; Moore, R.J. NetB, a new toxin that is associated with avian necrotic enteritis caused by Clostridium perfringens. PLoS Pathog. 2008, 4, e26. [Google Scholar] [CrossRef] [PubMed]
- Mehdizadeh Gohari, I.; Parreira, V.R.; Timoney, J.F.; Fallon, L.; Slovis, N.; Prescott, J.F. NetF-positive Clostridium perfringens in neonatal foal necrotising enteritis in kentucky. Vet. Rec. 2016, 178, 216. [Google Scholar] [CrossRef] [PubMed]
- Yonogi, S.; Matsuda, S.; Kawai, T.; Yoda, T.; Harada, T.; Kumeda, Y.; Gotoh, K.; Hiyoshi, H.; Nakamura, S.; Kodama, T.; et al. BEC, a novel enterotoxin of Clostridium perfringens found in human clinical isolates from acute gastroenteritis outbreaks. Infect Immun. 2014, 82, 2390–2399. [Google Scholar] [CrossRef] [PubMed]
- Irikura, D.; Monma, C.; Suzuki, Y.; Nakama, A.; Kai, A.; Fukui-Miyazaki, A.; Horiguchi, Y.; Yoshinari, T.; Sugita-Konishi, Y.; Kamata, Y. Identification and characterization of a new enterotoxin produced by Clostridium perfringens isolated from food poisoning outbreaks. PLoS ONE 2015, 10, e0138183. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, S.; Uzal, F.A.; Fisher, D.J.; Saputo, J.; Vidal, J.E.; Chen, Y.; Gupta, P.; Rood, J.I.; McClane, B.A. Beta toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol. Microbiol. 2008, 67, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.P.; Adams, V.; Beingesser, J.; Hughes, M.L.; Poon, R.; Lyras, D.; Hill, A.; McClane, B.A.; Rood, J.I.; Uzal, F.A. Epsilon toxin is essential for the virulence of Clostridium perfringens type D infection in sheep, goats and mice. Infect Immun. 2013, 81, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Gurjar, A.; Theoret, J.R.; Garcia, J.P.; Beingesser, J.; Freedman, J.C.; Fisher, D.J.; McClane, B.A.; Uzal, F.A. Synergistic effects of Clostridium perfringens enterotoxin and beta toxin in rabbit small intestinal loops. Infect Immun. 2014, 82, 2958–2970. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Li, J.; McClane, B.A. Genotypic and phenotypic characterization of Clostridium perfringens isolates from darmbrand cases in post-World War II Germany. Infect Immun. 2012, 80, 4354–4363. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, G.W. The pathogenesis of enteritis necroticans. In The Clostridia: Molecular Genetics and Pathogenesis; Rood, J.I., McClane, B.A., Songer, J.G., Titball, R.W., Eds.; Academic Press: London, UK, 1997; pp. 198–207. [Google Scholar]
- Johnson, S.; Gerding, D.N. Enterotoxemic infections. In The Clostridia: Molecular Biology and Pathogenesis; Rood, J.I., McClane, B.A., Songer, J.G., Titball, R.W., Eds.; Academic Press: London, UK, 1997; pp. 117–140. [Google Scholar]
- Freedman, J.C.; Shrestha, A.; McClane, B.A. Clostridium perfringens enterotoxin: Action, genetics, and translational applications. Toxins 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Posting date. CDC estimates of foodborne illness in the United States: Clostridium perfringens. Available online: http://www.cdc.gov/foodsafety/diseases/clostridium-perfringens.html (accessed on 28 July 2016).
- Caserta, J.A.; Robertson, S.L.; Saputo, J.; Shrestha, A.; McClane, B.A.; Uzal, F.A. Development and application of a mouse intestinal loop model to study the in vivo action of Clostridium perfringens enterotoxin. Infect. Immun. 2011, 79, 3020–3027. [Google Scholar] [CrossRef] [PubMed]
- Carman, R.J. Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea of man and other animals. Rev. Med. Microbiol. 1997, 8 (Suppl. S1), S43–S45. [Google Scholar] [CrossRef]
- Li, J.; Paredes-Sabja, D.; Sarker, M.; Mcclane, B. Clostridium perfringens sporulation and sporulation-associated toxin production. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Li, J.; McClane, B.A. A novel small acid soluble protein variant is important for spore resistance of most Clostridium perfringens food poisoning isolates. PLoS Pathog. 2008, 4, e1000056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severi, E.; Hood, D.W.; Thomas, G.H. Sialic acid utilization by bacterial pathogens. Microbiology 2007, 153, 2817–2822. [Google Scholar] [CrossRef] [PubMed]
- Traving, C.; Schauer, R. Structure, function and metabolism of sialic acids. Cell. Mol. Life Sci. 1998, 54, 1330–1349. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.L.; Lewis, W.G. Host sialoglycans and bacterial sialidases: A mucosal perspective. Cell. Microbiol. 2012, 14, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Vimr, E.R.; Kalivada, K.A.; Deszo, E.L.; Steenbergen, S.M. Diversity of microbial sialic acid metabolism. Microbiol. Molec Biol. Rev. 2004, 68, 132–153. [Google Scholar] [CrossRef]
- Rohmer, L.; Hocquet, D.; Miller, S.I. Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends Microb. 2011, 19, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McClane, B.A. The sialidases of Clostridium perfringens type D strain CN3718 differ in their properties and sensitivities to inhibitors. Appl. Environ. Microbiol. 2014, 80, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sayeed, S.; Robertson, S.; Chen, J.; McClane, B.A. Sialidases affect the host cell adherence and epsilon toxin-induced cytotoxicity of Clostridium perfringens type D strain CN3718. PLoS Pathog. 2011, 7, e1002429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boraston, A.B.; Ficko-Blean, E.; Healey, M. Carbohydrate recognition by a large sialidase toxin from Clostridium perfringens. Biochemistry 2007, 46, 11352–11360. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McClane, B.A. Contributions of NanI sialidase to Caco-2 cell adherence by Clostridium perfringens type A and C strains causing human intestinal disease. Infect Immun. 2014, 82, 4620–4630. [Google Scholar] [CrossRef] [PubMed]
- Chiarezza, M.; Lyras, D.; Pidot, S.J.; Flore-Diaz, M.; Awad, M.M.; Kennedy, C.L.; Cordner, L.M.; Phumoonna, T.; Poon, R.; Hughes, M.L.; et al. The NanI and NanJ sialidases of Clostridium perfringens are not essential for virulence. Infect Immun. 2009, 77, 4421–4428. [Google Scholar] [CrossRef] [PubMed]
- Myers, G.S.; Rasko, D.A.; Cheung, J.K.; Ravel, J.; Seshadri, R.; DeBoy, R.T.; Ren, Q.; Varga, J.; Awad, M.M.; Brinkac, L.M.; et al. Skewed genomic variability in strains of the toxigenic bacterial pathogen, Clostridium perfringens. Genome Res. 2006, 16, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Ohtani, K.; Hirakawa, H.; Ohshima, K.; Yamashita, A.; Shiba, T.; Ogasawara, N.; Hattori, M.; Kuhara, S.; Hayashi, H. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc. Natl. Acad. Sci. USA 2002, 99, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Therit, B.; Cheung, J.K.; Rood, J.I.; Melville, S.B. NanR, a transcriptional regulator that binds to the promoters of genes involved in sialic acid metabolism in the anaerobic pathogen Clostridium perfringens. PLoS ONE 2015, 10, e0133217. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, K.; Hirakawa, H.; Tashiro, K.; Yoshizawa, S.; Kuhara, S.; Shimizu, T. Identification of a two-component VirR/VirS regulon in Clostridium perfringens. Anaerobe 2010, 16, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, K.; Shimizu, T. Regulation of toxin gene expression in Clostridium perfringens. Res. Microbiol. 2014, 166, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Hiscox, T.J.; Harrison, P.F.; Chakravorty, A.; Choo, J.M.; Ohtani, K.; Shimizu, T.; Cheung, J.K.; Rood, J.I. Regulation of sialidase production in Clostridium perfringens by the orphan sensor histidine kinase ReeS. PLoS ONE 2013, 8, e73525. [Google Scholar] [CrossRef] [PubMed]
- Hiscox, T.J.; Chakravorty, A.; Choo, J.M.; Ohtani, K.; Shimizu, T.; Cheung, J.K.; Rood, J.I. Regulation of virulence by the RevR response regulator in Clostridium perfringens. Infect. Immun. 2011, 79, 2145–2153. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, M.; Sarker, M.R.; McClane, B.A. CodY is a global regulator of virulence-associated properties for Clostridium perfringens type D strain CN3718. mBio 2013, 4, e00770–e00713. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.M.; Stirewalt, V.L.; Melville, S.B. Cloning, sequence, and transcriptional regulation of the operon encoding a putative N-acetyl-mannosamine-6-phosphate epimerase (nanE) and sialic acid lyase (nanA) in Clostridium perfringens. J. Bacteriol. 1999, 181, 4526–4532. [Google Scholar] [PubMed]
- Olson, M.E.; King, J.M.; Yahr, T.L.; Horswill, A.R. Sialic acid catabolism in Staphylococcus aureus. J. Bacteriol. 2013, 195, 1779–1788. [Google Scholar] [CrossRef] [PubMed]
- Almagro-Moreno, S.; Boyd, E.F. Bacterial catabolism of nonulosonic (sialic) acid and fitness in the gut. Gut Microbes 2010, 1, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Almagro-Moreno, S.; Boyd, E.F. Sialic acid catabolism confers a competitive advantage to pathogenic Vibrio cholerae in the mouse intestine. Infect. Immun. 2009, 77, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.M.; Ferreyra, J.A.; Higginbottom, S.K.; Lynch, J.B.; Kashyap, P.C.; Gopinath, S.; Naidu, N.; Choudhury, B.; Weimer, B.C.; Monack, D.M.; et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 2013, 502, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E. Harnessing microbiota to kill a pathogen: The sweet tooth of Clostridium difficile. Nat. Med. 2014, 20, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Brittan, J.; Bucheridge, T.; Finn, A.; Kadioglu, A.; Jenkinson, H. Pneumococcal neuraminidase A: An essential upper airway colonization factor for Streptococcus pneumoniae. Mol. Oral Microb. 2012, 27, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Singleton, J.; Lyras, D. The sialidase NanS enhances non-TcsL mediated cytotoxicity of Clostridium sordellii. Toxins 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.E.; Smalley, D.J.; Tucker, D.L.; Leatham, M.P.; Norris, W.E.; Stevenson, S.J.; Anderson, A.B.; Grissom, J.E.; Laux, D.C.; Cohen, P.S.; et al. Carbon nutrition of Escherichia coli in the mouse intestine. Proc. Natl. Acad. Sci. USA 2004, 101, 7427–7432. [Google Scholar] [CrossRef] [PubMed]
- Galen, J.E.; Ketley, J.M.; Fasano, A.; Richardson, S.H.; Wasserman, S.S.; Kaper, J.B. Role of Vibrio cholerae neuraminidase in the fuction of cholera toxin. Infect. Immun. 1992, 60, 406–415. [Google Scholar] [PubMed]
- Jeong, H.G.; Oh, M.H.; Kim, B.S.; Lee, M.Y.; Han, H.J.; Choi, S.H. The capability of catabolic utilization of N-acetylneuraminic acid, a sialic acid, is essential for Vibrio vulnificus pathogenesis. Infect. Immun. 2009, 77, 3209–3217. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Freedman, J.C.; McClane, B.A. NanI sialidase, CcpA, and CodY work together to regulate epsilon toxin production by Clostridium perfringens type D strain CN3718. J. Bacteriol. 2015, 197, 3339–3353. [Google Scholar] [CrossRef] [PubMed]
- Shimanoto, S.; Tamai, E.; Matsushita, O.; Minami, J.; Okabe, A.; Miyata, S. Changes in ganglioside content affect the binding of Clostridium perfringens epsilon-toxin to detergent-resistant membranes of Madin-Darby canine kidney cells. Microbiol. Immun. 2005, 49, 245–253. [Google Scholar] [CrossRef]
- Nagahama, M.; Sakurai, J. High-affinity binding of Clostridium perfingens epsilon-toxin to rat brain. Infect. Immun. 1992, 60, 1237–1240. [Google Scholar] [PubMed]
- Jost, B.H.; Billington, S.J.; Trinh, H.T.; Songer, J.G. Association of genes encoding beta2 toxin and a collagen binding protein in Clostridium perfringens isolates of porcine origin. Vet. Microbiol. 2006, 115, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Hitsumoto, Y.; Morita, N.; Yamazoe, R.; Tagomori, M.; Yamasaki, T.; Katayama, S. Adhesive properties of Clostridium perfringens to extracellular matrix proteins collagens and fibronectin. Anaerobe 2014, 25, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Katayama, S.; Tagomori, M.; Morita, N.; Yamasaki, T.; Nariya, H.; Okada, M.; Watanabe, M.; Hitsumoto, Y. Determination of the Clostridium perfringens-binding site on fibronectin. Anaerobe 2015, 34, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Wade, B.; Keyburn, A.L.; Haring, V.; Ford, M.; Rood, J.I.; Moore, R.J. The adherent abilities of Clostridium perfringens strains are critical for the pathogenesis of avian necrotic enteritis. Vet. Microbiol. 2016, 197, 53–61. [Google Scholar] [CrossRef]
- Cioffi, D.L.; Pandey, S.; Alvarez, D.F.; Cioffi, E.A. Terminal sialic acids are an important determinant of pulmonary endothelial barrier integrity. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.C.; Li, J.; Uzal, F.A.; McClane, B.A. Proteolytic processing and activation of Clostridium perfringens epsilon toxin by caprine small intestinal contents. mBio 2014, 5, e01994–e01914. [Google Scholar] [CrossRef] [PubMed]
- Hanna, P.C.; Wieckowski, E.U.; Mietzner, T.A.; McClane, B.A. Mapping functional regions of Clostridium perfringens type A enterotoxin. Infect. Immun. 1992, 60, 2110–2114. [Google Scholar] [PubMed]
- Streicher, H. Inhibition of microbial sialidases-what has happened beyond the influenza virus? Curr. Med. Chem. Anti-Infect. Agents 2004, 3, 149–161. [Google Scholar] [CrossRef]
- Roy, S.; Honma, K.; Douglas, C.W.; Sharma, A.; Stafford, G.P. Role of sialidase in glycoprotein utilization by Tannerella forsythia. Microbiology 2011, 157, 3195–3202. [Google Scholar] [CrossRef] [PubMed]
- Holzer, C.T.; von Itzstein, M.; Jin, B.; Pegg, M.S.; Stewart, W.P.; Wu, W.Y. Inhibition of sialidases from viral, bacterial and mammalian sources by analogues of 2-deoxy-2,3-didehydro-N-acetylneuraminic acid modified at the C-4 position. Glycoconj. J. 1993, 10, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; Zhao, D. Synthesis of the sialidase inhibitor siastatin B. Org. Lett. 2000, 2, 4037–4040. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Toxin Production a | |||
|---|---|---|---|---|
| α | β | ε | ι | |
| A | + | − | − | − |
| B | + | + | + | − |
| C | + | + | − | − |
| D | + | − | + | − |
| E | + | − | − | + |
| Toxinotypes a | Subtype | Most Significant Diseases b |
|---|---|---|
| A | No CPE c or NetB production | Human and animal myonecrosis (gas gangrene) |
| NetB-producing | Necrotic enteritis of poultry | |
| CPE-producing | Human food poisoning and non-foodborne gastrointestinal disease | |
| B | Necro-hemorrhagic enteritis of sheep (lamb dysentery) | |
| C | Human enteritis necroticans (Darmbrand, pigbel); necrotic enteritis of neonatal individuals of several animal species (e.g., cattle, sheep, pigs) | |
| D | Enterotoxemia of sheep and goats | |
| E | Suspected association with gastrointestinal disease of cattle, sheep and rabbits |
| Sample | NADNA (IC50) | Siastatin B (IC50) |
|---|---|---|
| CN3718 | 18.9 µM | 42.2 µM |
| ENanJ | 12.4 µM | 15.1 µM |
| ENanI | 13.4 µM | 27.5 µM |
| ENanH | 44.6 µM | 50.9 µM |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Uzal, F.A.; McClane, B.A. Clostridium perfringens Sialidases: Potential Contributors to Intestinal Pathogenesis and Therapeutic Targets. Toxins 2016, 8, 341. https://doi.org/10.3390/toxins8110341
Li J, Uzal FA, McClane BA. Clostridium perfringens Sialidases: Potential Contributors to Intestinal Pathogenesis and Therapeutic Targets. Toxins. 2016; 8(11):341. https://doi.org/10.3390/toxins8110341
Chicago/Turabian StyleLi, Jihong, Francisco A. Uzal, and Bruce A. McClane. 2016. "Clostridium perfringens Sialidases: Potential Contributors to Intestinal Pathogenesis and Therapeutic Targets" Toxins 8, no. 11: 341. https://doi.org/10.3390/toxins8110341
APA StyleLi, J., Uzal, F. A., & McClane, B. A. (2016). Clostridium perfringens Sialidases: Potential Contributors to Intestinal Pathogenesis and Therapeutic Targets. Toxins, 8(11), 341. https://doi.org/10.3390/toxins8110341