
Shiga Toxins as Multi-Functional Proteins: Induction of Host Cellular Stress Responses, Role in Pathogenesis and Therapeutic Applications


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
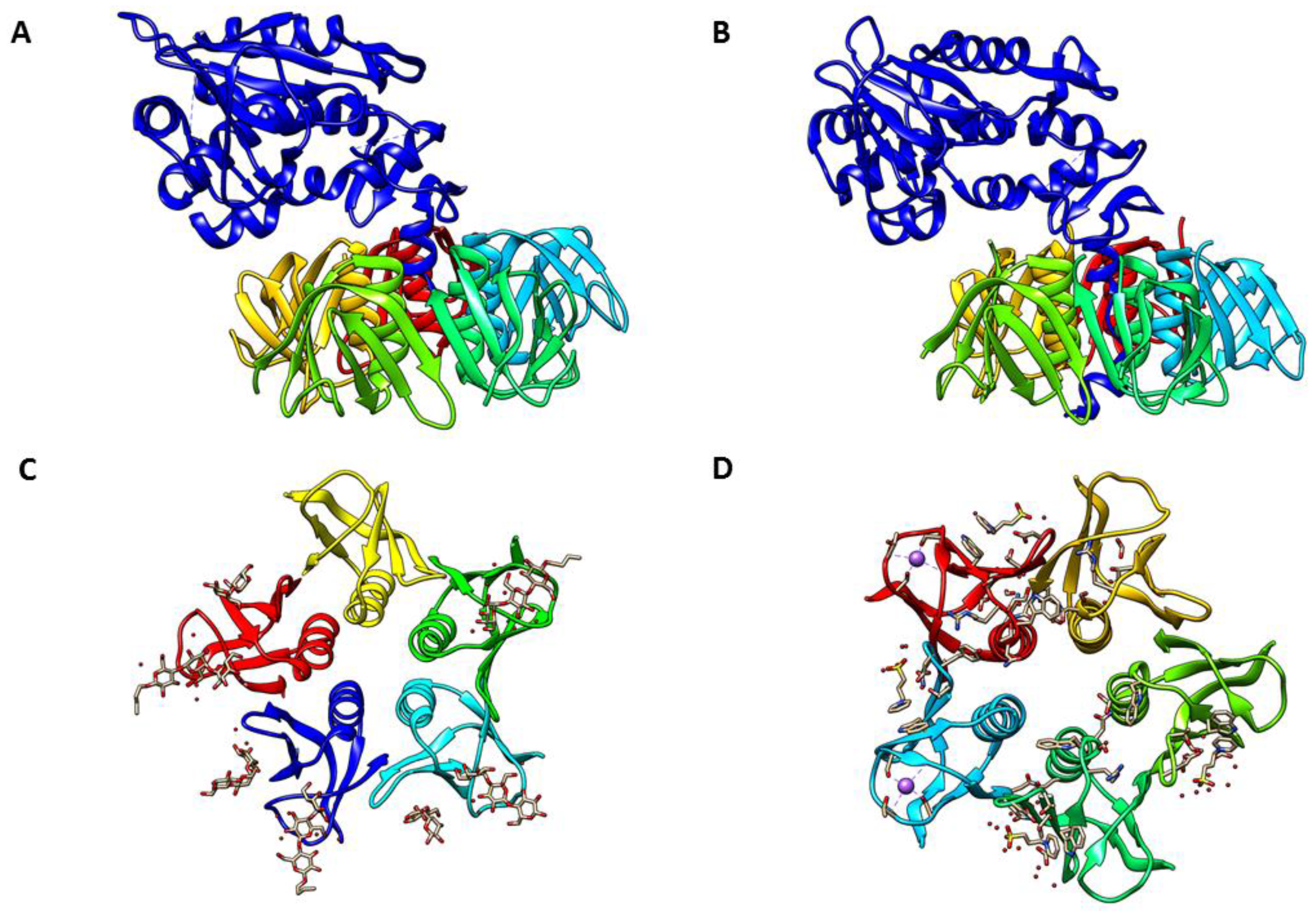
2. Stx Structure and Receptor Interaction
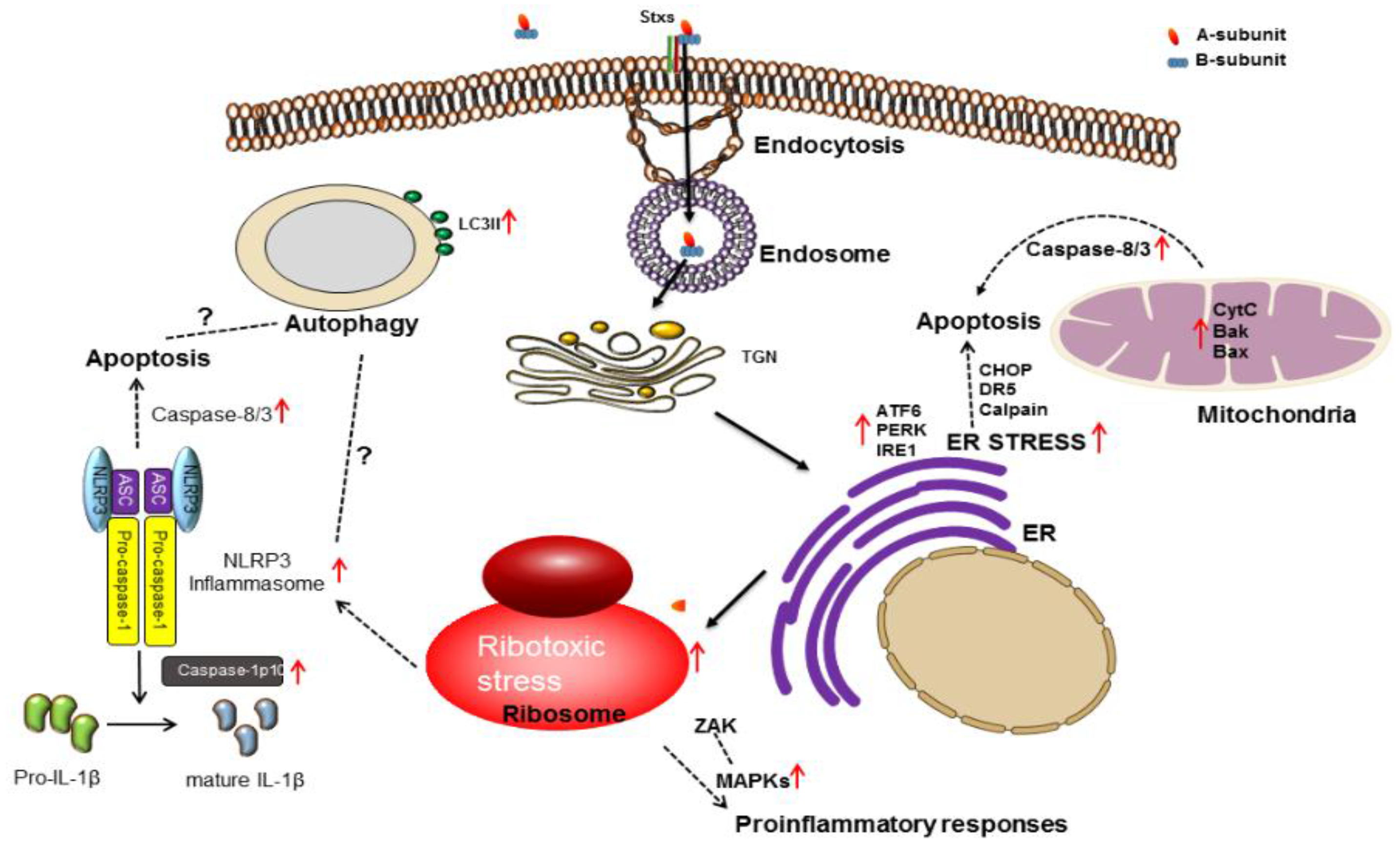
3. Stx Induces Multiple Signaling Pathways
3.1. Ribotoxic Stress Response
3.2. ER Stress
3.3. Apoptosis
3.4. Autophagy
3.5. Inflammatory Response
4. Toxin Engineering for Therapeutics
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Trofa, A.F.; Ueno-Olsen, H.; Oiwa, R.; Yoshikawa, M. Dr. Kiyoshi Shiga: Discoverer of the dysentery bacillus. Clin. Infect. Dis. 1999, 29, 1303–1306. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, A.D.; LaVeck, G.D.; Griffin, D.E.; Thompson, M.R. Characterization of Shigella dysenteriae 1 (Shiga) toxin purified by anti-Shiga toxin affinity chromatography. Infect. Immun. 1980, 30, 170–179. [Google Scholar] [PubMed]
- Olsnes, S.; Eiklid, K. Isolation and characterization of Shigella shigae cytotoxin. J. Biol. Chem. 1980, 255, 284–289. [Google Scholar] [PubMed]
- Konowalchuk, J.; Speirs, J.I.; Stavric, S. Vero response to a cytotoxin of Escherichia coli. Infect. Immun. 1977, 18, 775–779. [Google Scholar] [PubMed]
- O’Brien, A.D.; Lively, T.A.; Chen, M.E.; Rothman, S.W.; Formal, S.B. Escherichia coli O157:H7 strains associated with haemorrhagic colitis in the United States produce a Shigella dysenteriae 1 (Shiga) like cytotoxin. Lancet 1983, 1, 702. [Google Scholar] [CrossRef]
- Mora, A.; Herrera, A.; Lopez, C.; Dahbi, G.; Mamani, R.; Pita, J.M.; Alonso, M.P.; Llovo, J.; Bernardez, M.I.; Blanco, J.E.; et al. Characteristics of the Shiga-toxin-producing enteroaggregative Escherichia coli O104:H4 German outbreak strain and of STEC strains isolated in Spain. Int. Microbiol. 2011, 14, 121–141. [Google Scholar] [PubMed]
- Kaper, J.B.; O’Brien, A.D. Overview and historical perspectives. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Proulx, F.; Tesh, V.L. Renal diseases in the pediatric intensive care unit: Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenia purpura. In Pediatric Care Medicine: Basic Science and Clinical Evidence; Wheeler, D.S., Wong, H.R., Shanley, T.P., Eds.; Springer-Verlag: London, UK, 2007; pp. 1189–1203. [Google Scholar]
- Gould, L.H.; Demma, L.; Jones, T.F.; Hurd, S.; Vugia, D.J.; Smith, K.; Shiferaw, B.; Segler, S.; Palmer, A.; Zansky, S.; et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000–2006. Clin. Infect. Dis. 2009, 49, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005, 365, 1073–1086. [Google Scholar] [CrossRef]
- Tarr, P.I. Shiga toxin-associated hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: Distinct mechanisms of pathogenesis. Kidney Int. Suppl. 2009, S29–S32. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.L.; Leibowitz, C.S.; Kurosawa, S.; Stearns-Kurosawa, D.J. Shiga toxins and the pathophysiology of hemolytic uremic syndrome in humans and animals. Toxins 2012, 4, 1261–1287. [Google Scholar] [CrossRef] [PubMed]
- Gyles, C.L. Shiga toxin-producing Escherichia coli: An overview. J. Anim. Sci. 2007, 85, E45–E62. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.C.; Doyle, M.P. Food as a vehicle for transmission of Shiga toxin-producing Escherichia coli. J. Food Prot. 2007, 70, 2426–2449. [Google Scholar] [PubMed]
- Rangel, J.M.; Sparling, P.H.; Crowe, C.; Griffin, P.M.; Swerdlow, D.L. Epidemiology of Escherichia coli O157:H7 outbreaks, United States, 1982–2002. Emerg. Infect. Dis. 2005, 11, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Mead, P.S.; Slutsker, L.; Dietz, V.; McCaig, L.F.; Bresee, J.S.; Shapiro, C.; Griffin, P.M.; Tauxe, R.V. Food-related illness and death in the United States. Emerg. Infect. Dis. 1999, 5, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Scallan, E.; Mahon, B.E.; Hoekstra, R.M.; Griffin, P.M. Estimates of illnesses, hospitalizations and deaths caused by major bacterial enteric pathogens in young children in the United States. Pediatr. Infect. Dis. J. 2013, 32, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, U.; Bernard, H.; Werber, D.; Bohmer, M.M.; Remschmidt, C.; Wilking, H.; Delere, Y.; an der Heiden, M.; Adlhoch, C.; Dreesman, J.; et al. German outbreak of Escherichia coli O104:H4 associated with sprouts. N. Engl. J. Med. 2011, 365, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Mellmann, A.; Zhang, W.; Kock, R.; Fruth, A.; Bauwens, A.; Peters, G.; Karch, H. Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: A microbiological study. Lancet Infect. Dis. 2011, 11, 671–676. [Google Scholar] [CrossRef]
- Frenzen, P.D.; Drake, A.; Angulo, F.J.; Emerging Infections Program FoodNet Working Group. Economic cost of illness due to Escherichia coli O157 infections in the United States. J. Food Prot. 2005, 68, 2623–2630. [Google Scholar] [PubMed]
- Kotloff, K.L.; Winickoff, J.P.; Ivanoff, B.; Clemens, J.D.; Swerdlow, D.L.; Sansonetti, P.J.; Adak, G.K.; Levine, M.M. Global burden of Shigella infections: Implications for vaccine development and implementation of control strategies. Bull. World Health Organ. 1999, 77, 651–666. [Google Scholar] [PubMed]
- Nicolas, X.; Granier, H.; le Guen, P. Shigellosis or bacillary dysentery. Presse Med. 2007, 36, 1606–1618. [Google Scholar] [CrossRef] [PubMed]
- Tilden, J., Jr.; Young, W.; McNamara, A.M.; Custer, C.; Boesel, B.; Lambert-Fair, M.A.; Majkowski, J.; Vugia, D.; Werner, S.B.; Hollingsworth, J.; et al. A new route of transmission for Escherichia coli: Infection from dry fermented salami. Am. J. Public Health 1996, 86, 1142–1145. [Google Scholar] [CrossRef] [PubMed]
- DuPont, H.L.; Levine, M.M.; Hornick, R.B.; Formal, S.B. Inoculum size in shigellosis and implications for expected mode of tranmission. J. Infect. Dis. 1989, 159, 1126–1128. [Google Scholar] [CrossRef] [PubMed]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Pierard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef] [PubMed]
- Strockbine, N.A.; Marques, L.R.; Newland, J.W.; Smith, H.W.; Holmes, R.K.; O’Brien, A.D. Two toxin-converting phages from Escherichia coli O157:H7 strain 933 encode antigenically distinct toxins with similar biologic activities. Infect. Immun. 1986, 53, 135–140. [Google Scholar] [PubMed]
- Thorpe, C.M.; Hurley, B.P.; Acheson, D.W. Shiga toxin interactions with the intestinal epithelium. Methods Mol. Med. 2003, 73, 263–273. [Google Scholar] [PubMed]
- Schuller, S. Shiga toxin interaction with human intestinal epithelium. Toxins 2011, 3, 626–639. [Google Scholar] [CrossRef] [PubMed]
- Brigotti, M.; Carnicelli, D.; Ravanelli, E.; Barbieri, S.; Ricci, F.; Bontadini, A.; Tozzi, A.E.; Scavia, G.; Caprioli, A.; Tazzari, P.L. Interactions between Shiga toxins and human polymorphonuclear leukocytes. J. Leukoc. Biol. 2008, 84, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Brigotti, M.; Tazzari, P.L.; Ravanelli, E.; Carnicelli, D.; Barbieri, S.; Rocchi, L.; Arfilli, V.; Scavia, G.; Ricci, F.; Bontadini, A.; et al. Endothelial damage induced by Shiga toxins delivered by neutrophils during transmigration. J. Leukoc. Biol. 2010, 88, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Patzelt, W.J. Reflexion contrast, a new microscopic technic. Naturwissenschaften 1976, 63, 535. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.L.; Sartz, L.; Karpman, D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood 2011, 117, 5503–5513. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Hertel, B.; Emden, S.H.; Beneke, J.; Menne, J.; Haller, H.; von Vietinghoff, S. Microparticle generation and leucocyte death in Shiga toxin-mediated HUS. Nephrol. Dial. Transpl. 2012, 27, 2768–2775. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.L.; Arvidsson, I.; Johansson, K.E.; Chromek, M.; Rebetz, J.; Loos, S.; Kristoffersson, A.C.; Bekassy, Z.D.; Morgelin, M.; Karpman, D. A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathog. 2015, 11, e1004619. [Google Scholar] [CrossRef] [PubMed]
- Boyd, B.; Lingwood, C. Verotoxin receptor glycolipid in human renal tissue. Nephron 1989, 51, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Bergan, J.; Kavaliauskiene, S.; Skotland, T. Lipid requirements for entry of protein toxins into cells. Prog. Lipid Res. 2014, 54, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Romer, W.; Berland, L.; Chambon, V.; Gaus, K.; Windschiegl, B.; Tenza, D.; Aly, M.R.; Fraisier, V.; Florent, J.C.; Perrais, D.; et al. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature 2007, 450, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Garred, O.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; van Deurs, B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 1992, 358, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Garred, O.; van Deurs, B.; Sandvig, K. Furin-induced cleavage and activation of Shiga toxin. J. Biol. Chem. 1995, 270, 10817–10821. [Google Scholar] [PubMed]
- Garred, O.; Dubinina, E.; Polesskaya, A.; Olsnes, S.; Kozlov, J.; Sandvig, K. Role of the disulfide bond in Shiga toxin A-chain for toxin entry into cells. J. Biol. Chem. 1997, 272, 11414–11419. [Google Scholar] [PubMed]
- Garred, O.; Dubinina, E.; Holm, P.K.; Olsnes, S.; van Deurs, B.; Kozlov, J.V.; Sandvig, K. Role of processing and intracellular transport for optimal toxicity of Shiga toxin and toxin mutants. Exp. Cell Res. 1995, 218, 39–49. [Google Scholar] [CrossRef] [PubMed]
- LaPointe, P.; Wei, X.; Gariepy, J. A role for the protease-sensitive loop region of Shiga-like toxin 1 in the retrotranslocation of its A1 domain from the endoplasmic reticulum lumen. J. Biol. Chem. 2005, 280, 23310–23318. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Haslam, D.B. Shiga toxin is transported from the endoplasmic reticulum following interaction with the luminal chaperone HEDJ/ERdj3. Infect. Immun. 2005, 73, 2524–2532. [Google Scholar] [CrossRef] [PubMed]
- Falguieres, T.; Johannes, L. Shiga toxin B-subunit binds to the chaperone BiP and the nucleolar protein B23. Biol. Cell 2006, 98, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Tam, P.J.; Lingwood, C.A. Membrane cytosolic translocation of verotoxin A1 subunit in target cells. Microbiology 2007, 153, 2700–2710. [Google Scholar] [CrossRef] [PubMed]
- Menikh, A.; Saleh, M.T.; Gariepy, J.; Boggs, J.M. Orientation in lipid bilayers of a synthetic peptide representing the C-terminus of the A1 domain of Shiga toxin. A polarized ATR-FTIR study. Biochemistry 1997, 36, 15865–15872. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.T.; Ferguson, J.; Boggs, J.M.; Gariepy, J. Insertion and orientation of a synthetic peptide representing the C-terminus of the A1 domain of Shiga toxin into phospholipid membranes. Biochemistry 1996, 35, 9325–9334. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.A.; Lord, J.M. How ricin and Shiga toxin reach the cytosol of target cells: Retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol. 2012, 357, 19–40. [Google Scholar] [PubMed]
- Hazes, B.; Read, R.J. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 1997, 36, 11051–11054. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Romer, W. Shiga toxins-from cell biology to biomedical applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Janssen, K.P.; Vignjevic, D.; Boisgard, R.; Falguieres, T.; Bousquet, G.; Decaudin, D.; Dolle, F.; Louvard, D.; Tavitian, B.; Robine, S.; et al. In vivo tumor targeting using a novel intestinal pathogen-based delivery approach. Cancer Res 2006, 66, 7230–7236. [Google Scholar] [CrossRef] [PubMed]
- Distler, U.; Souady, J.; Hulsewig, M.; Drmic-Hofman, I.; Haier, J.; Friedrich, A.W.; Karch, H.; Senninger, N.; Dreisewerd, K.; Berkenkamp, S.; et al. Shiga toxin receptor Gb3Cer/CD77: Tumor-association and promising therapeutic target in pancreas and colon cancer. PLoS ONE 2009, 4, e6813. [Google Scholar] [CrossRef] [PubMed]
- Farkas-Himsley, H.; Hill, R.; Rosen, B.; Arab, S.; Lingwood, C.A. The bacterial colicin active against tumor cells in vitro and in vivo is verotoxin 1. Proc. Natl. Acad. Sci. USA 1995, 92, 6996–7000. [Google Scholar] [CrossRef] [PubMed]
- Maak, M.; Nitsche, U.; Keller, L.; Wolf, P.; Sarr, M.; Thiebaud, M.; Rosenberg, R.; Langer, R.; Kleeff, J.; Friess, H.; et al. Tumor-specific targeting of pancreatic cancer with Shiga toxin B-subunit. Mol. Cancer Ther. 2011, 10, 1918–1928. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.E.; Fujinaga, M.; Cherney, M.M.; Melton-Celsa, A.R.; Twiddy, E.M.; O’Brien, A.D.; James, M.N. Structure of Shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J. Biol. Chem. 2004, 279, 27511–27517. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.E.; Chernaia, M.M.; Kozlov, Y.V.; James, M.N. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 Å resolution. Nat. Struct. Biol. 1994, 1, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K. Shiga toxins. Toxicon 2001, 39, 1629–1635. [Google Scholar] [CrossRef]
- Beddoe, T.; Paton, A.W.; le Nours, J.; Rossjohn, J.; Paton, J.C. Structure, biological functions and applications of the AB5 toxins. Trends Biochem. Sci. 2010, 35, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Merritt, E.A.; Pronk, S.E.; Sixma, T.K.; Kalk, K.H.; van Zanten, B.A.; Hol, W.G. Structure of partially-activated E. coli heat-labile enterotoxin (LT) at 2.6 Å resolution. FEBS Lett. 1994, 337, 88–92. [Google Scholar] [CrossRef]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.K.; Ackerman, E.J. Microinjected oligonucleotides complementary to the alpha-sarcin loop of 28 S RNA abolish protein synthesis in Xenopus oocytes. J. Biol. Chem. 1990, 265, 3263–3269. [Google Scholar] [PubMed]
- Hovde, C.J.; Calderwood, S.B.; Mekalanos, J.J.; Collier, R.J. Evidence that glutamic acid 167 is an active-site residue of Shiga-like toxin I. Proc. Natl. Acad. Sci. USA 1988, 85, 2568–2572. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.E.; Cherney, M.M.; Marcato, P.; Mulvey, G.L.; Armstrong, G.D.; James, M.N. Binding of adenine to Stx2, the protein toxin from Escherichia coli O157:H7. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, C.A. Shiga toxin receptor glycolipid binding. Pathology and utility. Methods Mol. Med. 2003, 73, 165–186. [Google Scholar] [PubMed]
- Nakajima, H.; Kiyokawa, N.; Katagiri, Y.U.; Taguchi, T.; Suzuki, T.; Sekino, T.; Mimori, K.; Ebata, T.; Saito, M.; Nakao, H.; et al. Kinetic analysis of binding between Shiga toxin and receptor glycolipid Gb3Cer by surface plasmon resonance. J. Biol. Chem. 2001, 276, 42915–42922. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, K.M.; Conrady, D.G.; Karve, S.S.; Gunasekera, T.S.; Herr, A.B.; Weiss, A.A. Shiga toxin binding to glycolipids and glycans. PLoS ONE 2012, 7, e30368. [Google Scholar] [CrossRef] [PubMed]
- Brigotti, M.; Carnicelli, D.; Arfilli, V.; Tamassia, N.; Borsetti, F.; Fabbri, E.; Tazzari, P.L.; Ricci, F.; Pagliaro, P.; Spisni, E.; et al. Identification of TLR4 as the receptor that recognizes Shiga toxins in human neutrophils. J. Immunol. 2013, 191, 4748–4758. [Google Scholar] [CrossRef] [PubMed]
- Waddell, T.; Cohen, A.; Lingwood, C.A. Induction of verotoxin sensitivity in receptor-deficient cell lines using the receptor glycolipid globotriosylceramide. Proc. Natl. Acad. Sci. USA 1990, 87, 7898–7901. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Boodhoo, A.; Hazes, B.; Cummings, M.D.; Armstrong, G.D.; Brunton, J.L.; Read, R.J. Structure of the Shiga-like toxin I B-pentamer complexed with an analogue of its receptor Gb3. Biochemistry 1998, 37, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- St Hilaire, P.M.; Boyd, M.K.; Toone, E.J. Interaction of the Shiga-like toxin type 1 B-subunit with its carbohydrate receptor. Biochemistry 1994, 33, 14452–14463. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.E.; Boodhoo, A.; Tyrrell, G.J.; Brunton, J.L.; Read, R.J. Crystal structure of the cell-binding B oligomer of verotoxin-1 from E. coli. Nature 1992, 355, 748–750. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Field, R.A.; Homans, S.W.; Donohue-Rolfe, A. Solution structure of the complex between the B-subunit homopentamer of verotoxin VT-1 from Escherichia coli and the trisaccharide moiety of globotriaosylceramide. Biochemistry 1998, 37, 11078–11082. [Google Scholar] [CrossRef] [PubMed]
- Bast, D.J.; Banerjee, L.; Clark, C.; Read, R.J.; Brunton, J.L. The identification of three biologically relevant globotriaosyl ceramide receptor binding sites on the verotoxin 1 B subunit. Mol. Microbiol. 1999, 32, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Kitova, E.N.; Kitov, P.I.; Paszkiewicz, E.; Kim, J.; Mulvey, G.L.; Armstrong, G.D.; Bundle, D.R.; Klassen, J.S. Affinities of Shiga toxins 1 and 2 for univalent and oligovalent pk-trisaccharide analogs measured by electrospray ionization mass spectrometry. Glycobiology 2007, 17, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, G.; Mobassaleh, M.; Donohue-Rolfe, A.; Montgomery, R.K.; Grand, R.J.; Keusch, G.T. Pathogenesis of Shigella diarrhea: Rabbit intestinal cell microvillus membrane binding site for Shigella toxin. Infect. Immun. 1986, 53, 372–377. [Google Scholar] [PubMed]
- Jacobson, J.M.; Yin, J.; Kitov, P.I.; Mulvey, G.; Griener, T.P.; James, M.N.; Armstrong, G.; Bundle, D.R. The crystal structure of Shiga toxin type 2 with bound disaccharide guides the design of a heterobifunctional toxin inhibitor. J. Biol. Chem. 2014, 289, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Conrady, D.G.; Flagler, M.J.; Friedmann, D.R.; Vander Wielen, B.D.; Kovall, R.A.; Weiss, A.A.; Herr, A.B. Molecular basis of differential B-pentamer stability of Shiga toxins 1 and 2. PLoS ONE 2010, 5, e15153. [Google Scholar] [CrossRef] [PubMed]
- Karve, S.S.; Weiss, A.A. Glycolipid binding preferences of Shiga toxin variants. PLoS ONE 2014, 9, e101173. [Google Scholar] [CrossRef] [PubMed]
- Bergan, J.; Dyve Lingelem, A.B.; Simm, R.; Skotland, T.; Sandvig, K. Shiga toxins. Toxicon 2012, 60, 1085–1107. [Google Scholar] [CrossRef] [PubMed]
- Malyukova, I.; Murray, K.F.; Zhu, C.; Boedeker, E.; Kane, A.; Patterson, K.; Peterson, J.R.; Donowitz, M.; Kovbasnjuk, O. Macropinocytosis in Shiga toxin 1 uptake by human intestinal epithelial cells and transcellular transcytosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G78–G92. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Decaudin, D. Protein toxins: Intracellular trafficking for targeted therapy. Gene Ther. 2005, 12, 1360–1368. [Google Scholar] [CrossRef]
- Engedal, N.; Skotland, T.; Torgersen, M.; Sandvig, K. Shiga toxin and its use in targeted cancer therapy and imaging. Microb. Biotechnol. 2011, 4, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Stimmer, L.; Dehay, S.; Nemati, F.; Massonnet, G.; Richon, S.; Decaudin, D.; Klijanienko, J.; Johannes, L. Human breast cancer and lymph node metastases express Gb3 and can be targeted by StxB-vectorized chemotherapeutic compounds. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.M.; Lord, J.M. Ribosome-inactivating proteins: Entry into mammalian cells and intracellular routing. Mini Rev. Med. Chem. 2004, 4, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Chen, S.L.; Magun, B.E. Ribotoxic stress response: Activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the α-sarcin/ricin loop in the 28S rRNA. Mol. Cell. Biol. 1997, 17, 3373–3381. [Google Scholar] [CrossRef] [PubMed]
- Cherla, R.P.; Lee, S.Y.; Mees, P.L.; Tesh, V.L. Shiga toxin 1-induced cytokine production is mediated by MAP kinase pathways and translation initiation factor eIF4E in the macrophage-like THP-1 cell line. J. Leukoc. Biol. 2006, 79, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Foster, G.H.; Tesh, V.L. Shiga toxin 1-induced activation of c-Jun NH2-terminal kinase and p38 in the human monocytic cell line THP-1: Possible involvement in the production of TNF-α. J. Leukoc. Biol. 2002, 71, 107–114. [Google Scholar] [PubMed]
- Smith, W.E.; Kane, A.V.; Campbell, S.T.; Acheson, D.W.; Cochran, B.H.; Thorpe, C.M. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 2003, 71, 1497–1504. [Google Scholar] [CrossRef] [PubMed]
- Nallagatla, S.R.; Toroney, R.; Bevilacqua, P.C. Regulation of innate immunity through RNA structure and the protein kinase PKR. Curr. Opin. Struct. Biol. 2011, 21, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.R.; Lau, A.S.; Pestka, J.J. Role of double-stranded RNA-activated protein kinase R (PKR) in deoxynivalenol-induced ribotoxic stress response. Toxicol. Sci. 2003, 74, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.; Gray, J.S.; Li, M.; Vines, L.; Kim, J.; Pestka, J.J. Hematopoietic cell kinase associates with the 40S ribosomal subunit and mediates the ribotoxic stress response to deoxynivalenol in mononuclear phagocytes. Toxicol. Sci. 2010, 115, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Langland, J.O.; Jacobs, B.L.; Samuel, C.E. Protein kinase PKR-dependent activation of mitogen-activated protein kinases occurs through mitochondrial adapter IPS-1 and is antagonized by vaccinia virus E3L. J. Virol. 2009, 83, 5718–5725. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, D.M.; Ahluwalia, A.; Obrig, T.; Thorpe, C.M. ZAK: A MAP3Kinase that transduces Shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell. Microbiol. 2008, 10, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Garibal, J.; Hollville, E.; Renouf, B.; Tetaud, C.; Wiels, J. Caspase-8-mediated cleavage of Bid and protein phosphatase 2a-mediated activation of Bax are necessary for verotoxin-1-induced apoptosis in Burkitt’s lymphoma cells. Cell. Signal. 2010, 22, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, H.; Nishitoh, H.; Ichijo, H. Survival and apoptosis signals in ER stress: The role of protein kinases. J. Chem. Neuroanat. 2004, 28, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.A.; Walters, K.A.; Lamb, S.E.; Yeh, M.M.; Zhu, L.F.; Kneteman, N.; Doyle, J.S.; Katze, M.G.; Tyrrell, D.L. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 2009, 5, e1000291. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.W. Unfolded protein response in Hepatitis C Virus infection. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Celli, J.; Tsolis, R.M. Bacteria, the endoplasmic reticulum and the unfolded protein response: Friends or foes? Nat. Rev. Microbiol. 2015, 13, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Lee, M.S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell. Microbiol. 2008, 10, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Lentz, E.K.; Leyva-Illades, D.; Lee, M.S.; Cherla, R.P.; Tesh, V.L. Differential response of the human renal proximal tubular epithelial cell line HK-2 to Shiga toxin types 1 and 2. Infect. Immun. 2011, 79, 3527–3540. [Google Scholar] [CrossRef] [PubMed]
- Parello, C.S.; Mayer, C.L.; Lee, B.C.; Motomochi, A.; Kurosawa, S.; Stearns-Kurosawa, D.J. Shiga toxin 2-induced endoplasmic reticulum stress is minimized by activated protein C but does not correlate with lethal kidney injury. Toxins 2015, 7, 170–186. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Li, Q.; Zhao, X.H.; Wang, H.G.; Li, N.; Fang, Y.; Wang, K.; Jia, Y.P.; Zhu, P.; Gu, J.; et al. Shiga toxins induce autophagic cell death in intestinal epithelial cells via the endoplasmic reticulum stress pathway. Autophagy 2015, 11, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Mele, C.; Remuzzi, G.; Noris, M. Hemolytic uremic syndrome. Semin. Immunopathol. 2014, 36, 399–420. [Google Scholar] [CrossRef] [PubMed]
- Inward, C.D.; Williams, J.; Chant, I.; Crocker, J.; Milford, D.V.; Rose, P.E.; Taylor, C.M. Verocytotoxin-1 induces apoptosis in Vero cells. J. Infect. 1995, 30, 213–218. [Google Scholar] [CrossRef]
- Karpman, D.; Hakansson, A.; Perez, M.T.; Isaksson, C.; Carlemalm, E.; Caprioli, A.; Svanborg, C. Apoptosis of renal cortical cells in the hemolytic-uremic syndrome: In vivo and in vitro studies. Infect. Immun. 1998, 66, 636–644. [Google Scholar] [PubMed]
- Te Loo, D.M.; Monnens, L.A.; van der Velden, T.J.; Vermeer, M.A.; Preyers, F.; Demacker, P.N.; van den Heuvel, L.P.; van Hinsbergh, V.W. Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood 2000, 95, 3396–3402. [Google Scholar] [PubMed]
- Lee, M.S.; Cherla, R.P.; Leyva-Illades, D.; Tesh, V.L. Bcl-2 regulates the onset of Shiga toxin 1-induced apoptosis in THP-1 cells. Infect. Immun. 2009, 77, 5233–5244. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Deng, X.; Carr, B.; May, W.S. Bcl-2 phosphorylation required for anti-apoptosis function. J. Biol. Chem. 1997, 272, 11671–11673. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Xiao, L.; Lang, W.; Gao, F.; Ruvolo, P.; May, W.S., Jr. Novel role for JNK as a stress-activated Bcl2 kinase. J. Biol. Chem. 2001, 276, 23681–23688. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Cherla, R.P.; Lentz, E.K.; Leyva-Illades, D.; Tesh, V.L. Signaling through C/EBP homologous protein and death receptor 5 and calpain activation differentially regulate THP-1 cell maturation-dependent apoptosis induced by Shiga toxin type 1. Infect. Immun. 2010, 78, 3378–3391. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Gunji, Y.; Sonoda, H.; Oshikawa, S.; Shimono, M.; Horie, A.; Ito, K.; Yamasaki, S. Inhibitory effect of tyrphostin 47 on Shiga toxin-induced cell death. Eur. J. Pharmacol. 2006, 546, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.L.; Islur, A.; Haq, R.; Mascarenhas, M.; Karmali, M.A.; Perdue, M.H.; Zanke, B.W.; Sherman, P.M. Escherichia coli Shiga toxins induce apoptosis in epithelial cells that is regulated by the BCL-2 family. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G811–G819. [Google Scholar] [PubMed]
- Fujii, J.; Matsui, T.; Heatherly, D.P.; Schlegel, K.H.; Lobo, P.I.; Yutsudo, T.; Ciraolo, G.M.; Morris, R.E.; Obrig, T. Rapid apoptosis induced by Shiga toxin in HeLa cells. Infect. Immun. 2003, 71, 2724–2735. [Google Scholar] [CrossRef] [PubMed]
- Erwert, R.D.; Eiting, K.T.; Tupper, J.C.; Winn, R.K.; Harlan, J.M.; Bannerman, D.D. Shiga toxin induces decreased expression of the anti-apoptotic protein Mcl-1 concomitant with the onset of endothelial apoptosis. Microb. Pathog. 2003, 35, 87–93. [Google Scholar] [CrossRef]
- Kiyokawa, N.; Mori, T.; Taguchi, T.; Saito, M.; Mimori, K.; Suzuki, T.; Sekino, T.; Sato, N.; Nakajima, H.; Katagiri, Y.U.; et al. Activation of the caspase cascade during Stx1-induced apoptosis in Burkitt’s lymphoma cells. J. Cell. Biochem. 2001, 81, 128–142. [Google Scholar] [CrossRef]
- Mangeney, M.; Lingwood, C.A.; Taga, S.; Caillou, B.; Tursz, T.; Wiels, J. Apoptosis induced in Burkitt’s lymphoma cells via Gb3/CD77, a glycolipid antigen. Cancer Res. 1993, 53, 5314–5319. [Google Scholar] [PubMed]
- Tetaud, C.; Falguieres, T.; Carlier, K.; Lecluse, Y.; Garibal, J.; Coulaud, D.; Busson, P.; Steffensen, R.; Clausen, H.; Johannes, L.; et al. Two distinct Gb3/CD77 signaling pathways leading to apoptosis are triggered by anti-Gb3/CD77 mab and verotoxin-1. J. Biol. Chem. 2003, 278, 45200–45208. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Kiyokawa, N.; Ohtomo, Y.; Nagaoka, R.; Yamashiro, Y.; Taguchi, T.; Mori, T.; Fujimoto, J.; Takeda, T. Apoptosis of renal tubular cells in Shiga-toxin-mediated hemolytic uremic syndrome. Nephron 2001, 87, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Te Loo, D.M.; Monnens, L.A.; van den Heuvel, L.P.; Gubler, M.C.; Kockx, M.M. Detection of apoptosis in kidney biopsies of patients with D+ hemolytic uremic syndrome. Pediatr. Res. 2001, 49, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Psotka, M.A.; Obata, F.; Kolling, G.L.; Gross, L.K.; Saleem, M.A.; Satchell, S.C.; Mathieson, P.W.; Obrig, T.G. Shiga toxin 2 targets the murine renal collecting duct epithelium. Infect. Immun. 2009, 77, 959–969. [Google Scholar] [CrossRef] [PubMed]
- DesRochers, T.M.; Kimmerling, E.P.; Jandhyala, D.M.; El-Jouni, W.; Zhou, J.; Thorpe, C.M.; Leong, J.M.; Kaplan, D.L. Effects of Shiga toxin type 2 on a bioengineered three-dimensional model of human renal tissue. Infect. Immun. 2015, 83, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Funata, N.; Ikuta, F.; Sato, S. Neuronal apoptosis and inflammatory responses in the central nervous system of a rabbit treated with Shiga toxin-2. J. Neuroinflamm. 2008, 5. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. The induction of apoptosis by Shiga toxins and ricin. Curr. Top. Microbiol. Immunol. 2012, 357, 137–178. [Google Scholar] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, W.; User, S.D.; El-Gazzah, M.; Jaksik, R.; Sajjadi, E.; Rzeszowska-Wolny, J.; Los, M.J. Autophagy, apoptosis, mitoptosis and necrosis: Interdependence between those pathways and effects on cancer. Arch. Immunol. Ther. Exp. 2013, 61, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Moretti, L.; Cha, Y.I.; Niermann, K.J.; Lu, B. Switch between apoptosis and autophagy: Radiation-induced endoplasmic reticulum stress? Cell Cycle 2007, 6, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Che, X.; Zheng, Q.; Wu, A.; Pan, K.; Shao, A.; Wu, Q.; Zhang, J.; Hong, Y. Caspases: A molecular switch node in the crosstalk between autophagy and apoptosis. Int. J. Biol. Sci. 2014, 10, 1072–1083. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.A.; Tavallai, S.; Hamed, H.A.; Cruickshanks, N.; Dent, P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell. Signal. 2014, 26, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; van Deurs, B. Toxin-induced cell lysis: Protection by 3-methyladenine and cycloheximide. Exp. Cell Res. 1992, 200, 253–262. [Google Scholar] [CrossRef]
- Lee, M.S.; Cherla, R.P.; Jenson, M.H.; Leyva-Illades, D.; Martinez-Moczygemba, M.; Tesh, V.L. Shiga toxins induce autophagy leading to differential signalling pathways in toxin-sensitive and toxin-resistant human cells. Cell. Microbiol. 2011, 13, 1479–1496. [Google Scholar] [CrossRef] [PubMed]
- Weston, R.T.; Puthalakath, H. Endoplasmic reticulum stress and BCL-2 family members. Adv. Exp. Med. Biol. 2010, 687, 65–77. [Google Scholar] [PubMed]
- Hoyer-Hansen, M.; Jaattela, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Leyva-Illades, D.; Cherla, R.P.; Lee, M.S.; Tesh, V.L. Regulation of cytokine and chemokine expression by the ribotoxic stress response elicited by Shiga toxin type 1 in human macrophage-like THP-1 cells. Infect. Immun. 2012, 80, 2109–2120. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. Shiga toxins—Not just cytotoxins anymore. Trends Microbiol. 2001, 9, 584–585. [Google Scholar] [CrossRef]
- Foster, G.H.; Armstrong, C.S.; Sakiri, R.; Tesh, V.L. Shiga toxin-induced tumor necrosis factor alpha expression: Requirement for toxin enzymatic activity and monocyte protein kinase C and protein tyrosine kinases. Infect. Immun. 2000, 68, 5183–5189. [Google Scholar] [CrossRef] [PubMed]
- Broggi, A.; Granucci, F. Microbe- and danger-induced inflammation. Mol. Immunol. 2015, 63, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Wiersinga, W.J.; Leopold, S.J.; Cranendonk, D.R.; van der Poll, T. Host innate immune responses to sepsis. Virulence 2014, 5, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Girardis, M.; Cossarizza, A. Early alterations of B cells in patients with septic shock: Another piece in the complex puzzle of the immune response in sepsis. Crit. Care 2013, 17. [Google Scholar] [CrossRef] [PubMed]
- Gustot, T. Multiple organ failure in sepsis: Prognosis and role of systemic inflammatory response. Curr. Opin. Crit. Care 2011, 17, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Van de Kar, N.C.; Monnens, L.A.; Karmali, M.A.; van Hinsbergh, V.W. Tumor necrosis factor and interleukin-1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: Implications for the pathogenesis of the hemolytic uremic syndrome. Blood 1992, 80, 2755–2764. [Google Scholar] [PubMed]
- Ramegowda, B.; Samuel, J.E.; Tesh, V.L. Interaction of Shiga toxins with human brain microvascular endothelial cells: Cytokines as sensitizing agents. J. Infect. Dis. 1999, 180, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Stricklett, P.K.; Hughes, A.K.; Ergonul, Z.; Kohan, D.E. Molecular basis for up-regulation by inflammatory cytokines of Shiga toxin 1 cytotoxicity and globotriaosylceramide expression. J. Infect. Dis. 2002, 186, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Raqib, R.; Wretlind, B.; Andersson, J.; Lindberg, A.A. Cytokine secretion in acute shigellosis is correlated to disease activity and directed more to stool than to plasma. J. Infect. Dis. 1995, 171, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nagayama, K.; Satomura, K.; Honda, T.; Okada, S. Increased serum IL-10 and endothelin levels in hemolytic uremic syndrome caused by Escherichia coli O157. Nephron 2000, 84, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.M.; van Haaften, W.C.; Tesh, V.L. Regulation of proinflammatory cytokine expression by Shiga toxin 1 and/or lipopolysaccharides in the human monocytic cell line THP-1. Infect. Immun. 2004, 72, 2618–2627. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.M.; van den Hoogen, C.; van Haaften, W.C.E.; Tesh, V.L. Chemokine expression in the monocytic cell line THP-1 in response to purified Shiga toxin 1 and/or lipopolysaccharides. Infect. Immun. 2005, 73, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.M.; Cherla, R.P.; van den Hoogen, C.; van Haaften, W.C.; Lee, S.Y.; Tesh, V.L. Comparative evaluation of apoptosis induced by Shiga toxin 1 and/or lipopolysaccharides in human monocytic and macrophage-like cells. Microb. Pathog. 2005, 38, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Van Setten, P.A.; Monnens, L.A.; Verstraten, R.G.; van den Heuvel, L.P.; van Hinsbergh, V.W. Effects of verocytotoxin-1 on nonadherent human monocytes: Binding characteristics, protein synthesis, and induction of cytokine release. Blood 1996, 88, 174–183. [Google Scholar] [PubMed]
- Falguieres, T.; Mallard, F.; Baron, C.; Hanau, D.; Lingwood, C.; Goud, B.; Salamero, J.; Johannes, L. Targeting of Shiga toxin B-subunit to retrograde transport route in association with detergent-resistant membranes. Mol. Biol. Cell 2001, 12, 2453–2468. [Google Scholar] [CrossRef] [PubMed]
- Ramegowda, B.; Tesh, V.L. Differentiation-associated toxin receptor modulation, cytokine production, and sensitivity to Shiga-like toxins in human monocytes and monocytic cell lines. Infect. Immun. 1996, 64, 1173–1180. [Google Scholar] [PubMed]
- Smith, D.C.; Sillence, D.J.; Falguieres, T.; Jarvis, R.M.; Johannes, L.; Lord, J.M.; Platt, F.M.; Roberts, L.M. The association of Shiga-like toxin with detergent-resistant membranes is modulated by glucosylceramide and is an essential requirement in the endoplasmic reticulum for a cytotoxic effect. Mol. Biol. Cell 2006, 17, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Higuchi, T.; Takada, K.; Oida, K.; Horie, S.; Ishii, H. Verotoxin-1 stimulation of macrophage-like THP-1 cells up-regulates tissue factor expression through activation of c-Yes tyrosine kinase: Possible signal transduction in tissue factor up-regulation. Biochim. Biophys. Acta 2006, 1762, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kwon, H.; Lee, E.Y.; Kim, D.J.; Park, J.H.; Tesh, V.L.; Oh, T.K.; Kim, M.H. Shiga toxins activate the NLRP3 inflammasome pathway to promote both production of the proinflammatory cytokine interleukin-1β and apoptotic cell death. Infect. Immun. 2016, 84, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Philpott, D.J.; Ackerley, C.A.; Kiliaan, A.J.; Karmali, M.A.; Perdue, M.H.; Sherman, P.M. Translocation of verotoxin-1 across T84 monolayers: Mechanism of bacterial toxin penetration of epithelium. Am. J. Physiol. 1997, 273, G1349–G1358. [Google Scholar] [PubMed]
- Bellmeyer, A.; Cotton, C.; Kanteti, R.; Koutsouris, A.; Viswanathan, V.K.; Hecht, G. Enterohemorrhagic Escherichia coli suppresses inflammatory response to cytokines and its own toxin. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G576–G581. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, D.M.; Rogers, T.J.; Kane, A.; Paton, A.W.; Paton, J.C.; Thorpe, C.M. Shiga toxin 2 and flagellin from Shiga-toxigenic Escherichia coli superinduce interleukin-8 through synergistic effects on host stress-activated protein kinase activation. Infect. Immun. 2010, 78, 2984–2994. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Iimura, M.; Kaper, J.B.; Torres, A.G.; Kagnoff, M.F. Role of Shiga toxin versus H7 flagellin in enterohaemorrhagic Escherichia coli signalling of human colon epithelium in vivo. Cell. Microbiol. 2006, 8, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Keepers, T.R.; Gross, L.K.; Obrig, T.G. Monocyte chemoattractant protein 1, macrophage inflammatory protein 1 alpha, and RANTES recruit macrophages to the kidney in a mouse model of hemolytic-uremic syndrome. Infect. Immun. 2007, 75, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Kurosawa, D.J.; Collins, V.; Freeman, S.; Tesh, V.L.; Kurosawa, S. Distinct physiologic and inflammatory responses elicited in baboons after challenge with Shiga toxin type 1 or 2 from enterohemorrhagic Escherichia coli. Infect. Immun. 2010, 78, 2497–2504. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Kurosawa, D.J.; Oh, S.Y.; Cherla, R.P.; Lee, M.S.; Tesh, V.L.; Papin, J.; Henderson, J.; Kurosawa, S. Distinct renal pathology and a chemotactic phenotype after enterohemorrhagic Escherichia coli Shiga toxins in non-human primate models of hemolytic uremic syndrome. Am. J. Pathol. 2013, 182, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Cherla, R.P.; Lee, S.Y.; Mulder, R.A.; Lee, M.S.; Tesh, V.L. Shiga toxin 1-induced proinflammatory cytokine production is regulated by the Phosphatidylinositol 3-kinase/Akt/Mammalian Target of Rapamycin signaling pathway. Infect. Immun. 2009, 77, 3919–3931. [Google Scholar] [CrossRef] [PubMed]
- Lentz, E.K.; Cherla, R.P.; Jaspers, V.; Weeks, B.R.; Tesh, V.L. Role of tumor necrosis factor alpha in disease using a mouse model of Shiga toxin-mediated renal damage. Infect. Immun. 2010, 78, 3689–3699. [Google Scholar] [CrossRef] [PubMed]
- Ergonul, Z.; Hughes, A.K.; Kohan, D.E. Induction of apoptosis of human brain microvascular endothelial cells by Shiga toxin 1. J. Infect. Dis. 2003, 187, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Hagel, C.; Krasemann, S.; Loffler, J.; Puschel, K.; Magnus, T.; Glatzel, M. Upregulation of Shiga toxin receptor CD77/Gb3 and interleukin-1beta expression in the brain of EHEC patients with hemolytic uremic syndrome and neurologic symptoms. Brain Pathol. 2015, 25, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Landoni, V.I.; Schierloh, P.; de Campos Nebel, M.; Fernandez, G.C.; Calatayud, C.; Lapponi, M.J.; Isturiz, M.A. Shiga toxin 1 induces on lipopolysaccharide-treated astrocytes the release of tumor necrosis factor-alpha that alter brain-like endothelium integrity. PLoS Pathog. 2012, 8, e1002632. [Google Scholar] [CrossRef] [PubMed]
- Van Setten, P.A.; van Hinsbergh, V.W.; van der Velden, T.J.; van de Kar, N.C.; Vermeer, M.; Mahan, J.D.; Assmann, K.J.; van den Heuvel, L.P.; Monnens, L.A. Effects of TNF alpha on verocytotoxin cytotoxicity in purified human glomerular microvascular endothelial cells. Kidney Int. 1997, 51, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, P.B.; Jacewicz, M.S.; Conn, K.J.; Koul, O.; Wells, J.M.; Fine, R.E.; Newburg, D.S. Escherichia coli Shiga toxin 1 and TNF-alpha induce cytokine release by human cerebral microvascular endothelial cells. Microb. Pathog. 2004, 36, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kim, M.H.; Tesh, V.L. Shiga toxins expressed by human pathogenic bacteria induce immune responses in host cells. J. Microbiol. 2013, 51, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Kovbasnjuk, O.; Mourtazina, R.; Baibakov, B.; Wang, T.; Elowsky, C.; Choti, M.A.; Kane, A.; Donowitz, M. The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 19087–19092. [Google Scholar] [CrossRef] [PubMed]
- Falguieres, T.; Maak, M.; von Weyhern, C.; Sarr, M.; Sastre, X.; Poupon, M.F.; Robine, S.; Johannes, L.; Janssen, K.P. Human colorectal tumors and metastases express Gb3 and can be targeted by an intestinal pathogen-based delivery tool. Mol. Cancer Ther. 2008, 7, 2498–2508. [Google Scholar] [CrossRef] [PubMed]
- LaCasse, E.C.; Bray, M.R.; Patterson, B.; Lim, W.M.; Perampalam, S.; Radvanyi, L.G.; Keating, A.; Stewart, A.K.; Buckstein, R.; Sandhu, J.S.; et al. Shiga-like toxin-1 receptor on human breast cancer, lymphoma, and myeloma and absence from CD34(+) hematopoietic stem cells: Implications for ex vivo tumor purging and autologous stem cell transplantation. Blood 1999, 94, 2901–2910. [Google Scholar] [PubMed]
- Johansson, D.; Kosovac, E.; Moharer, J.; Ljuslinder, I.; Brannstrom, T.; Johansson, A.; Behnam-Motlagh, P. Expression of verotoxin-1 receptor Gb3 in breast cancer tissue and verotoxin-1 signal transduction to apoptosis. BMC Cancer 2009, 9. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, M.; Johansson, M.; Bergstrom, P.; Bergenheim, A.T.; Henriksson, R. Effects of the VEGFR inhibitor ZD6474 in combination with radiotherapy and temozolomide in an orthotopic glioma model. J. Neurooncol. 2008, 88, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Heath-Engel, H.M.; Lingwood, C.A. Verotoxin sensitivity of ECV304 cells in vitro and in vivo in a xenograft tumour model: VT1 as a tumour neovascular marker. Angiogenesis 2003, 6, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Salhia, B.; Rutka, J.T.; Lingwood, C.; Nutikka, A.; van Furth, W.R. The treatment of malignant meningioma with verotoxin. Neoplasia 2002, 4, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Ishitoya, S.; Kurazono, H.; Nishiyama, H.; Nakamura, E.; Kamoto, T.; Habuchi, T.; Terai, A.; Ogawa, O.; Yamamoto, S. Verotoxin induces rapid elimination of human renal tumor xenografts in scid mice. J. Urol. 2004, 171, 1309–1313. [Google Scholar] [CrossRef] [PubMed]
- Arab, S.; Rutka, J.; Lingwood, C. Verotoxin induces apoptosis and the complete, rapid, long-term elimination of human astrocytoma xenografts in nude mice. Oncol. Res. 1999, 11, 33–39. [Google Scholar] [PubMed]
- Amessou, M.; Carrez, D.; Patin, D.; Sarr, M.; Grierson, D.S.; Croisy, A.; Tedesco, A.C.; Maillard, P.; Johannes, L. Retrograde delivery of photosensitizer (TPPp-O-β-GluOH)3 selectively potentiates its photodynamic activity. Bioconj. Chem. 2008, 19, 532–538. [Google Scholar] [CrossRef] [PubMed]
- El Alaoui, A.; Schmidt, F.; Amessou, M.; Sarr, M.; Decaudin, D.; Florent, J.C.; Johannes, L. Shiga toxin-mediated retrograde delivery of a topoisomerase I inhibitor prodrug. Angew. Chem. Int. Ed. Engl. 2007, 46, 6469–6472. [Google Scholar] [CrossRef] [PubMed]
- Batisse, C.; Dransart, E.; Ait Sarkouh, R.; Brulle, L.; Bai, S.K.; Godefroy, S.; Johannes, L.; Schmidt, F. A new delivery system for auristatin in StxB-drug conjugate therapy. Eur. J. Med. Chem. 2015, 95, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Viel, T.; Dransart, E.; Nemati, F.; Henry, E.; Theze, B.; Decaudin, D.; Lewandowski, D.; Boisgard, R.; Johannes, L.; Tavitian, B. In vivo tumor targeting by the B-subunit of Shiga toxin. Mol. Imaging 2008, 7, 239–247. [Google Scholar] [PubMed]
- Qin, S.; Caskey, C.F.; Ferrara, K.W. Ultrasound contrast microbubbles in imaging and therapy: Physical principles and engineering. Phys. Med. Biol. 2009, 54, R27–R57. [Google Scholar] [CrossRef] [PubMed]
- Couture, O.; Dransart, E.; Dehay, S.; Nemati, F.; Decaudin, D.; Johannes, L.; Tanter, M. Tumor delivery of ultrasound contrast agents using Shiga toxin B subunit. Mol. Imaging 2011, 10, 135–143. [Google Scholar] [PubMed]
- Bray, M.R.; Bisland, S.; Perampalam, S.; Lim, W.M.; Gariepy, J. Probing the surface of eukaryotic cells using combinatorial toxin libraries. Curr. Biol. 2001, 11, 697–701. [Google Scholar] [CrossRef]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.-S.; Koo, S.; Jeong, D.G.; Tesh, V.L. Shiga Toxins as Multi-Functional Proteins: Induction of Host Cellular Stress Responses, Role in Pathogenesis and Therapeutic Applications. Toxins 2016, 8, 77. https://doi.org/10.3390/toxins8030077
Lee M-S, Koo S, Jeong DG, Tesh VL. Shiga Toxins as Multi-Functional Proteins: Induction of Host Cellular Stress Responses, Role in Pathogenesis and Therapeutic Applications. Toxins. 2016; 8(3):77. https://doi.org/10.3390/toxins8030077
Chicago/Turabian StyleLee, Moo-Seung, Sunwoo Koo, Dae Gwin Jeong, and Vernon L. Tesh. 2016. "Shiga Toxins as Multi-Functional Proteins: Induction of Host Cellular Stress Responses, Role in Pathogenesis and Therapeutic Applications" Toxins 8, no. 3: 77. https://doi.org/10.3390/toxins8030077