
Liquid Chromatography with a Fluorimetric Detection Method for Analysis of Paralytic Shellfish Toxins and Tetrodotoxin Based on a Porous Graphitic Carbon Column



Abstract
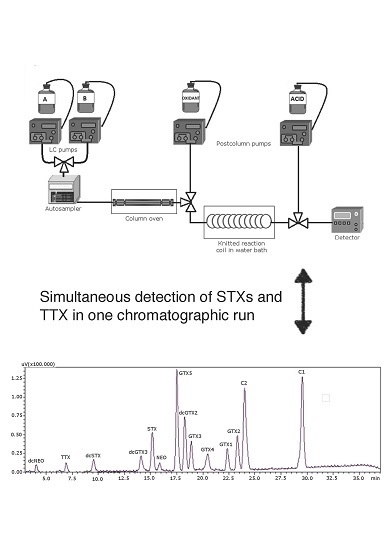
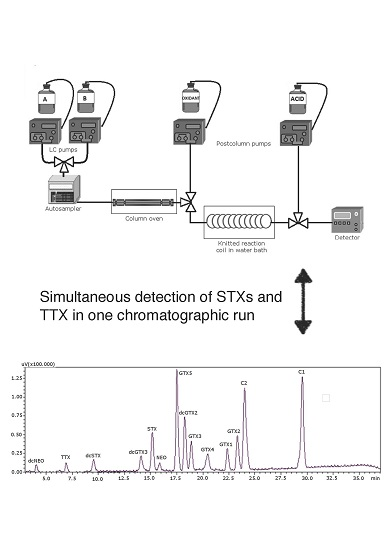
:
1. Introduction
2. Results and Discussion
2.1. Chromatographic Conditions
2.1.1. Development of TFA as an Ion Pairing Agent
2.1.2. Detector Outflow pH
2.1.3. Reaction Temperature
2.1.4. Column Temperature
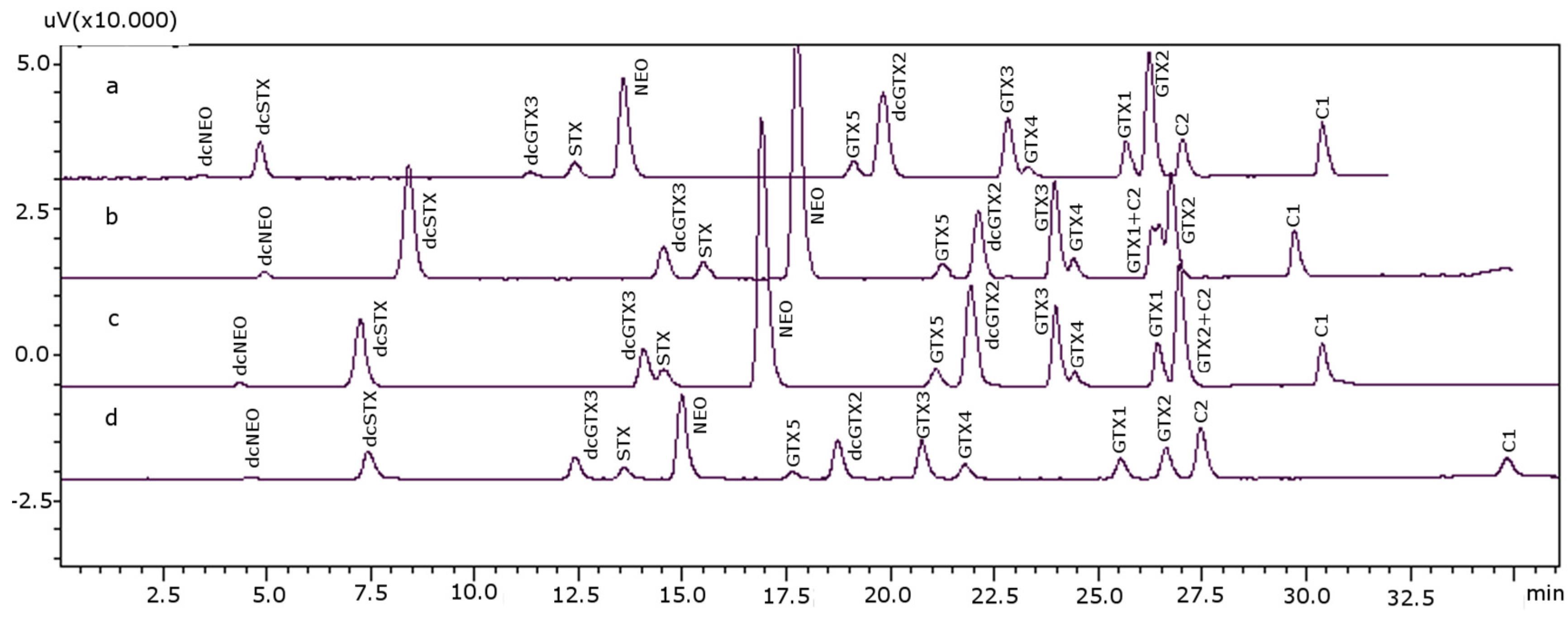
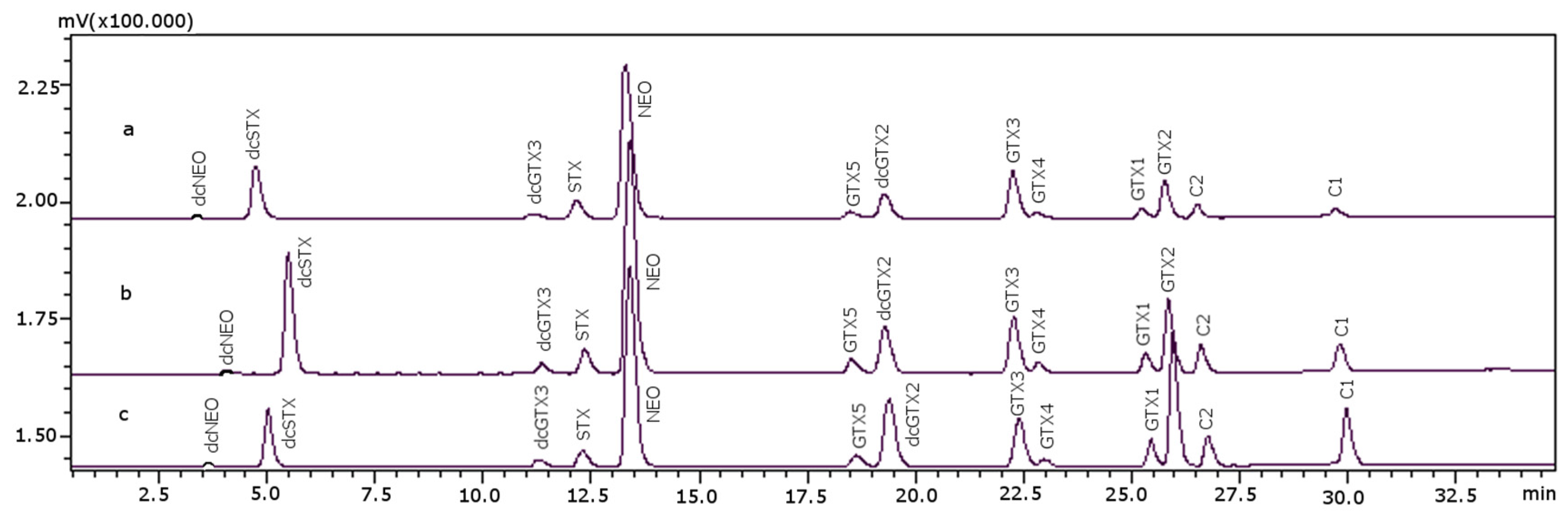
2.2. Extraction and Sample Clean-Up
2.3. Validation Parameters
3. Conclusions
4. Materials and Methods
4.1. Chemicals
4.2. Instrumentation
4.3. Samples Extraction and Clean-Up
4.4. Development of HPLC and PCOX Fluorescence Detection
4.5. Method Validation
4.6. Regeneration of Hypercarb Column and SPE Cartridges
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Daneshian, M.; Botana, L.M.; Dechraoui Bottein, M.Y.; Buckland, G.; Campas, M.; Dennison, N.; Dickey, R.W.; Diogene, J.; Fessard, V.; Hartung, T.; et al. A roadmap for hazard monitoring and risk assessment of marine biotoxins on the basis of chemical and biological test systems. ALTEX 2013, 30, 487–545. [Google Scholar] [CrossRef] [PubMed]
- Van Dolah, F.M. Marine algal toxins: Origins, health effects, and their increased occurrence. Environ. Health Perspect. 2000, 108, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Bricelj, V.M.; Shumway, S.E. Paralytic Shellfish Toxins in Bivalve Molluscs: Occurrence, Transfer Kinetics, and Biotransformation. Rev. Fish. Sci. 1998, 6, 315–383. [Google Scholar] [CrossRef]
- Vlamis, A.; Katikou, P. Ecobiology and geographical distribution of potentially toxic marine dinoflagellates. In Seafood and Freshwater Toxins: Pharmacology, Physiology and Detection, 3rd ed.; Botana, L.M, Ed.; CRC Press: Boca Raton, FL, USA, 2014; pp. 569–625. [Google Scholar]
- Dell’Aversano, C.; Walter, J.A.; Burton, I.W.; Stirling, D.J.; Fattorusso, E.; Quilliam, M.A. Isolation and structure elucidation of new and unusual saxitoxin analogues from mussels. J. Nat. Prod. 2008, 71, 1518–1523. [Google Scholar] [CrossRef] [PubMed]
- Wiese, M.; D’ Agostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic Alkaloids: Saxitoxin and its Analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.J.; Wekell, M.M. Determination of paralytic shellfish poisoning toxins by high pressure liquid chromatography. In Seafood Toxins; Ragelis, E.P., Ed.; American Chemical Society: Washington, DC, USA, 1984. [Google Scholar]
- Humpage, A.R.; Magalhaes, V.F.; Froscio, S.M. Comparison of analytical tools and biological assays for detection of paralytic shellfish poisoning toxins. Anal. Bioanal. Chem. 2010, 397, 1655–1671. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.F.; Niedzwiadek, B. Quantitative Determination of Paralytic Shellfish Poisoning Toxins in Shellfish by Using Prechromatographic Oxidation and Liquid Chromatography with Fluorescence Detection. J. AOAC Int. 2001, 84, 1099–1108. [Google Scholar] [PubMed]
- Thomas, K.M.; Chung, S.; Ku, J.; Reeves, K.; Quilliam, M.A. Analysis of PSP toxins by liquid chromatography with post column oxidation and fluorescence detection. In Molluscan Shellfish Safety; Henshilwood, K., McMahon, B.D.T., Cusack, C., Keaveney, S., Silke, J., O’Cinneide, M., Lyons, D., Hess, P., Eds.; The Marine Institute: Galway, Ireland, 2006; pp. 63–122. [Google Scholar]
- Lawrence, J.F.; Niedzwiadek, B.; Menard, C. Quantitative Determination of Paralytic Shellfish Poisoning Toxins in Shellfish Using Prechromatographic Oxidation and Liquid Chromatography with Fluorescence Detection: Collaborative Study. J. AOAC Int. 2005, 88, 1714–1732. [Google Scholar] [PubMed]
- AOAC. Method 2005.06: Paralytic Shellfish Poisoning Toxins in Shellfish. Prechromatographic Oxidation and Liquid Chromatography with Fluorescence Detection. In Official Methods of Analysis of the Association of Official Analytical Chemists, 1st ed.; AOAC: Gaithersburg, MD, USA, 2005. [Google Scholar]
- Turner, A.D.; Hatfield, R.G. Refinement of AOAC Official Method 2005.06 liquid chromatography-fluorescence detection method to improve performance characteristics for the determination of paralytic shellfish toxins in king and queen scallops. J. AOAC Int. 2012, 95, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.D.; Hatfield, R.G.; Rapkova-Dhanji, M.; Norton, D.M.; Algoet, M.; Lees, D.N. Single-laboratory validation of a refined AOAC HPLC method 2005.06 for oysters, cockles, and clams in U.K. shellfish. J. AOAC Int. 2010, 93, 1482–1493. [Google Scholar] [PubMed]
- Ben-Gigirey, B.; Rodriguez-Velasco, M.L.; Gago-Martinez, A. Extension of the validation of AOAC Official Method 2005.06 for dc-GTX2,3: Interlaboratory study. J. AOAC Int. 2012, 95, 111–121. [Google Scholar] [CrossRef] [PubMed]
- DeGrasse, S.L.; van de Riet, J.; Hatfield, R.; Turner, A. Pre- versus pos-tcolumn oxidation liquid chromatography fluorescence detection of paralytic shellfish toxins. Toxicon 2010, 57, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Vale, C.; Alfonso, A.; Vieytes, M.R.; Romaris, X.M.; Arevalo, F.; Botana, A.M.; Botana, L.M. In vitro and in vivo evaluation of paralytic shellfish poisoning toxin potency and the influence of the pH of extraction. Anal. Chem. 2008, 80, 1770–1176. [Google Scholar] [CrossRef] [PubMed]
- Alonso, E.; Alfonso, A.; Vieytes, M.R.; Botana, L.M. Evaluation of toxicity equivalent factors of paralytic shellfish poisoning toxins in seven human sodium channels types by an automated high throughput electrophysiology system. Arch. Toxicol. 2015, 90, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.; Vale, C.; Botana, A.M.; Alonso, E.; Vieytes, M.R.; Botana, L.M. Determination of Toxicity Equivalent Factors for Paralytic Shellfish Toxins by Electrophysiological Measurements in Cultured Neurons. Chem. Res. Toxicol. 2011, 24, 1153–1157. [Google Scholar] [CrossRef] [PubMed]
- Oshima, Y. Postcolumn derivatization liquid chromatographic method for paralytic shellfish toxins. J. AOAC Int. 1995, 78, 528–532. [Google Scholar]
- Rourke, W.A.; Murphy, C.J.; Pitcher, G.; Van de Riet, J.M.; Burns, B.G.; Thomas, K.M.; Quilliam, M.A. Rapid postcolumn methodology for determination of paralytic shellfish toxins in shellfish tissue. J. AOAC Int. 2008, 91, 589–597. [Google Scholar] [PubMed]
- Van de Riet, J.M.; Gibbs, R.S.; Chou, F.W.; Muggah, P.M.; Rourke, W.A.; Burns, G.; Thomas, K.M.; Quilliam, M.A. Liquid chromatographic post-column oxidation method for analysis of paralytic shellfish toxins in mussels, clams, scallops, and oysters: Single-laboratory validation. J. AOAC Int. 2009, 92, 1690–1704. [Google Scholar] [PubMed]
- Van de Riet, J.M.; Gibbs, R.S.; Muggah, P.M.; Rourke, W.A.; MacNeil, J.D.; Quilliam, M.A. Liquid chromatography post-column oxidation (PCOX) method for the determination of paralytic shellfish toxins in mussels, clams, oysters, and scallops: Collaborative study. J. AOAC Int. 2010, 94, 1154–1176. [Google Scholar]
- AOAC. Official method 2011.02 Determination of paralytic shellfish poisoning toxins in mussels, clams, oysters and Scallops. In Post-Column Oxidation Method (PCOX); First Action 2011; AOAC International: Gaithersburg, MD, USA, 2011. [Google Scholar]
- Biré, R.; Krys, S.; Frémy, J.M.; Dragacci, S. Improved Solid-Phase Extraction Procedure in the Analysis of Paralytic Shellfish Poisoning Toxins by Liquid Chromatography with Fluorescence Detection. J. Agric. Food. Chem. 2003, 51, 6386–6390. [Google Scholar] [CrossRef] [PubMed]
- Boundy, M.J.; Selwood, A.I.; Harwood, D.T.; McNabb, P.S.; Turner, A.D. Development of a sensitive and selective liquid chromatography–mass spectrometry method for high throughput analysis of paralytic shellfish toxins using graphitised carbon solid phase extraction. J. Chromatogr. A 2015, 1387, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vlamis, A.; Katikou, P.; Rodriguez, I.; Rey, V.; Alfonso, A.; Papazachariou, A.; Zacharaki, T.; Botana, A.M.; Botana, L.M. First Detection of Tetrodotoxin in Greek Shellfish by UPLC-MS/MS Potentially Linked to the Presence of the Dinoflagellate Prorocentrum. minimum. Toxins 2015, 7, 1779–1807. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.D.; Powell, A.; Schofield, A.; Lees, D.N.; Baker-Austin, C. Detection of the pufferfish toxin tetrodotoxin in European bivalves, England, 2013 to 2014. Euro Surveill. 2015, 20, 1–33. [Google Scholar] [CrossRef]
- Bane, V.; Lehane, M.; Dikshit, M.; O’Riordan, A.; Furey, A. Tetrodotoxin: Chemistry, toxicity, source, distribution and detection. Toxins 2014, 6, 693–755. [Google Scholar] [CrossRef] [PubMed]
- Saoudi, M.; Rabeh, F.B.; Jammoussi, K.; Abdelmouleh, A.; Belbahri, L.; El Feki, A. Biochemical and physiological responses in Wistar rat after administration of puffer fish (Lagocephalus lagocephalus) flesh. J. Food Agric. Environ. 2007, 5, 107–111. [Google Scholar]
- Noguchi, T.; Ebesu, J.S.M. Puffer poisoning: Epidemiology and treatment. Toxin Rev. 2001, 20, 1–10. [Google Scholar] [CrossRef]
- Bentur, Y.; Ashkar, J.; Lurie, Y.; Levy, Y.; Azzam, Z.S.; Litmanovich, M.; Golik, M.; Gurevych, B.; Golani, D.; Eisenman, A. Lessepsian migration and tetrodotoxin poisoning due to Lagocephalus sceleratus in the eastern Mediterranean. Toxicon 2008, 52, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.; Alfonso, A.; Vale, C.; Alfonso, C.; Vale, P.; Tellez, A.; Botana, L.M. First toxicity report of tetrodotoxin and 5, 6, 11-trideoxyTTX in the trumpet shell Charonia lampas lampas in Europe. Anal. Chem. 2008, 80, 5622–5629. [Google Scholar] [CrossRef] [PubMed]
- Shiu, Y.C.; Lu, Y.H.; Tsai, Y.H.; Chen, S.K.; Hwang, D.F. Occurrence of tetrodotoxin in the causative gastropod Polinices didyma and another gastropod Natica lineata collected from western Taiwan. J. Food Drug Anal. 2003, 11, 159–163. [Google Scholar]
- Yasumoto, T.; Tooru, M. Fluorometric determination of tetrodotoxin by high performance liquid chromatography. Agric. Biol. Chem. 1985, 49, 3077–3080. [Google Scholar]
- Wingerd, J.S.; Vetter, I.; Lewis, R.J. Voltage-Gated Sodium Channels as Therapeutic Targets. In Therapeutic Targets: Modulation, Inhibition, and Activation; Botana, L.M., Loza, M., Eds.; Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 63–122. [Google Scholar]
- Kawabata, T. Assay method for tetrodotoxin. J. Food Hyg. Soc. Jpn. 1978, 2, 232–240. [Google Scholar]
- Gessner, B.D.; McLaughlin, J.B. Epidemiologic Impact of Toxic Episodes: Neurotoxic Toxins. In Seafood and Freshwater Toxins: Pharmacology, Physiology, and Detection; Botana, L.M., Ed.; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2008; pp. 77–103. [Google Scholar]
- Cohen, N.J.; Deeds, J.R.; Wong, E.S.; Hanner, R.H.; Yancy, H.F.; White, K.D.; Thompson, T.M.; Wahl, M.; Pham, T.D.; Guichard, F.M. Public health response to puffer fish (tetrodotoxin) poisoning from mislabeled product. J. Food Prot. 2009, 72, 810–817. [Google Scholar] [PubMed]
- Scientific, T. Method Development Guide for Hypercarb Columns; Thermo Scientific: Runcorn, UK, 2007. [Google Scholar]
- AOAC. Official method 959.08. Paralytic shellfish poison. Biological method. Final action. In AOAC Official Methods for Analysis; AOAC: Gaithersburg, MD, USA, 2005; pp. 79–80. [Google Scholar]
- Botana, L.M.; Vieytes, M.R.; Alfonso, A.; Louzao, M.C. Phycotoxins. Paralytic Shellfish Poisoning. Diarrhetic Shellfish Poisoning. In Handbook of Food Analysis; Nollet, L., Ed.; Marcel Dekker Inc.: London, UK, 1996; pp. 1147–1170. [Google Scholar]
- Hummert, C.; Ritscher, M.; Reinhardt, R.; Luckas, B. Analysis of the characteristic PSP profiles of Pyrodinium bahamense and several strains of Alexandrium by HPLC based on ion-pair chromatographic separation, post-column oxidation, and fluorescence detection. Chromatographia 1997, 45, 312–316. [Google Scholar] [CrossRef]
- Luckas, B. Chemical Analysis of PSP Toxins. In Seafood and Freshwater Toxins: Pharmacology, Physiology and Detection, 2nd ed.; Botana, L.M., Ed.; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2000; pp. 173–186. [Google Scholar]
- Ben-Gigirey, B.; Rodriguez-Velasco, M.L.; Otero, A.; Vieites, J.M.; Cabado, A.G. A comparative study for PSP toxins quantification by using MBA and HPLC official methods in shellfish. Toxicon 2012, 60, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Rey, V.; Alfonso, A.; Botana, L.M.; Botana, A.M. Influence of Different Shellfish Matrices on the Separation of PSP Toxins Using a Postcolumn Oxidation Liquid Chromatography Method. Toxins 2015, 7, 1324–1340. [Google Scholar] [CrossRef] [PubMed]
- IUPAC. Guidelines for Collaborative Study of Procedure to Validate Characteristics of a Method of Analysis. J. AOAC Int. 1989, 72, 694–704. [Google Scholar]
- Peters, F.T.; Drummer, O.H.; Musshoff, F. Validation of new methods. Forensic Sci. Int. 2007, 165, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Lindner, W.; Wainer, I.W. Requirements for initial assay validation and publication in J. Chromatography B. J. Chromatogr. B 1998, 707, 1–2. [Google Scholar]
- Alder, L.; Holland, P.T.; Lantos, J.; Lee, M.; McNeil, J.D.; O’Rangers, J.; Van Zoonen, P.; Ambrus, A. Guidelines for Single Laboratory Validation of Analytical Methods for Trace Level Concentrations of Organic Chemical. In Principles and Practices of Method Validation; Fajgelj, A.A.A., Ed.; The Royal Society of Chemistry: Cambridge, UK, 2000; p. 18. [Google Scholar]
- Hwang, D.F.; Cheng, C.A.; Tsai, H.T.; Shih, D.Y.C.; Ko, H.C.; Yang, R.Z.; Jeng, S.S. Identification of Tetrodotoxin and Paralytic Shellfish Toxins in Marine Gastropods Implicated in Food Poisoning. Fish. Sci. 1995, 61, 675–679. [Google Scholar]
- Nagashima, Y.; Maruyama, J.; Noguchi, T.; Hashimoto, K. Analysis of paralytic shellfish poison and tetrodotoxin (in shellfish and fish) by ion-pairing high performance liquid chromatography. Bull. Jpn. Soc. Sci. Fish. 1987, 53, 819–823. [Google Scholar] [CrossRef]
- Martin-Smith, M.; Rudd, D.R. The importance of proper validation of the analytical methods employed in the quality control pharmaceuticals. Acta Pharm. Jugosl. 1990, 40, 7–19. [Google Scholar]
- Camacho Sanchez, M.A.; Torres Suarez, A.I.; Gil Alegre, M.E.; Obregon Sanchez, M.M.; Ruz Palomar, V. Validation protocol of analytical methods for finished pharmaceutical products. STP Pharma Prat. 1993, 3, 197–202. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Range in mg·kg−1 | Range in mg STX∙diHCl kg−1 | Correlation (r) | ||
|---|---|---|---|---|---|
| Lower | Upper | Lower | Upper | ||
| dcNEO | 0.021 | 1.33 | 0.009 | 0.575 | 0.9935 |
| dcSTX | 0.003 | 3.28 | 0.002 | 2.443 | 0.9849 |
| dcGTX3 | 0.004 | 1.03 | 0.001 | 0.408 | 0.9987 |
| STX | 0.007 | 3.81 | 0.007 | 3.812 | 0.9950 |
| NEO | 0.052 | 1.66 | 0.046 | 1.816 | 0.9937 |
| GTX5 | 0.160 | 2.62 | 0.010 | 0.165 | 0.9937 |
| dcGTX2 | 0.002 | 4.37 | 0.0003 | 0.710 | 0.9982 |
| GTX3 | 0.032 | 1.02 | 0.019 | 0.612 | 0.9986 |
| GTX4 | 0.0007 | 1.28 | 0.0005 | 0.841 | 0.9911 |
| GTX1 | 0.004 | 3.95 | 0.004 | 3.554 | 0.9994 |
| C2 | 0.006 | 1.44 | 0.0005 | 0.109 | 0.9969 |
| GTX2 | 0.043 | 2.77 | 0.014 | 0.934 | 0.9981 |
| C1 | 0.157 | 5.01 | 0.0007 | 0.024 | 0.9980 |
| Matrix | dcNEO | dcSTX | dcGTX3 | STX | NEO | GTX5 | dcGTX2 | GTX3 | GTX4 | GTX1 | C2 | GTX2 | C1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mussel | LOD | 0.0045 | 0.0004 | 0.0006 | 0.0044 | 0.0093 | 0.0027 | 0.0001 | 0.0012 | 0.0015 | 0.0037 | 0.0003 | 0.0001 | 0.0002 |
| LOQ | 0.0070 | 0.0007 | 0.0019 | 0.0055 | 0.0163 | 0.0099 | 0.0003 | 0.0113 | 0.0059 | 0.0117 | 0.0007 | 0.0138 | 0.0005 | |
| Clam | LOD | 0.0030 | 0.0003 | 0.0035 | 0.0041 | 0.0059 | 0.0007 | 0.0007 | 0.0114 | 0.0010 | 0.0008 | 0.0024 | 0.0043 | 0.0004 |
| LOQ | 0.0040 | 0.0004 | 0.0035 | 0.0047 | 0.0078 | 0.0012 | 0.0014 | 0.0136 | 0.0026 | 0.0032 | 0.0027 | 0.0070 | 0.0005 | |
| Scallop | LOD | 0.0344 | 0.0003 | 0.0002 | 0.0038 | 0.0054 | 0.0002 | 0.0004 | 0.0007 | 0.0007 | 0.0005 | 0.0001 | 0.0005 | 0.0001 |
| LOQ | 0.0750 | 0.0004 | 0.0005 | 0.0039 | 0.0065 | 0.0007 | 0.0010 | 0.0018 | 0.0019 | 0.0011 | 0.0001 | 0.0012 | 0.0001 | |
| Oyster | LOD | 0.0330 | 0.0003 | 0.0106 | 0.0039 | 0.0053 | 0.0069 | 0.0253 | 0.0062 | 0.0004 | 0.0016 | 0.0035 | 0.0366 | 0.0008 |
| LOQ | 0.0750 | 0.0004 | 0.0111 | 0.0042 | 0.0065 | 0.0087 | 0.0258 | 0.0074 | 0.0009 | 0.0043 | 0.0037 | 0.0420 | 0.0008 | |
| mg STX∙diHCl kg−1 | Matrix | dcNEO | dcSTX | dcGTX3 | STX | NEO | GTX5 | dcGTX2 | GTX3 | GTX4 | GTX1 | C2 | GTX2 | C1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.2 | Mussel | 7.7 | 5.1 | 2.7 | 4.7 | 8.7 | 6.4 | 5.9 | 3.8 | 2.4 | 2.9 | 4.0 | 4.6 | 5.6 |
| Clam | 6.8 | 3.9 | 2.0 | 4.7 | 5.7 | 6.2 | 5.1 | 5.7 | 3.6 | 3.6 | 6.5 | 6.9 | 5.5 | |
| Scallop | 8.4 | 5.8 | 2.3 | 6.3 | 5.7 | 6.0 | 1.4 | 3.9 | 6.4 | 8.0 | 9.4 | 4.9 | 6.5 | |
| Oyster | 7.3 | 4.6 | 5.8 | 7.8 | 9.0 | 9.1 | 1.8 | 4.9 | 5.0 | 4.7 | 6.5 | 4.3 | 5.2 | |
| 0.8 | Mussel | 3.9 | 4.6 | 3.6 | 5.3 | 4.8 | 2.8 | 5.7 | 4.4 | 5.5 | 2.7 | 6.1 | 5.5 | 4.1 |
| Clam | 6.0 | 6.4 | 3.6 | 5.3 | 5.8 | 6.7 | 3.1 | 7.0 | 4.7 | 2.0 | 3.6 | 5.8 | 3.4 | |
| Scallop | 6.2 | 3.2 | 1.8 | 5.5 | 2.1 | 6.0 | 5.4 | 5.2 | 5.5 | 4.8 | 7.8 | 3.3 | 2.2 | |
| Oyster | 8.6 | 1.6 | 1.7 | 7.1 | 6.6 | 8.8 | 6.4 | 4.5 | 2.6 | 5.3 | 3.3 | 4.3 | 2.7 | |
| 1.6 | Mussel | 2.3 | 4.3 | 6.4 | 5.6 | 4.5 | 1.7 | 2.6 | 4.7 | 5.4 | 2.1 | 4.6 | 2.7 | 3.7 |
| Clam | 5.2 | 3.3 | 4.6 | 3.9 | 5.1 | 2.9 | 2.9 | 6.0 | 5.6 | 2.4 | 4.2 | 6.3 | 4.1 | |
| Scallop | 4.4 | 4.9 | 5.4 | 6.8 | 4.4 | 2.4 | 3.7 | 3.4 | 6.1 | 2.6 | 3.9 | 5.4 | 3.6 | |
| Oyster | 2.5 | 2.4 | 3.5 | 5.9 | 5.2 | 2.5 | 2.7 | 2.8 | 3.1 | 2.0 | 6.5 | 4.6 | 5.6 |
| mg STX∙diHCl kg−1 | Matrix | dcNEO | dcSTX | dcGTX3 | STX | NEO | GTX5 | dcGTX2 | GTX3 | GTX4 | GTX1 | C2 | GTX2 | C1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.2 | Mussel | 0.8 | 0.6 | 0.3 | 0.8 | 0.8 | 0.7 | 0.6 | 0.5 | 0.4 | 0.3 | 0.4 | 0.4 | 0.2 |
| Clam | 0.5 | 0.4 | 0.8 | 0.7 | 0.7 | 0.3 | 0.5 | 0.5 | 0.5 | 0.2 | 0.4 | 0.3 | 0.3 | |
| Scallop | 0.9 | 0.2 | 0.3 | 0.2 | 0.3 | 0.3 | 0.2 | 0.3 | 0.6 | 0.3 | 0.2 | 0.2 | 0.2 | |
| Oyster | 0.5 | 0.2 | 0.3 | 0.3 | 0.3 | 0.3 | 0.2 | 0.2 | 0.2 | 0.3 | 0.2 | 0.2 | 0.2 | |
| 0.8 | Mussel | 0.6 | 0.5 | 0.4 | 0.1 | 0.2 | 0.3 | 0.3 | 0.3 | 0.3 | 0.6 | 0.2 | 0.2 | 0.5 |
| Clam | 0.8 | 0.6 | 0.4 | 0.5 | 0.5 | 0.3 | 0.3 | 0.3 | 0.3 | 0.5 | 0.2 | 0.2 | 0.4 | |
| Scallop | 0.3 | 0.5 | 0.5 | 0.4 | 0.4 | 0.3 | 0.3 | 0.3 | 0.4 | 0.5 | 0.2 | 0.3 | 0.4 | |
| Oyster | 0.7 | 0.8 | 0.7 | 0.7 | 0.8 | 0.8 | 0.7 | 0.6 | 0.4 | 0.6 | 0.3 | 0.1 | 0.4 | |
| 1.6 | Mussel | 0.7 | 0.6 | 0.4 | 0.5 | 0.4 | 0.7 | 0.8 | 0.4 | 0.7 | 0.5 | 0.5 | 0.5 | 0.8 |
| Clam | 0.9 | 0.4 | 0.4 | 0.8 | 0.7 | 0.3 | 0.7 | 0.5 | 0.5 | 0.9 | 0.5 | 0.8 | 0.8 | |
| Scallop | 0.3 | 0.4 | 0.3 | 0.4 | 0.5 | 0.7 | 0.2 | 0.5 | 0.6 | 0.5 | 0.7 | 0.6 | 0.8 | |
| Oyster | 0.8 | 0.7 | 0.6 | 0.8 | 0.2 | 0.6 | 0.8 | 0.8 | 0.8 | 0.8 | 0.5 | 0.6 | 0.8 |
| Spiked Matrix | dcNEO | dcSTX | dcGTX3 | STX | NEO | GTX5 | dcGTX2 | GTX3 | GTX4 | GTX1 | C2 | GTX2 | C1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Matrix | Spiked level, mg·kg−1 | 0.08 | 0.03 | 0.02 | 0.04 | 0.09 | 0.16 | 0.08 | 0.05 | 0.02 | 0.06 | 0.03 | 0.13 | 0.10 |
| Mussel | Recovery% | 65.8 | 82.4 | 65.2 | 83.4 | 72 | 85.8 | 87.8 | 84.2 | 76.2 | 79.2 | 76 | 73.2 | 78 |
| %RSD | 4.7 | 5.4 | 3.0 | 7.0 | 6.4 | 7.7 | 9.3 | 9.3 | 1.6 | 1.5 | 8.9 | 5.4 | 2.9 | |
| Clam | Recovery% | 63.6 | 92.6 | 67.8 | 78.8 | 72.8 | 86.2 | 84 | 77.6 | 73 | 75.4 | 68.2 | 83 | 74.6 |
| %RSD | 2.9 | 3.6 | 1.9 | 2.6 | 2.6 | 4.0 | 8.8 | 4.0 | 2.9 | 3.5 | 2.2 | 4.2 | 3.8 | |
| Scallop | Recovery% | 72 | 88.2 | 69 | 80.2 | 72 | 84.6 | 118.6 | 78.6 | 74.4 | 73.8 | 75.2 | 82.6 | 73.8 |
| %RSD | 2.1 | 1.3 | 1.2 | 1.9 | 2.5 | 2.9 | 1.7 | 1.5 | 3.6 | 4.1 | 2.9 | 4.0 | 2.5 | |
| Oyster | Recovery% | 68.6 | 78.2 | 63.6 | 79.2 | 74.4 | 72.4 | 80 | 69.8 | 73.2 | 76 | 66.2 | 67.2 | 62.6 |
| %RSD | 2.5 | 1.9 | 5.8 | 4.6 | 4.3 | 3.5 | 1.9 | 3.1 | 1.6 | 4.0 | 3.0 | 2.6 | 2.2 | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rey, V.; Botana, A.M.; Alvarez, M.; Antelo, A.; Botana, L.M. Liquid Chromatography with a Fluorimetric Detection Method for Analysis of Paralytic Shellfish Toxins and Tetrodotoxin Based on a Porous Graphitic Carbon Column. Toxins 2016, 8, 196. https://doi.org/10.3390/toxins8070196
Rey V, Botana AM, Alvarez M, Antelo A, Botana LM. Liquid Chromatography with a Fluorimetric Detection Method for Analysis of Paralytic Shellfish Toxins and Tetrodotoxin Based on a Porous Graphitic Carbon Column. Toxins. 2016; 8(7):196. https://doi.org/10.3390/toxins8070196
Chicago/Turabian StyleRey, Veronica, Ana M. Botana, Mercedes Alvarez, Alvaro Antelo, and Luis M. Botana. 2016. "Liquid Chromatography with a Fluorimetric Detection Method for Analysis of Paralytic Shellfish Toxins and Tetrodotoxin Based on a Porous Graphitic Carbon Column" Toxins 8, no. 7: 196. https://doi.org/10.3390/toxins8070196
APA StyleRey, V., Botana, A. M., Alvarez, M., Antelo, A., & Botana, L. M. (2016). Liquid Chromatography with a Fluorimetric Detection Method for Analysis of Paralytic Shellfish Toxins and Tetrodotoxin Based on a Porous Graphitic Carbon Column. Toxins, 8(7), 196. https://doi.org/10.3390/toxins8070196