
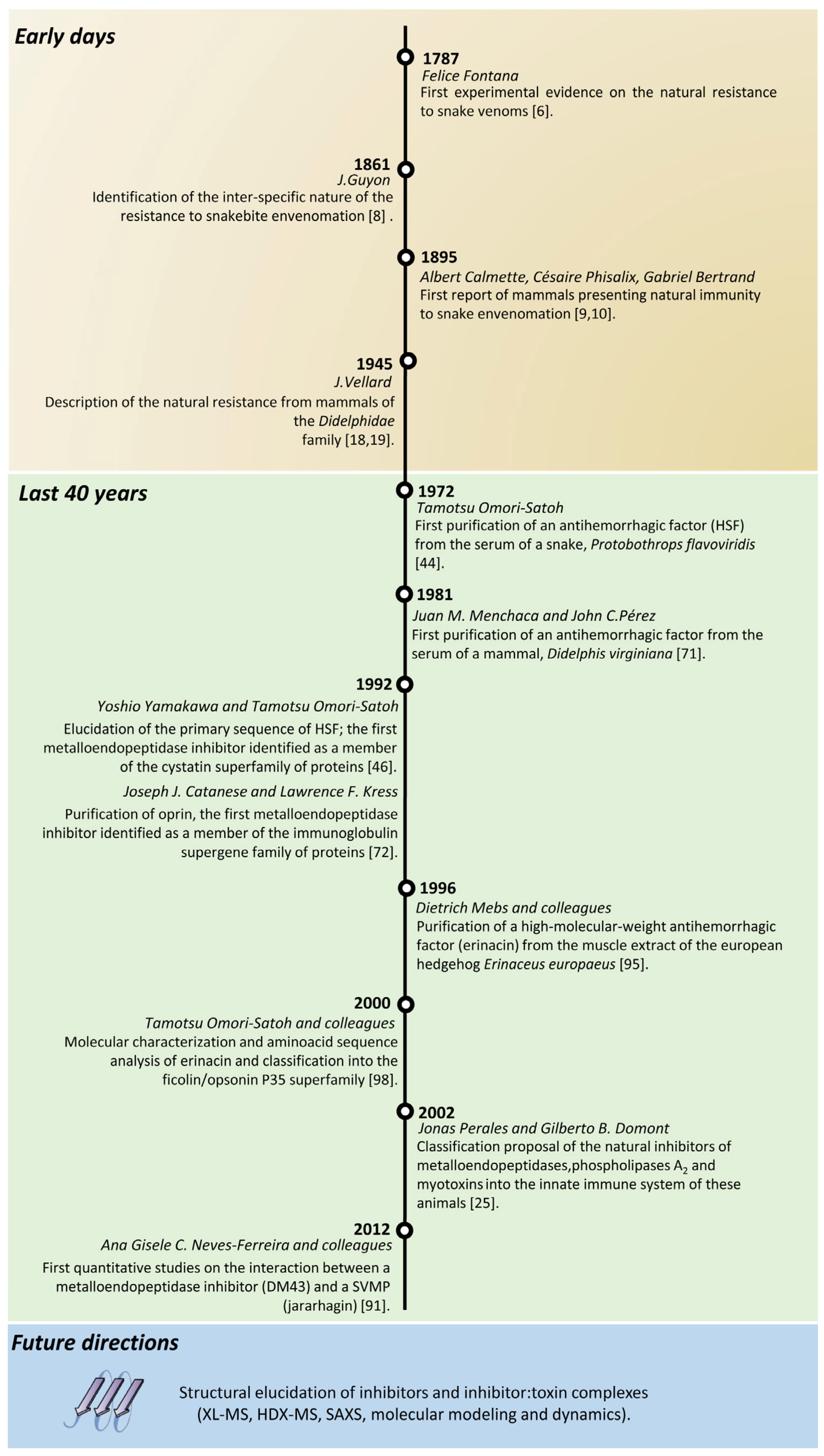
Natural Inhibitors of Snake Venom Metalloendopeptidases: History and Current Challenges


Abstract
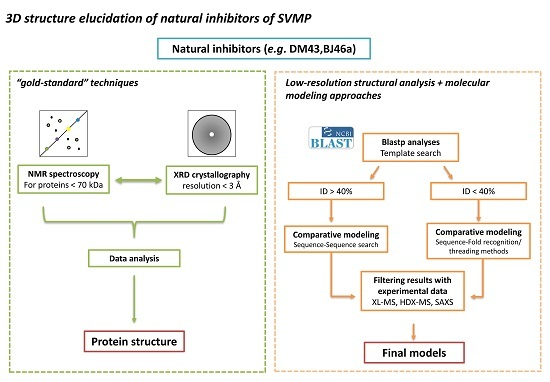
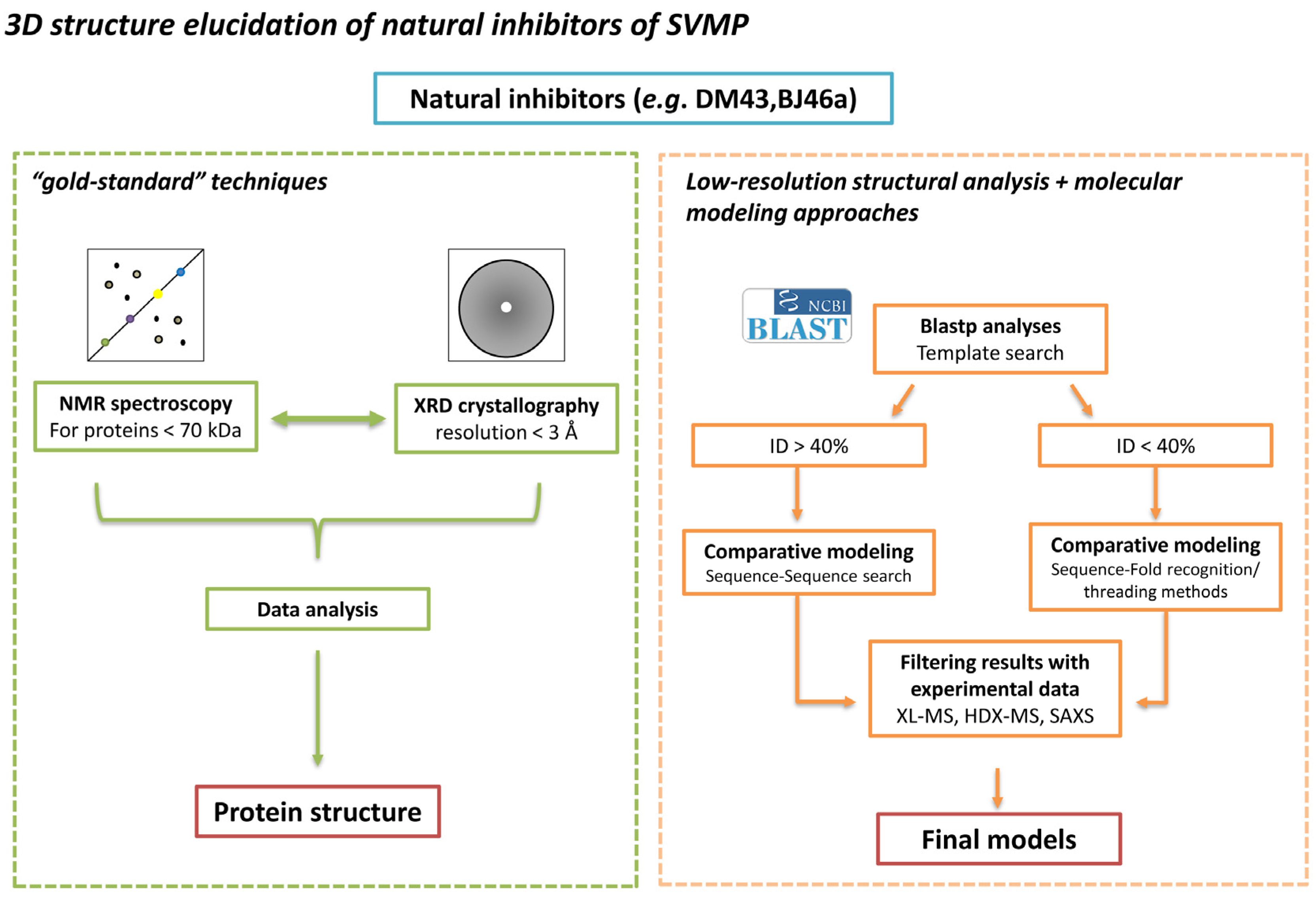
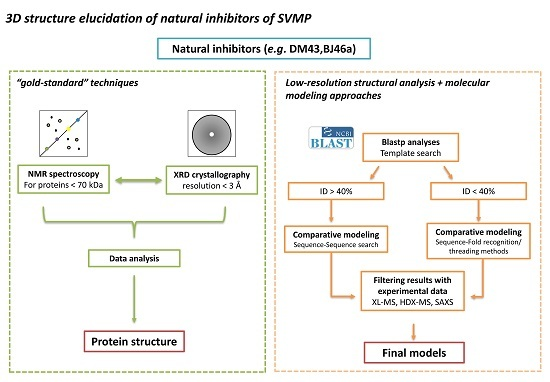
:
1. Introduction
2. Biochemical Background
2.1. Snake Venom Metalloendopeptidases (Metalloproteinases)
2.2. SVMPIs Isolated from Snakes
2.2.1. Cystatin Superfamily (Fetuin-Like Proteins)
2.2.2. Undetermined Protein Family
2.3. SVMPIs Isolated from Mammals
2.3.1. Immunoglobulin Supergene Family
2.3.2. Ficolin/Opsonin P35 Family
3. Possible Therapeutic Applications
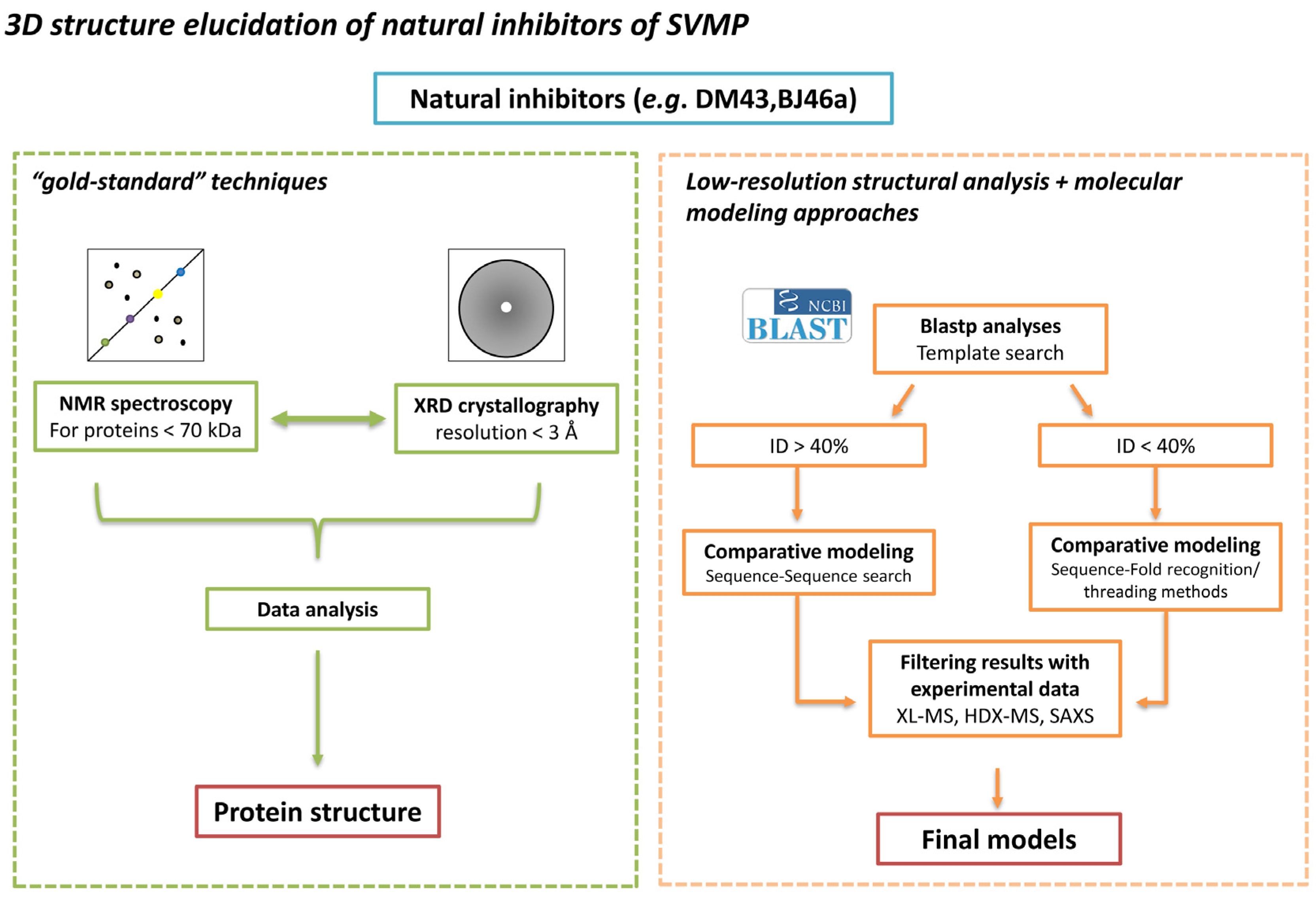
4. Status Quo and Perspectives on Three-Dimensional Structure Determination for SVMPIs
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ABC | AntiBothropic Complex |
| ABF | AntiBothropic Fraction |
| ADAM | A Disintegrin And Metalloendopeptidase |
| ADAMTS | ADAM with ThromboSpondin motifs |
| BPP | Bradikynin-Potentiating Peptide |
| HDX-MS | Hydrogen/Deuterium eXchange MS |
| HLP | Habu-Like Protein |
| HSF | Habu Serum Factor |
| Ig | Immunoglobulin |
| MALDI TOF | Matrix-Assisted Laser/Desorption Ionization |
| MS | Mass Spectrometry |
| MMP | Matrix MetalloendoPeptidase |
| PDB ID | Protein Data Bank IDentifier |
| PLI | PhosphoLipase A2 Inhibitor |
| NMR | Nuclear Magnetic Resonance |
| SAXS | Small-Angle X-ray Scattering |
| SDS-PAGE | Sodium Dodecyl Sulfate PolyAcrylamide Gel Electrophoresis |
| SSP | Small Serum Protein |
| SVMP | Snake Venom MetalloendoPeptidase |
| SVMPI | SVMP Inhibitor |
| TIMP | Tissue Inhibitor of MetalloendoPeptidase |
| 2D-PAGE | Two-Dimensional PolyAcrilamide Gel Electrophoresis |
| XL-MS | Cross-Linking MS |
| XRD | X-ray Diffraction |
References
- Fraser, T. Serpent’s Venom: Artificial and Natural Immunity; Antidotal Properties of the Blood-Serum of Immunized Animals and of Venomous Serpents; Neill and Company: Edinburgh, UK, 1895; p. 42. [Google Scholar]
- Karaberopoulos, D.; Karamanou, M.; Androutsos, G. The theriac in antiquity. Lancet 2012, 379, 1942–1943. [Google Scholar] [CrossRef]
- Parojcic, D.; Stupar, D.; Mirica, M. Theriac: Medicine and antidote. Vesalius 2003, 9, 28–32. [Google Scholar] [PubMed]
- Garrison, F.H. Felice Fontana: A forgotten physiologist of the Trentino. Bull. N. Y. Acad. Med. 1935, 11, 117–122. [Google Scholar] [PubMed]
- Hawgood, B.J. Abbé Felice Fontana (1730–1805): Founder of Modern Toxinology. Toxicon 1995, 33, 591–601. [Google Scholar] [CrossRef]
- Fontana, F. Ricerche Fisiche Sopra il Veleno della Vipera con Alcune Offervazioni Sopra la Anguillette dele Grano Sperone; Jacopo Giusti: Lucca, Italy, 1767; p. 170. (In Italian) [Google Scholar]
- Fontana, F. Treatise on the Venom of the Viper; on the American Poisons; and on the Cherry Laurel; and Some Other Vegetable Poisons; Jamie Murray: London, UK, 1787; Volume 1, p. 447. [Google Scholar]
- Guyon, J. Le Venin de Serpents Exerce-t-il sur Eux-mêmes l’Action Qu’il Exerce sur D’autres Animaux? In Animaux Venimeux et Venins: La Fonction Venimeuse chez tous les Animaux; les Appareils Venimeux; les Venins et leurs Propriétés; les Fonctions et Usages des Venins; L’envenimation et son Traitement; Phisalix, M., Ed.; Masson & Cie.: Paris, France, 1861; Volume 2, p. 744. (In French) [Google Scholar]
- Calmette, A. Le Venin des Serpents: Physiologie de l’Envenimation, Traitement des Morsures Venimeuses; Société d’Éditions Scientifiques: Paris, France, 1896; Volume 1, p. 90. (In French) [Google Scholar]
- Phisalix, C.; Bertrand, G. Recherches sur l’immunité du hérisson contre le venin de vipère. C. R. Soc. Biol. 1895, 47, 639–641. (In French) [Google Scholar]
- Gloyd, H.K. On the effects of the mocassin venom upon a rattlesnake. Science 1933, 78, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Keegan, H.L.; Andrews, T.F. Effects of crotalid venom on North American snakes. Copeia 1942, 4, 251–254. [Google Scholar] [CrossRef]
- Swanson, P.L. Effects of snake venoms on snakes. Copeia 1946, 4, 242–249. [Google Scholar] [CrossRef]
- Philpot, V.B.; Smith, R.G. Neutralization of pit viper venom by king snake serum. Proc. Soc. Exp. Biol. Med. 1950, 75, 521–523. [Google Scholar] [CrossRef]
- Abalos, J.W. The Ophiofagus habits of Pseudoboa cloelia. Toxicon 1963, 1, 90–91. [Google Scholar] [CrossRef]
- Straight, R.; Glenn, J.L.; Snyder, C.C. Antivenom activity of rattlesnake blood plasma. Nature 1976, 261, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Clark, W.C.; Voris, H.K. Venom neutralization by rattlesnake serum albumin. Science 1969, 164, 1402–1404. [Google Scholar] [CrossRef] [PubMed]
- Vellard, J. Resistencia de los “Didelphis” (Zarigueya) a los venenos ofidicos (Nota Previa). Rev. Bras. Biol. 1945, 5, 463–467. (In Spanish) [Google Scholar] [PubMed]
- Vellard, J. Investigaciones sobre imunidad natural contra los venenos de serpientes. I. Publ. Mus. Hist. Nat. Lima Ser. A Zool. 1949, 1, 1–61. (In Spanish) [Google Scholar]
- Domont, G.B.; Perales, J.; Moussatché, H. Natural anti-snake venom proteins. Toxicon 1991, 29, 1183–1194. [Google Scholar] [CrossRef]
- Perales, J.; Neves-Ferreira, A.G.C.; Valente, R.H.; Domont, G.B. Natural inhibitors of snake venom hemorrhagic metalloproteinases. Toxicon 2005, 45, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Fortes-Dias, C.L. Endogenous inhibitors of snake venom phospholipases A(2) in the blood plasma of snakes. Toxicon 2002, 40, 481–484. [Google Scholar] [CrossRef]
- Lizano, S.; Domont, G.; Perales, J. Natural phospholipase A2 myotoxin inhibitor proteins from snakes mammals and plants. Toxicon 2003, 42, 963–977. [Google Scholar] [CrossRef] [PubMed]
- Neves-Ferreira, A.G.C.; Valente, R.H.; Perales, J.; Domont, G.B. Natural Inhibitors—Innate Immunity to Snake Venoms. In Handbook of Venoms and Toxins of Reptiles; Mackessy, S.P., Ed.; CRC Press: Boca Raton, FL, USA, 2010; Volume 1, pp. 259–284. [Google Scholar]
- Perales, J.; Domont, G.B. Are Inhibitors of Metalloproteinases, Phospholipases A2 and Myotoxins Members of the Innate Immune System? In Perspectives in Molecular Toxinology; Ménez, A., Ed.; John Wiley & Sons: Chichester, UK, 2002; pp. 435–456. [Google Scholar]
- Thwin, M.M.; Gopalakrishnakone, P. Snake envenomation and protective natural endogenous proteins: A mini review of the recent developments (1991–1997). Toxicon 1998, 36, 1471–1482. [Google Scholar] [CrossRef]
- Perez, J.C.; Sanchez, E.E. Natural protease inhibitors to hemorrhagins in snake venoms and their potential use in medicine. Toxicon 1999, 37, 703–728. [Google Scholar] [CrossRef]
- Valente, R.H.; Neves-Ferreira, A.G.C.; Caffarena, E.R.; Domont, G.B.; Perales, J. Snake Venom Metalloproteinase Inhibitors—An Overview And Future Perspectives. In Animal Toxins—State of the Art. Perspectives in Health and Biotechnology; Lima, M.E., Ed.; Editora UFMG: Belo Horizonte, Brazil, 2009; pp. 547–558. [Google Scholar]
- Neves-Ferreira, A.G.C.; Valente, R.H.; Domont, G.B.; Perales, J. Natural Inhibitors of Snake Venom Metallopeptidases. In Toxins and Drug Discovery; Gopalakrishnakone, P., Ed.; Springer: Dordrecht, The Netherlands, 2016; Volume 1, pp. 1–23. [Google Scholar]
- De Lacerda, J.B. Leçons sur le Venin des Serpents du Brèsil et sur la Méthode de Traitement des Morsures Venimeuses par le Permanganate de Potasse; Livraria Lombaerts & C.: Rio de Janeiro, Brazil, 1884; p. 194. (In French) [Google Scholar]
- Flexner, S.; Noguchi, H. The constitution of snake venom and snake sera. J. Pathol. Bacteriol. 1903, 8, 379–410. [Google Scholar] [CrossRef]
- Maeno, H.; Mitsuhashi, S.; Sato, R. Studies on habu snake venom. 2c. Studies on H beta-proteinase of habu venom. Jpn. J. Microbiol. 1960, 4, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Ohsaka, A. Fractionation of habu snake venom by chromatography on cm-cellulose with special reference to biological activities. Jpn. J. Med. Sci. Biol. 1960, 13, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Ohsaka, A.; Ikezawa, H.; Kondo, H.; Kondo, S.; Uchida, N. Haemorrhagic activities of habu snake venom, and their relations to lethal toxicity, proteolytic activities and other pathological activities. Br. J. Exp. Pathol. 1960, 41, 478–486. [Google Scholar] [PubMed]
- Okonogi, T.; Hoshi, S.; Honma, M.; Mitsuhashi, S.; Maeno, H.; Sawai, Y. Studies on the habu snake venom. 3–2. A comparative study of histopathological changes caused by crude venom, purified Habu-proteinase and other proteinases. Jpn. J. Microbiol. 1960, 4, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Bjarnason, J.B.; Tu, A.T. Hemorrhagic toxins from Western diamondback rattlesnake (Crotalus atrox) venom: Isolation and characterization of five toxins and the role of zinc in hemorrhagic toxin e. Biochemistry 1978, 17, 3395–3404. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.W.; Serrano, S.M. Insights into and speculations about snake venom metalloproteinase (SVMP) synthesis, folding and disulfide bond formation and their contribution to venom complexity. FEBS J. 2008, 275, 3016–3030. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, J.M.; Rucavado, A. Snake venom metalloproteinases: Their role in the pathogenesis of local tissue damage. Biochimie 2000, 82, 841–850. [Google Scholar] [CrossRef]
- Tanjoni, I.; Weinlich, R.; Della-Casa, M.S.; Clissa, P.B.; Saldanha-Gama, R.F.; de Freitas, M.S.; Barja-Fidalgo, C.; Amarante-Mendes, G.P.; Moura-da-Silva, A.M. Jararhagin, a snake venom metalloproteinase, induces a specialized form of apoptosis (anoikis) selective to endothelial cells. Apoptosis 2005, 10, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, J.M.; Escalante, T.; Rucavado, A.; Herrera, C. Hemorrhage caused by snake venom metalloproteinases: A journey of discovery and understanding. Toxins 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Farsky, S.H.; Goncalves, L.R.; Cury, Y. Characterization of local tissue damage evoked by Bothrops jararaca venom in the rat connective tissue microcirculation: An intravital microscopic study. Toxicon 1999, 37, 1079–1083. [Google Scholar] [CrossRef]
- Warrell, D.A. Snake bite. Lancet 2010, 375, 77–88. [Google Scholar] [CrossRef]
- Gutiérrez, J.M.; Rucavado, A.; Escalante, T. Snake Venom Metalloproteinases—Biological Roles and Participation in the Pathophysiology of Envenomation. In Handbook of Venoms and Toxins of Reptiles; Mackessy, S.P., Ed.; CRC Press: Boca Raton, FL, USA, 2010; pp. 115–138. [Google Scholar]
- Omori-Satoh, T.; Sadahiro, S.; Ohsaka, A.; Murata, R. Purification and characterization of an antihemorrhagic factor in the serum of Trimeresurus flavoviridis, a crotalid. Biochim. Biophys. Acta 1972, 285, 414–426. [Google Scholar] [CrossRef]
- Omori-Satoh, T. Antihemorrhagic factor as a proteinase inhibitor isolated from the serum of Trimeresurus flavoviridis. Biochim. Biophys. Acta 1977, 495, 93–98. [Google Scholar] [CrossRef]
- Yamakawa, Y.; Omori-Satoh, T. Primary structure of the antihemorrhagic factor in serum of the Japanese habu: A snake venom metalloproteinase inhibitor with a double-headed cystatin domain. J. Biochem. 1992, 112, 583–589. [Google Scholar] [PubMed]
- Deshimaru, M.; Tanaka, C.; Tokunaga, A.; Goto, M.; Terada, S. Efficient Purification of an Antihemorrhagic factor in the serum of Japanese Habu (Trimeresurus flavoviridis). Fukuoka Univ. Sci. Rep. 2003, 33, 45–53. [Google Scholar]
- Deshimaru, M.; Tanaka, C.; Fujino, K.; Aoki, N.; Terada, S.; Hattori, S.; Ohno, M. Properties and cDNA cloning of an antihemorrhagic factor (HSF) purified from the serum of Trimeresurus flavoviridis. Toxicon 2005, 46, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Shioi, N.; Narazaki, M.; Terada, S. Novel function of antihemorrhagic factor HSF as an SSP-binding protein in habu (Trimeresurus flavoviridis) serum. Fukuoka Univ. Sci. Rep. 2011, 41, 177–184. [Google Scholar]
- Shioi, N.; Ogawa, E.; Mizukami, Y.; Abe, S.; Hayashi, R.; Terada, S. Small serum protein-1 changes the susceptibility of an apoptosis-inducing metalloproteinase HV1 to a metalloproteinase inhibitor in habu snake (Trimeresurus flavoviridis). J. Biochem. 2012, 153, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Aoki, N.; Deshimaru, M.; Terada, S. Active fragments of the antihemorrhagic protein HSF from serum of habu (Trimeresurus flavoviridis). Toxicon 2007, 49, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Fujimura, Y.; Miura, S.; Shima, H.; Yoshida, E.; Yoshioka, A.; Hirano, K.; Suzuki, M.; Titani, K. A 28 kDa-protein with disintegrin-like structure (jararhagin-C) purified from Bothrops jararaca venom inhibits collagen- and ADP-induced platelet aggregation. Biochem. Biophys. Res. Commun. 1994, 201, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Valente, R.H.; Dragulev, B.; Perales, J.; Fox, J.W.; Domont, G.B. BJ46a, a snake venom metalloproteinase inhibitor. Isolation, characterization, cloning and insights into its mechanism of action. Eur. J. Biochem. 2001, 268, 3042–3052. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ji, M.-K.; Xu, J.-W.; Lin, X.; Lin, J.-Y. High-level expression, purification, characterization and structural prediction of a snake venom metalloproteinase inhibitor in Pichia pastoris. Protein J. 2012, 31, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.-K.; Shi, Y.; Xu, J.-W.; Lin, X.; Lin, J.-Y. Recombinant snake venom metalloproteinase inhibitor BJ46A inhibits invasion and metastasis of B16F10 and MHCC97H cells through reductions of matrix metalloproteinases 2 and 9 activities. Anticancer Drugs 2013, 24, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Aoki, N.; Tsutsumi, K.; Deshimaru, M.; Terada, S. Properties and cDNA cloning of antihemorrhagic factors in sera of Chinese and Japanese mamushi (Gloydius blomhoffi). Toxicon 2008, 51, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Shioi, N.; Deshimaru, M.; Terada, S. Structural analysis and characterization of new small serum proteins from the serum of a venomous snake (Gloydius blomhoffii). Biosci. Biotechnol. Biochem. 2014, 78, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Jahnen-Dechent, W.; Schinke, T.; Trindl, A.; Muller-Esterl, W.; Sablitzky, F.; Kaiser, S.; Blessing, M. Cloning and targeted deletion of the mouse fetuin gene. J. Biol. Chem. 1997, 272, 31496–31503. [Google Scholar] [CrossRef] [PubMed]
- Aoki, N.; Deshimaru, M.; Kihara, H.; Terada, S. Snake fetuin: Isolation and structural analysis of new fetuin family proteins from the sera of venomous snakes. Toxicon 2009, 54, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, S.A.; Lafaye, P.J.; Smith, L.A. Observations on a venom neutralizing fraction isolated from the serum of the Northern Copperhead. Agkistrodon Contortrix Mokasen Copeia 1991, 5, 777–786. [Google Scholar] [CrossRef]
- Borkow, G.; Gutierrez, J.M.; Ovadia, M. Isolation, characterization and mode of neutralization of a potent antihemorrhagic factor from the serum of the snake Bothrops asper. Biochim. Biophys. Acta 1995, 1245, 232–238. [Google Scholar] [CrossRef]
- Weissenberg, S.; Ovadia, M.; Fleminger, G.; Kochva, E. Antihemorrhagic factors in the blood serum of the western diamondback rattlesnake Crotalus atrox. Toxicon 1991, 29, 807–818. [Google Scholar] [CrossRef]
- Weissenberg, S.; Ovadia, M.; Kochva, E. Inhibition of the proteolytic activity of hemorrhagin-e from Crotalus atrox serum by antihemorrhagins from homologous serum. Toxicon 1992, 30, 591–597. [Google Scholar] [CrossRef]
- Tomihara, Y.; Kawamura, Y.; Yonaha, K.; Nozaki, M.; Yamakawa, M.; Yoshida, C. Neutralization of hemorrhagic snake venoms by sera of Trimeresurus flavoviridis (habu), Herpestes edwardsii (mongoose) and Dinodon semicarinatus (akamata). Toxicon 1990, 28, 989–991. [Google Scholar] [CrossRef]
- Borkow, G.; Gutierrez, J.M.; Ovadia, M. A potent antihemorrhagin in the serum of the non-poisonous water snake Natrix tessellata: Isolation, characterization and mechanism of neutralization. Biochim. Biophys. Acta 1994, 1201, 482–490. [Google Scholar] [CrossRef]
- Huang, K.-F.; Chow, L.-P.; Chiou, S.-H. Isolation and characterization of a novel proteinase inhibitor from the snake serum of Taiwan Habu (Trimeresurus mucrosquamatus). Biochem. Biophys. Res. Commun. 1999, 263, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Ovadia, M. Purification and characterization of an antihemorrhagic factor from the serum of the snake Vipera palaestinae. Toxicon 1978, 16, 661–672. [Google Scholar] [CrossRef]
- Azara, F.D. Apuntamientos para la Historia Natural de los Quadrúpedos del Paragüay y Río de la Plata; Imprenta de la Viuda de Ibarra: Madrid, Spain, 1802; p. 340. (In Spanish) [Google Scholar]
- Voss, R.S.; Jansa, S.A. Snake-venom resistance as a mammalian trophic adaptation: Lessons from didelphid marsupials. Biol. Rev. 2012, 87, 822–837. [Google Scholar] [CrossRef] [PubMed]
- Kilmon, J.A. High tolerance to snake venom by the Virginia opossum, Didelphis virginiana. Toxicon 1976, 14, 337–340. [Google Scholar] [CrossRef]
- Menchaca, J.M.; Perez, J.C. The purification and characterization of an antihemorrhagic factor in opossum (Didelphis virginiana) serum. Toxicon 1981, 19, 623–632. [Google Scholar] [CrossRef]
- Catanese, J.J.; Kress, L.F. Isolation from opossum serum of a metalloproteinase inhibitor homologous to human alpha 1B-glycoprotein. Biochemistry 1992, 31, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Tomihara, Y.; Yonaha, K.; Nozaki, M.; Yamakawa, M.; Kamura, T.; Toyama, S. Purification of three antihemorrhagic factors from the serum of a mongoose (Herpestes edwardsii). Toxicon 1987, 25, 685–689. [Google Scholar] [CrossRef]
- Qi, Z.-Q.; Yonaha, K.; Tomihara, Y.; Toyama, S. Characterization of the antihemorrhagic factors of mongoose (Herpestes edwardsii). Toxicon 1994, 32, 1459–1469. [Google Scholar] [CrossRef]
- Qi, Z.Q.; Yonaha, K.; Tomihara, Y.; Toyama, S. Isolation of peptides homologous to domains of human alpha 1B-glycoprotein from a mongoose antihemorrhagic factor. Toxicon 1995, 33, 241–245. [Google Scholar] [CrossRef]
- Perales, J.; Moussatché, H.; Marangoni, S.; Oliveira, B.; Domont, G.B. Isolation and partial characterization of an anti-bothropic complex from the serum of the south american Didelphidae. Toxicon 1994, 32, 1237–1249. [Google Scholar] [CrossRef]
- Jurgilas, P.B.; Neves-Ferreira, A.G.; Domont, G.B.; Perales, J. PO41, a snake venom metalloproteinase inhibitor isolated from Philander opossum serum. Toxicon 2003, 42, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Farah, M.D.F.L.; One, M.; Novello, J.C.; Toyama, M.H.; Perales, J.; Moussatché, H.; Domont, G.B.; Oliveira, B.; Marangoni, S. Isolation of protein factors from opossum (Didelphis albiventris) serum which protect against Bothrops jararaca venom. Toxicon 1996, 34, 1067–1071. [Google Scholar] [CrossRef]
- Moussatché, H.; Yates, A.; Leonardi, F.; Borche, L. Mechanisms of resistance of the opossum to some snake venoms. Toxicon 1979, 17, 130. [Google Scholar]
- Moussatché, H.; Yates, A.; Leonardi, F.; Borche, L. Obtención de una fracción del suero de Didelphis activa contra la acción tóxica del veneno de B. jararaca. Acta Cient. Venez. 1980, 31, 104. (In Spanish) [Google Scholar]
- Moussatché, H.; Leonardi, F.; Mandelbaum, F. Inhibición por una proteína aislada del suero de D. marsupialis a la acción hemorrhágica por una fracción del veneno de Bothrops jararaca. Acta Cient. Venez. 1981, 32, 173. (In Spanish) [Google Scholar]
- Perales, J.; Munoz, R.; Moussatché, H. Isolation and partial characterization of a protein fraction from the opossum (Didelphis marsupialis) serum, with protecting property against Bothrops jararaca venom. An. Acad. Bras. Cienc. 1986, 58, 155–162. [Google Scholar] [PubMed]
- Moussatché, H.; Perales, J. Factors underlying the natural resistance of animals against snake venoms. Mem. Inst. Oswaldo Cruz 1989, 84, 391–394. [Google Scholar] [CrossRef]
- Perales, J.; Amorim, C.Z.; Rocha, S.L.G.; Domont, G.B.; Moussatché, H. Neutralization of the oedematogenic activity of Bothrops jararaca venom on the mouse paw by a antibothropic fraction isolated from opossum (Didelphis marsupialis) serum. Agents Actions 1992, 37, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.L.; Frutuoso, V.S.; Domont, G.B.; Martins, M.A.; Moussatche, H.; Perales, J. Inhibition of the hyperalgesic activity of Bothrops jararaca venom by an antibothropic fraction isolated from opossum (Didelphis marsupialis) serum. Toxicon 2000, 38, 875–880. [Google Scholar] [CrossRef]
- Neves-Ferreira, A.G.C.; Perales, J.; Ovadia, M.; Moussatché, H.; Domont, G.B. Inhibitory properties of the antibothropic complex from the south american opossum (Didelphis marsupialis) serum. Toxicon 1997, 35, 849–863. [Google Scholar] [CrossRef]
- Jurgilas, P.B.; Neves-Ferreira, A.G.C.; Domont, G.B.; Moussatché, H.; Perales, J. Detection of an antibothropic fraction in opossum (Didelphis marsupialis) milk that neutralizes Bothrops jararaca venom. Toxicon 1999, 37, 167–172. [Google Scholar] [CrossRef]
- Neves-Ferreira, A.G.C.; Cardinale, N.; Rocha, S.L.G.; Perales, J.; Domont, G.B. Isolation and characterization of DM40 and DM43, two snake venom metalloproteinase inhibitors from Didelphis marsupialis serum. Biochim. Biophys. Acta 2000, 1474, 309–320. [Google Scholar] [CrossRef]
- Neves-Ferreira, A.G.C.; Perales, J.; Fox, J.W.; Shannon, J.D.; Makino, D.L.; Garratt, R.C.; Domont, G.B. Structural and functional analyses of DM43, a snake venom metalloproteinase inhibitor from Didelphis marsupialis serum. J. Biol. Chem. 2002, 277, 13129–13137. [Google Scholar] [CrossRef] [PubMed]
- Léon, I.R.; Neves-Ferreira, A.G.C.; Rocha, S.L.G.; Trugilho, M.R.O.; Perales, J.; Valente, R.H. Using mass spectrometry to explore the neglected glycan moieties of the antiophidic proteins DM43 and DM64. Proteomics 2012, 12, 2753–2765. [Google Scholar] [CrossRef] [PubMed]
- Brand, G.D.; Salbo, R.; Jorgensen, T.J.; Bloch, C., Jr.; Boeri Erba, E.; Robinson, C.V.; Tanjoni, I.; Moura-da-Silva, A.M.; Roepstorff, P.; Domont, G.B.; et al. The interaction of the antitoxin DM43 with a snake venom metalloproteinase analyzed by mass spectrometry and surface plasmon resonance. J. Mass Spectrom. 2012, 47, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Drake, A.W.; Myszka, D.G.; Klakamp, S.L. Characterizing high-affinity antigen/antibody complexes by kinetic- and equilibrium-based methods. Anal. Biochem. 2004, 328, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.L.; Neves-Ferreira, A.G.; Trugilho, M.R.; Chapeaurouge, A.; Leon, I.R.; Valente, R.H.; Domont, G.B.; Perales, J. Crotalid snake venom subproteomes unraveled by the antiophidic protein DM43. J. Proteome Res. 2009, 8, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Asega, A.F.; Oliveira, A.K.; Menezes, M.C.; Neves-Ferreira, A.G.; Serrano, S.M. Interaction of Bothrops jararaca venom metalloproteinases with protein inhibitors. Toxicon 2014, 80, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jurgilas, P.B.; de Meis, J.; Valente, R.H.; Neves-Ferreira, A.G.C.; da Cruz, D.A.M.; de Oliveira, D.A.F.; Savino, W.; Domont, G.B.; Perales, J. Use of DM43 and Its Fragments as Matrix Metalloproteinases Inhibitor. U.S. Patent 20080249005 A1, 9 October 2008. [Google Scholar]
- Mebs, D.; Omori-Satoh, T.; Yamakawa, M.; Nagaoka, Y. Erinacin, an antihemorrhagic factor from the european hedgehog, Erinaceus europaeus. Toxicon 1996, 34, 1313–1316. [Google Scholar] [CrossRef]
- Ohashi, T.; Erickson, H.P. Two oligomeric forms of plasma ficolin have differential lectin activity. J. Biol. Chem. 1997, 272, 14220–14226. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, R.; Yae, Y.; Akaiwa, M.; Kitajima, S.; Shibata, Y.; Sato, H.; Hirata, J.; Okochi, K.; Izuhara, K.; Hamasaki, N. Cloning and characterization of the Hakata antigen, a member of the ficolin/opsonin p35 lectin family. J. Biol. Chem. 1998, 273, 20721–20727. [Google Scholar] [CrossRef] [PubMed]
- Omori-Satoh, T.; Yamakawa, Y.; Mebs, D. The antihemorrhagic factor, erinacin, from the European hedgehog (Erinaceus europaeus), a metalloprotease inhibitor of large molecular size possessing ficolin/opsonin P35 lectin domains. Toxicon 2000, 38, 1561–1580. [Google Scholar] [CrossRef]
- Matsushita, M.; Endo, Y.; Taira, S.; Sato, Y.; Fujita, T.; Ichikawa, N.; Nakata, M.; Mizuochi, T. A novel human serum lectin with collagen- and fibrinogen-like domains that functions as an opsonin. J. Biol. Chem. 1996, 271, 2448–2454. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J. Evolutionary families of peptidases. Biochem. J. 1993, 290, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.W.; Serrano, S.M. Snake Venom Metalloproteinases. In Handbook of Venoms and Toxins of Reptiles; Mackessy, S.P., Ed.; CRC Press: Boca Raton, FL, USA, 2010; pp. 96–109. [Google Scholar]
- Khokha, R.; Murthy, A.; Weiss, A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Battellino, C.; Piazza, R.; Silva, A.M.M.; Cury, Y.; Farsky, S.H.P. Assessment of efficacy of bothropic antivenom therapy on microcirculatory effects induced by Bothrops jararaca snake venom. Toxicon 2003, 41, 583–593. [Google Scholar] [CrossRef]
- Gutierrez, J.M.; Leon, G.; Lomonte, B. Pharmacokinetic-pharmacodynamic relationships of immunoglobulin therapy for envenomation. Clin. Pharmacokinet. 2003, 42, 721–741. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, J.M.; Lomonte, B.; Leon, G.; Rucavado, A.; Chaves, F.; Angulo, Y. Trends in snakebite envenomation therapy: Scientific, technological and public health considerations. Curr. Pharm. Des. 2007, 13, 2935–2950. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.H.; Bartelt, D.C.; Greene, L.J. Isolation of bradykinin-potentiating peptides from Bothrops jararaca venom. Biochemistry 1970, 9, 2583–2593. [Google Scholar] [CrossRef] [PubMed]
- Ondetti, M.A.; Williams, N.J.; Sabo, E.F.; Pluscec, J.; Weaver, E.R.; Kocy, O. Angiotensin-converting enzyme inhibitors from the venom of Bothrops jararaca. Isolation, elucidation of structure, and synthesis. Biochemistry 1971, 10, 4033–4039. [Google Scholar] [CrossRef] [PubMed]
- Gavras, H.; Brunner, H.R.; Laragh, J.H.; Sealey, J.E.; Gavras, I.; Vukovich, R.A. An angiotensin converting-enzyme inhibitor to identify and treat vasoconstrictor and volume factors in hypertensive patients. N. Engl. J. Med. 1974, 291, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Cushman, D.W.; Ondetti, M.A. Design of angiotensin converting enzyme inhibitors. Nat. Med. 1999, 5, 1110–1113. [Google Scholar] [CrossRef] [PubMed]
- Cushman, D.W.; Cheung, H.S.; Sabo, E.F.; Ondetti, M.A. Design of potent competitive inhibitors of angiotensin-converting enzyme. Carboxyalkanoyl and mercaptoalkanoyl amino acids. Biochemistry 1977, 16, 5484–5491. [Google Scholar] [CrossRef] [PubMed]
- Ondetti, M.A.; Rubin, B.; Cushman, D.W. Design of specific inhibitors of angiotensin-converting enzyme: New class of orally active antihypertensive agents. Science 1977, 196, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Acharya, K.R.; Sturrock, E.D.; Riordan, J.F.; Ehlers, M.R. ACE revisited: A new target for structure-based drug design. Nat. Rev. Drug Discov. 2003, 2, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Abbenante, G.; Fairlie, D.P. Protease inhibitors in the clinic. Med. Chem. 2005, 1, 71–104. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, T.; Lemaitre, V.; D’Armiento, J.; Okada, Y. Matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs in non-neoplastic diseases. Pathol. Int. 2010, 60, 477–496. [Google Scholar] [CrossRef] [PubMed]
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Batra, J.; Robinson, J.; Soares, A.S.; Fields, A.P.; Radisky, D.C.; Radisky, E.S. Matrix metalloproteinase-10 (MMP-10) interaction with tissue inhibitors of metalloproteinases TIMP-1 and TIMP-2: Binding studies and crystal structure. J. Biol. Chem. 2012, 287, 15935–15946. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Minor, W.; Dauter, Z.; Jaskolski, M. Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures. FEBS J. 2008, 275, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Skrisovska, L.; Schubert, M.; Allain, F.H. Recent advances in segmental isotope labeling of proteins: NMR applications to large proteins and glycoproteins. J. Biomol. NMR 2010, 46, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Pervushin, K.; Riek, R.; Wider, G.; Wuthrich, K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA 1997, 94, 12366–12371. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.; Schneider, R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins 1991, 9, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S.E.; Chothia, C.; Hubbard, T.J. Assessing sequence comparison methods with reliable structurally identified distant evolutionary relationships. Proc. Natl. Acad. Sci. USA 1998, 95, 6073–6078. [Google Scholar] [CrossRef] [PubMed]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 1999, 12, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Bowie, J.U.; Luthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. A new approach to protein fold recognition. Nature 1992, 358, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 2008, 18, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Boratyn, G.M.; Schaffer, A.A.; Agarwala, R.; Altschul, S.F.; Lipman, D.J.; Madden, T.L. Domain enhanced lookup time accelerated BLAST. Biol. Direct. 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. GenTHREADER: An efficient and reliable protein fold recognition method for genomic sequences. J. Mol. Biol. 1999, 287, 797–815. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Kahraman, A.; Herzog, F.; Leitner, A.; Rosenberger, G.; Aebersold, R.; Malmstrom, L. Cross-link guided molecular modeling with ROSETTA. PLoS ONE 2013, 8, e73411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Majumder, E.L.; Yue, H.; Blankenship, R.E.; Gross, M.L. Structural analysis of diheme cytochrome c by hydrogen-deuterium exchange mass spectrometry and homology modeling. Biochemistry 2014, 53, 5619–5630. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Kim, S.J.; Sali, A. Integrative structural modeling with small angle X-ray scattering profiles. BMC Struct. Biol. 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Doniach, S. Protein structure prediction constrained by solution X-ray scattering data and structural homology identification. J. Mol. Biol. 2002, 316, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Hammel, M.; Tainer, J.A.; Sali, A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 2013, 105, 962–974. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Feature | Ig Supergene Family | Cystatin Superfamily | ||
|---|---|---|---|---|
| DM43 (291) | HSF (323) | BJ46a (322) | ||
| Template (PDB ID) | 1NKR | 5EIQ | 2KZX | 2WBK |
| Release date | 11 November 1998 | 25 November 2015 | 15 February 2012 | 4 April 2014 |
| Number of aligned residues | 193 | 192 | 98 | 55 |
| E-value | 4 × 10−5 | 7 × 10−18 | 1 × 10−1 | 5 × 10−1 |
| Identity | (50/193) 26% | (71/192) 37% | (26/98) 27% | (15/55) 33% |
| Positives | (78/193) 40% | (91/192) 47% | (42/98) 42% | (29/55) 52% |
| Gaps | (15/193) 7% | (14/192) 7% | (5/98) 5% | (1/55) 1% |
| Aligned region | 9–196 | 1–188 | 14–110 | 207–261 |
| Feature | HSF (323) | |||
|---|---|---|---|---|
| Template (PDB ID) | 4LZI | 3PS8 | 1R4C | 1G96 |
| Release date | 26 February 2014 | 21 December 2011 | 21 September 2004 | 6 April 2001 |
| Number of aligned residues | 222 | 115 | 107 | 115 |
| E-value | 4 × 10−55 | 6 × 10−35 | 8 × 10−36 | 2 × 10−34 |
| Identity | (31/222) 14% | (15/115) 13% | (17/107) 16% | (15/115) 13% |
| Positives | (67/222) 30% | (39/115) 33% | (32/107) 29% | (39/115) 33% |
| Gaps | (55/222) 24% | (8/115) 6% | (4/107) 3% | (8/115) 6% |
| Feature | BJ46a (322) | ||
|---|---|---|---|
| Template (PDB ID) | 4LZI | 3PS8 | 1G96 |
| Release date | 26 February 2014 | 21 December 2011 | 6 April 2001 |
| Number of aligned residues | 226 | 115 | 115 |
| E-value | 3 × 10−33 | 3 × 10−27 | 6 × 10−27 |
| Identity | (30/226) 13% | (14/115) 12% | (14/115) 12% |
| Positives | (65/226) 28% | (41/115) 35% | (41/115) 35% |
| Gaps | (55/226) 25% | (8/115) 6% | (8/115) 6% |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bastos, V.A.; Gomes-Neto, F.; Perales, J.; Neves-Ferreira, A.G.C.; Valente, R.H. Natural Inhibitors of Snake Venom Metalloendopeptidases: History and Current Challenges. Toxins 2016, 8, 250. https://doi.org/10.3390/toxins8090250
Bastos VA, Gomes-Neto F, Perales J, Neves-Ferreira AGC, Valente RH. Natural Inhibitors of Snake Venom Metalloendopeptidases: History and Current Challenges. Toxins. 2016; 8(9):250. https://doi.org/10.3390/toxins8090250
Chicago/Turabian StyleBastos, Viviane A., Francisco Gomes-Neto, Jonas Perales, Ana Gisele C. Neves-Ferreira, and Richard H. Valente. 2016. "Natural Inhibitors of Snake Venom Metalloendopeptidases: History and Current Challenges" Toxins 8, no. 9: 250. https://doi.org/10.3390/toxins8090250