
Early Activation of MAPK p44/42 Is Partially Involved in DON-Induced Disruption of the Intestinal Barrier Function and Tight Junction Network


Abstract
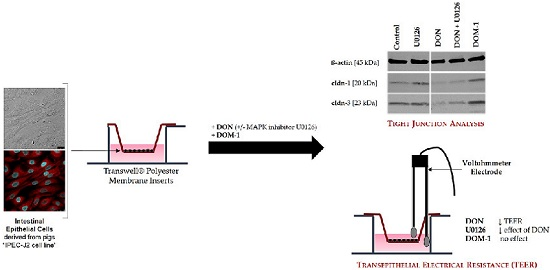
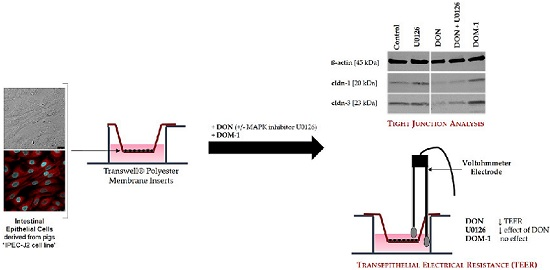
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
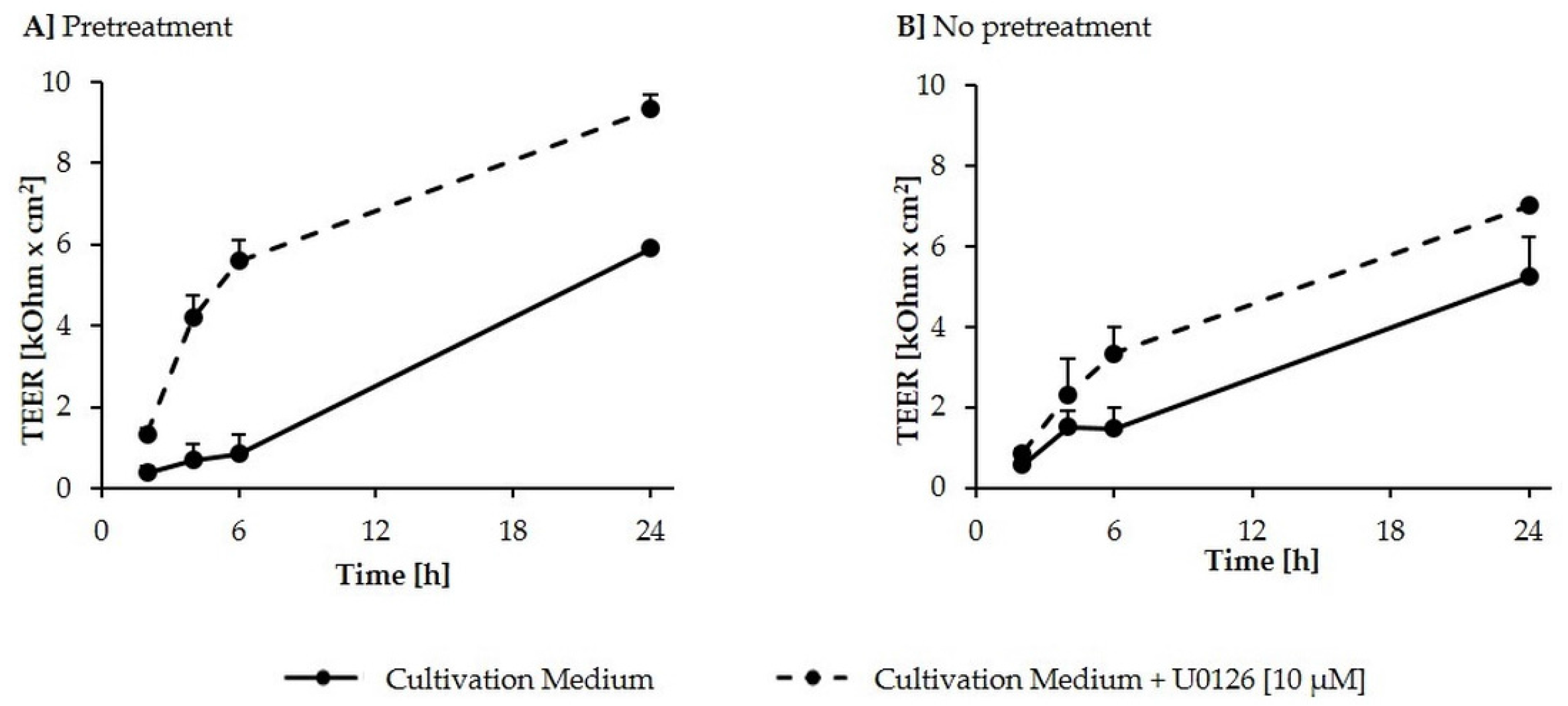
2.1. Formation of a Differentiated IPEC-J2 Monolayer
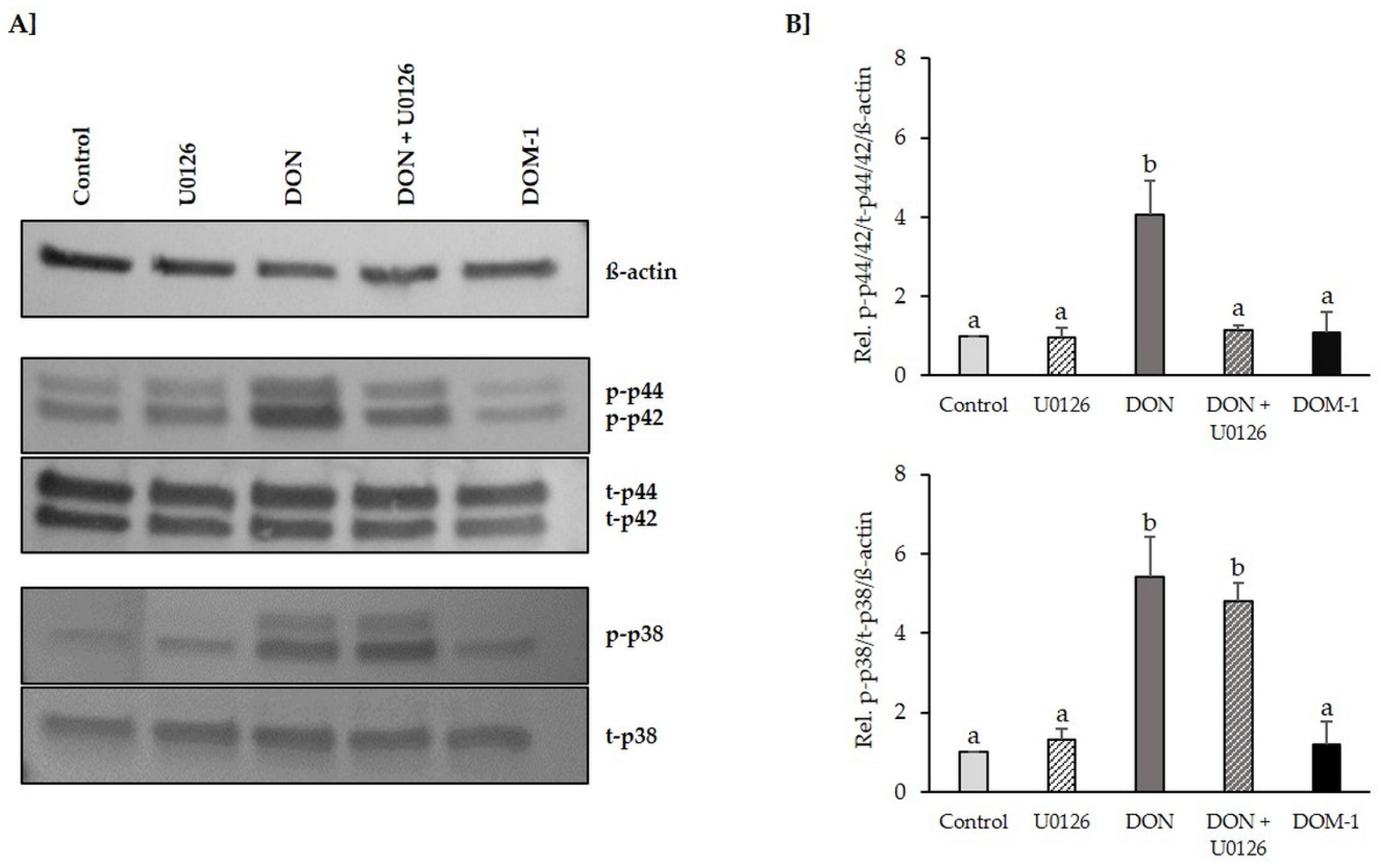
2.2. Inhibition of DON-Induced p44/42 (ERK1/2) Activation by MAPK Inhibitor U0126 Monoethanolate (U0126)
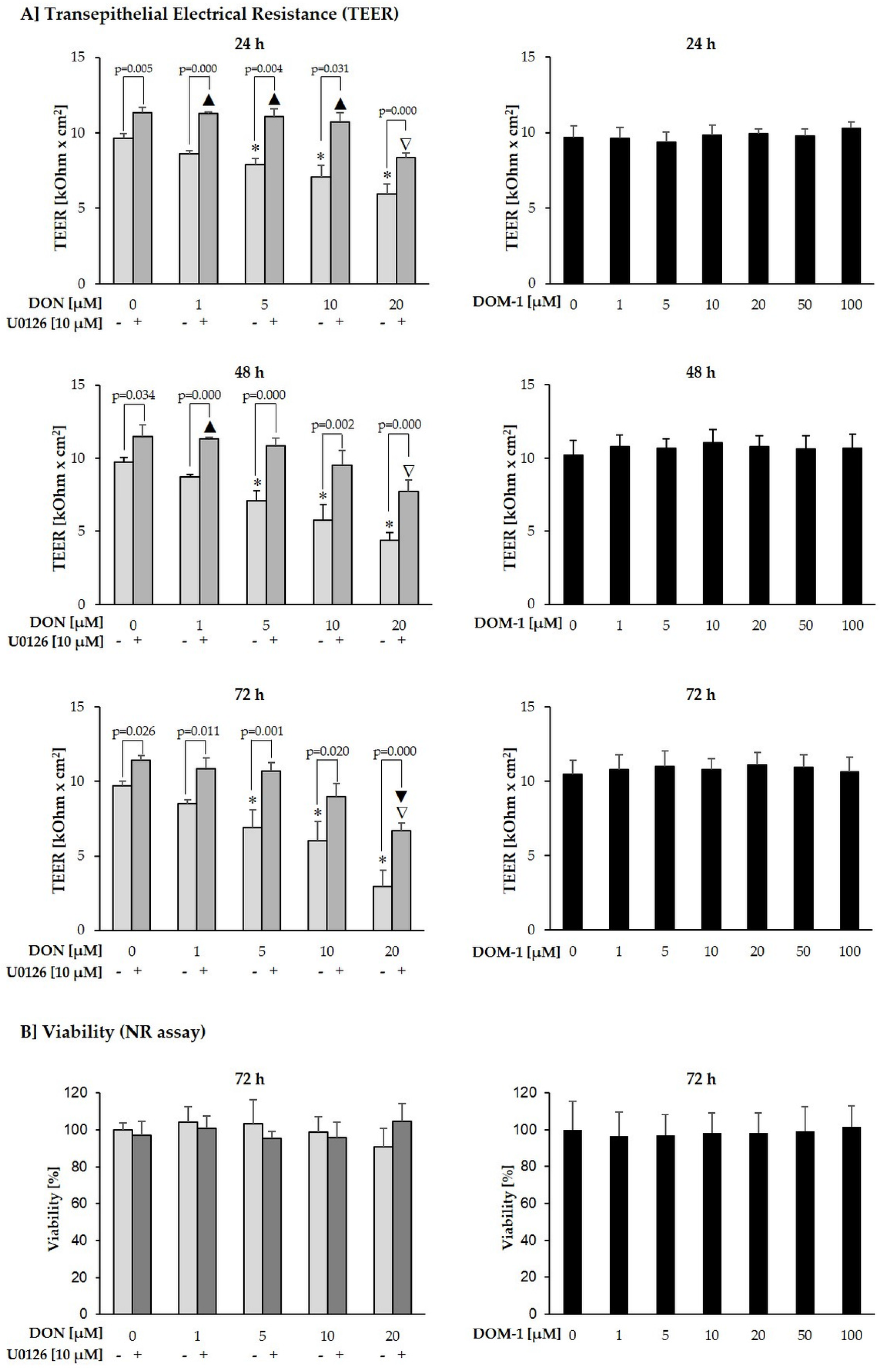
2.3. Effects of DON (+/− U0126) and DOM-1 on Intestinal Barrier Integrity
2.4. Calcium Switch Assay
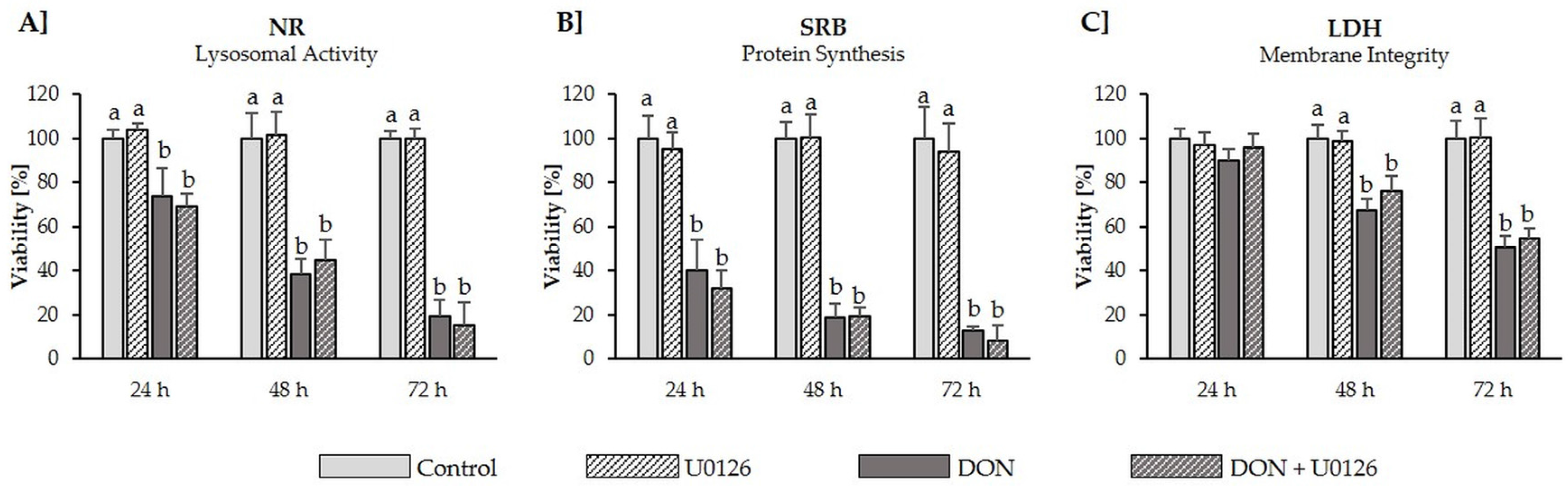
2.5. Cytotoxicity
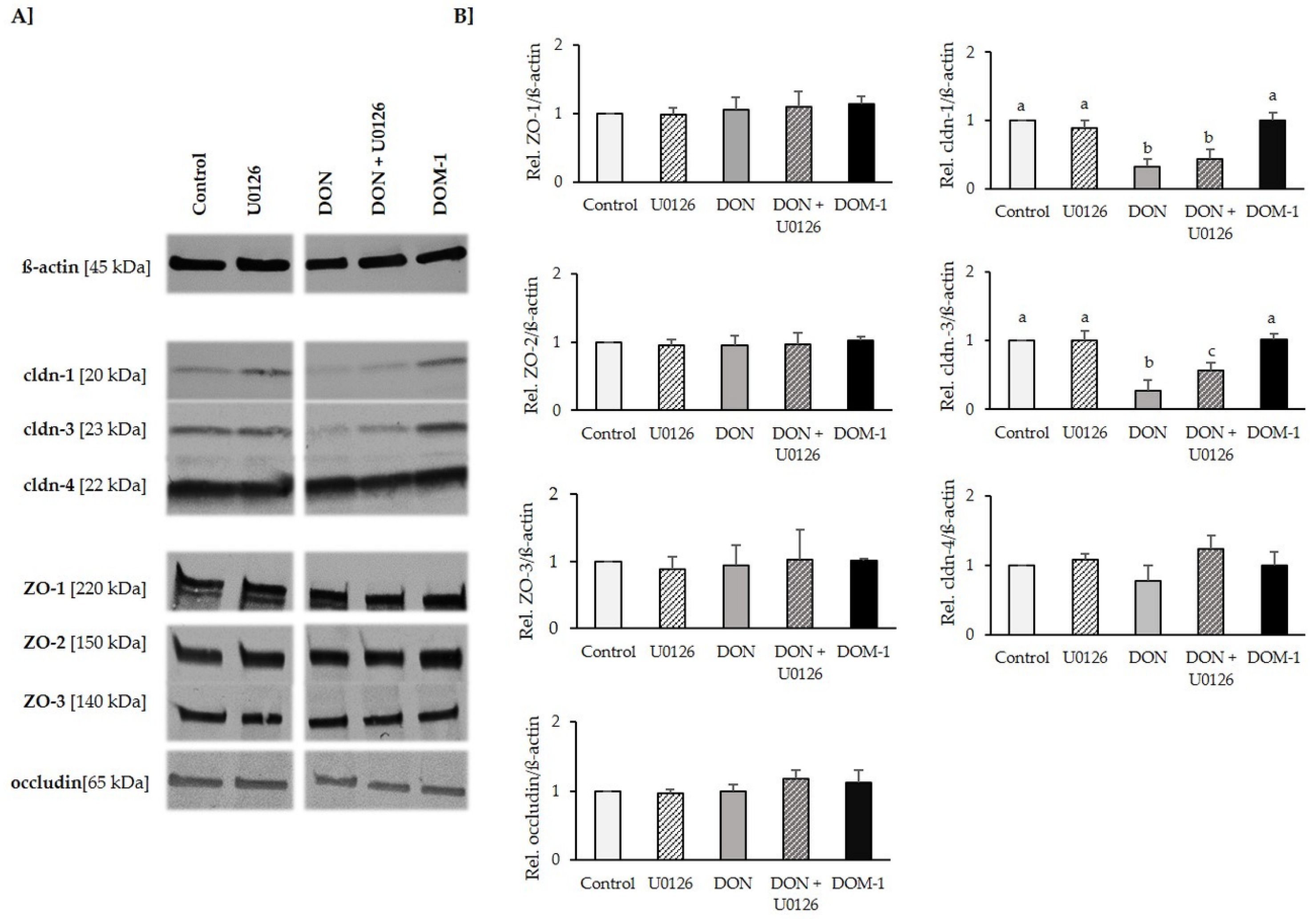
2.6. Tight Junctions
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Culture
5.2. Compounds
5.3. Measurement of Transepithelial Electrical Resistance (TEER)
5.4. Calcium Switch Assay
5.5. Cytotoxicity Assays
5.6. Cell Protein Extraction, SDS-PAGE and Western Blotting
5.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BCA | bicinchoninic acid |
| BCIP | 5-Bromo-4-chloro-3-indolyl phosphate |
| BSA | bovine serum albumin |
| Cldn | claudin |
| DF | dissociation factor |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMSO | dimethyl sulfoxide |
| DOM-1 | deepoxy deoxynivalenol |
| DON | deoxynivalenol |
| EGF | epidermal growth factor |
| EGTA | ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| EPEC | enteropathogenic Escherichia coli |
| ERS | Electrical Resistance System |
| FBS | fetal bovine serum |
| HBSS | Hank’s balanced salt solution |
| HGF | hepatocyte growth factor |
| HIV | human immunodeficiency |
| IL | interleukin |
| IPEC | Intestinal porcine epithelial cells |
| ITS | insulin-transferrin-selenium |
| EGF | epidermal growth factor |
| LDH | lactate dehydrogenase |
| MAPK | mitogen activated protein kinase |
| MMP | matrix metalloproteinases |
| NBT | Nitro blue tetrazolium |
| NR | neutral red |
| OD | optical density |
| PBS | phosphate buffered saline |
| PDGF-1 | platelet derived growth factor-1 |
| RIPA | Radioimmunoprecipitation assay |
| PVDF | Polyvinylidene fluoride |
| SDS | Sodium dodecyl sulfate |
| SRB | sulforhodamine B |
| TBS | Tris-buffered saline |
| TEER | transepithelial electrical resistance |
| TGF-ß | transforming growth factor-beta |
| VEGF | vascular endothelial growth factor |
| ZO | zona occludens |
References
- Odenwald, M.A.; Turner, J.R. Intestinal permeability defects: Is it time to treat? Clin. Gastroenterol. Hepatol. 2013, 11, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Menard, S.; Cerf-Bensussan, N.; Heyman, M. Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal Immunol. 2010, 3, 247–259. [Google Scholar] [CrossRef] [PubMed]
- EFSA. Deoxynivalenol in food and feed: Occurrence and exposure. EFSA J. 2013, 11, 319–324. [Google Scholar]
- Steed, E.; Balda, M.S.; Matter, K. Dynamics and functions of tight junctions. Trends Cell Biol. 2010, 20, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Streit, E.; Schatzmayr, G.; Tassis, P.; Tzika, E.; Marin, D.; Taranu, I.; Tabuc, C.; Nicolau, A.; Aprodu, I.; Puel, O.; et al. Current situation of mycotoxin contamination and co-occurrence in animal feed—Focus on Europe. Toxins (Basel) 2012, 4, 788–809. [Google Scholar] [CrossRef] [PubMed]
- Sugita-Konishi, Y.; Park, B.J.; Kobayashi-Hattori, K.; Tanaka, T.; Chonan, T.; Yoshikawa, K.; Kumagai, S. Effect of cooking process on the deoxynivalenol content and its subsequent cytotoxicity in wheat products. Biosci. Biotechnol. Biochem. 2006, 70, 1764–1768. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Dixit, S.; Dwivedi, P.D.; Pandey, H.P.; Das, M. Influence of temperature and pH on the degradation of deoxynivalenol (DON) in aqueous medium: Comparative cytotoxicity of DON and degraded product. Food Addit. Contam. Part A Chem. Anal. Control Expo Risk Assess. 2014, 31, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Nougayrede, J.P.; Del Rio, J.C.; Moreno, C.; Marin, D.E.; Ferrier, L.; Bracarense, A.P.; Kolf-Clauw, M.; Oswald, I.P. The food contaminant deoxynivalenol, decreases intestinal barrier permeability and reduces claudin expression. Toxicol. Appl. Pharmacol. 2009, 237, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinton, P.; Braicu, C.; Nougayrede, J.P.; Laffitte, J.; Taranu, I.; Oswald, I.P. Deoxynivalenol impairs porcine intestinal barrier function and decreases the protein expression of claudin-4 through a mitogen-activated protein kinase-dependent mechanism. J. Nutr. 2010, 140, 1956–1962. [Google Scholar] [CrossRef] [PubMed]
- Diesing, A.K.; Nossol, C.; Danicke, S.; Walk, N.; Post, A.; Kahlert, S.; Rothkotter, H.J.; Kluess, J. Vulnerability of polarised intestinal porcine epithelial cells to mycotoxin deoxynivalenol depends on the route of application. PLoS ONE 2011, 6, e17472. [Google Scholar] [CrossRef] [PubMed]
- Rotter, B.A.; Prelusky, D.B.; Pestka, J.J. Toxicology of deoxynivalenol (vomitoxin). J. Toxicol. Environ. Health 1996, 48, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Oshima, T.; Miwa, H.; Joh, T. Aspirin induces gastric epithelial barrier dysfunction by activating p38 MAPK via claudin-7. Am. J. Physiol. Cell Physiol. 2008, 295, C800–C806. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Suzuki, T.; Taylor, W.L.; Bhargava, A.; Rao, R.K. Contrasting effects of ERK on tight junction integrity in differentiated and under-differentiated Caco-2 cell monolayers. Biochem. J. 2011, 433, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Mariscal, L.; Tapia, R.; Chamorro, D. Crosstalk of tight junction components with signaling pathways. Biochim. Biophys. Acta 2008, 1778, 729–756. [Google Scholar] [CrossRef] [PubMed]
- Prelusky, D.B.; Gerdes, R.G.; Underhill, K.L.; Rotter, B.A.; Jui, P.Y.; Trenholm, H.L. Effects of low-level dietary deoxynivalenol on haematological and clinical parameters of the pig. Nat. Toxins 1994, 2, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Karlovsky, P. Biological detoxification of the mycotoxin deoxynivalenol and its use in genetically engineered crops and feed additives. Appl. Microbiol. Biotechnol. 2011, 91, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Crews, C.; Dall’Asta, C.; Saeger, S.D.; Haesaert, G.; Karlovsky, P.; Oswald, I.P.; Seefelder, W.; Speijers, G.; Stroka, J. Masked mycotoxins: A review. Mol. Nutr. Food Res. 2013, 57, 165–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz-Zimmermann, H.E.; Hametner, C.; Nagl, V.; Slavik, V.; Moll, W.D.; Berthiller, F. Deoxynivalenol (DON) sulfonates as major DON metabolites in rats: From identification to biomarker method development, validation and application. Anal. Bioanal. Chem. 2014, 406, 7911–7924. [Google Scholar] [CrossRef] [PubMed]
- Shima, J.; Takase, S.; Takahashi, Y.; Iwai, Y.; Fujimoto, H.; Yamazaki, M.; Ochi, K. Novel detoxification of the trichothecene mycotoxin deoxynivalenol by a soil bacterium isolated by enrichment culture. Appl. Environ. Microbiol. 1997, 63, 3825–3830. [Google Scholar] [PubMed]
- He, J.W.; Hassan, Y.I.; Perilla, N.; Li, X.Z.; Boland, G.J.; Zhou, T. Bacterial Epimerization as a Route for Deoxynivalenol Detoxification: The Influence of Growth and Environmental Conditions. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Ikunaga, Y.; Sato, I.; Grond, S.; Numaziri, N.; Yoshida, S.; Yamaya, H.; Hiradate, S.; Hasegawa, M.; Toshima, H.; Koitabashi, M.; et al. Nocardioides sp. strain WSN05-2, isolated from a wheat field, degrades deoxynivalenol, producing the novel intermediate 3-epi-deoxynivalenol. Appl. Microbiol. Biotechnol. 2011, 89, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Nagl, V.S.G. Deoxynivalenol and its masked forms in food and feed. Curr. Opin. Food Sci. 2015, 5, 43–49. [Google Scholar] [CrossRef]
- Maresca, M. From the gut to the brain: Journey and pathophysiological effects of the food-associated trichothecene mycotoxin deoxynivalenol. Toxins 2013, 5, 784–820. [Google Scholar] [CrossRef] [PubMed]
- European Commission (EC). Commission implementing regulation (EU) No 1016/2013 of 23 October 2013 concerning the authorisation of a preparation of mico-organism strain DSM 11798 of the Coriobacteriacae family as a feed additive for pigs. Off. J. Eur. Union 2013, 11, 36–38. [Google Scholar]
- Schierack, P.; Nordhoff, M.; Pollmann, M.; Weyrauch, K.D.; Amasheh, S.; Lodemann, U.; Jores, J.; Tachu, B.; Kleta, S.; Blikslager, A.; et al. Characterization of a porcine intestinal epithelial cell line for in vitro studies of microbial pathogenesis in swine. Histochem. Cell Biol. 2006, 125, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, M.C.; Bistritz, L.; Meddings, J.B. Alterations in intestinal permeability. Gut 2006, 55, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Akbari, P.; Braber, S.; Gremmels, H.; Koelink, P.J.; Verheijden, K.A.; Garssen, J.; Fink-Gremmels, J. Deoxynivalenol: A trigger for intestinal integrity breakdown. FASEB J. 2014, 28, 2414–2429. [Google Scholar] [CrossRef] [PubMed]
- Maresca, M.; Mahfoud, R.; Garmy, N.; Fantini, J. The mycotoxin deoxynivalenol affects nutrient absorption in human intestinal epithelial cells. J. Nutr. 2002, 132, 2723–2731. [Google Scholar] [PubMed]
- Trucksess, M.W.; Thomas, F.; Young, K.; Stack, M.E.; Fulgueras, W.J.; Page, S.W. Survey of deoxynivalenol in U.S. 1993 wheat and barley crops by enzyme-linked immunosorbent assay. J. AOAC Int. 1995, 78, 631–636. [Google Scholar] [PubMed]
- Chung, D.H.; Abouzied, M.M.; Pestka, J.J. Immunochemical assay applied to mycotoxin biosynthesis: ELISA comparison of sterigmatocystin production by Aspergillus versicolor and Aspergillus nidulans. Mycopathologia 1989, 107, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, V.; Croubels, S.; Martel, A.; Verbrugghe, E.; Goossens, J.; Van Deun, K.; Boyen, F.; Thomp son, A.; Shearer, N.; De Backer, P.; et al. The mycotoxin deoxynivalenol potentiates intestinal inflammation by Salmonella typhimurium in porcine ileal loops. PLoS ONE 2011, 6, e23871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EC. Collection of Occurrence Data of Fusarium Toxins in Food and Assessment of Dietary Intake by the Population of EU Member States; European Commission—Health and Consumers Protection Directorate-General: Brussels, Belgium, 2003; pp. 1–606. [Google Scholar]
- Gonzalez-Vallina, R.; Wang, H.; Zhan, R.; Berschneider, H.M.; Lee, R.M.; Davidson, N.O.; Black, D.D. Lipoprotein and apolipoprotein secretion by a newborn piglet intestinal cell line (IPEC-1). Am. J. Physiol. 1996, 271, G249–G259. [Google Scholar] [PubMed]
- Berschneider, H.M. Development of normal cultured small intestinal epithelial cell lines which transport Na and Cl (Abstract). Gastroenterology 1989, 96, A41. [Google Scholar]
- Kaeffer, B.; Bottreau, E.; Velge, P.; Pardon, P. Epithelioid and fibroblastic cell lines derived from the ileum of an adult histocompatible miniature boar (D/D haplotype) and immortalized by SV40 plasmid. Eur. J. Cell Biol. 1993, 62, 152–162. [Google Scholar] [PubMed]
- Wernersson, R.; Schierup, M.H.; Jorgensen, F.G.; Gorodkin, J.; Panitz, F.; Staerfeldt, H.H.; Christensen, O.F.; Mailund, T.; Hornshoj, H.; Klein, A.; et al. Pigs in sequence space: A 0.66X coverage pig genome survey based on shotgun sequencing. BMC Genom. 2005, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nossol, C.; Barta-Boszormenyi, A.; Kahlert, S.; Zuschratter, W.; Faber-Zuschratter, H.; Reinhardt, N.; Ponsuksili, S.; Wimmers, K.; Diesing, A.K.; Rothkotter, H.J. Comparing Two Intestinal Porcine Epithelial Cell Lines (IPECs): Morphological Differentiation, Function and Metabolism. PLoS ONE 2015, 10, e0132323. [Google Scholar] [CrossRef] [PubMed]
- Akbari, P.; Braber, S.; Alizadeh, A.; Verheijden, K.A.; Schoterman, M.H.; Kraneveld, A.D.; Garssen, J.; Fink-Gremmels, J. Galacto-oligosaccharides Protect the Intestinal Barrier by Maintaining the Tight Junction Network and Modulating the Inflammatory Responses after a Challenge with the Mycotoxin Deoxynivalenol in Human Caco-2 Cell Monolayers and B6C3F1 Mice. J. Nutr. 2015, 145, 1604–1613. [Google Scholar] [CrossRef] [PubMed]
- De Walle, J.V.; Sergent, T.; Piront, N.; Toussaint, O.; Schneider, Y.J.; Larondelle, Y. Deoxynivalenol affects in vitro intestinal epithelial cell barrier integrity through inhibition of protein synthesis. Toxicol. Appl. Pharmacol. 2010, 245, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Sergent, T.; Parys, M.; Garsou, S.; Pussemier, L.; Schneider, Y.J.; Larondelle, Y. Deoxynivalenol transport across human intestinal Caco-2 cells and its effects on cellular metabolism at realistic intestinal concentrations. Toxicol. Lett. 2006, 164, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Kadota, T.; Furusawa, H.; Hirano, S.; Tajima, O.; Kamata, Y.; Sugita-Konishi, Y. Comparative study of deoxynivalenol, 3-acetyldeoxynivalenol, and 15-acetyldeoxynivalenol on intestinal transport and IL-8 secretion in the human cell line Caco-2. Toxicol. In Vitro 2013, 27, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, F.; Hara-Kudo, Y.; Saito, N.; Kumagai, S.; Sugita-Konishi, Y. In vitro effect of deoxynivalenol on the differentiation of human colonic cell lines Caco-2 and T84. Mycopathologia 1998, 142, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Tsybulskyy, D.; Lucioli, J.; Laffitte, J.; Callu, P.; Lyazhri, F.; Grosjean, F.; Bracarense, A.P.; Kolf-Clauw, M.; Oswald, I.P. Toxicity of deoxynivalenol and its acetylated derivatives on the intestine: differential effects on morphology, barrier function, tight junction proteins, and mitogen-activated protein kinases. Toxicol. Sci. 2012, 130, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, G.D.; Silva, C.A.; Pinton, P.; Oswald, I.P.; Bracarense, A.P. Phytic acid protects porcine intestinal epithelial cells from deoxynivalenol (DON) cytotoxicity. Exp. Toxicol. Pathol. 2012, 64, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Kluess, J.W.; Kahlert, S.; Krober, A.; Diesing, A.K.; Rothkotter, H.J.; Wimmers, K.; Danicke, S. Deoxynivalenol, but not E. coli lipopolysaccharide, changes the response pattern of intestinal porcine epithelial cells (IPEC-J2) according to its route of application. Toxicol. Lett. 2015, 239, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Goossens, J.; Pasmans, F.; Verbrugghe, E.; Vandenbroucke, V.; De Baere, S.; Meyer, E.; Haesebrouck, F.; De Backer, P.; Croubels, S. Porcine intestinal epithelial barrier disruption by the Fusarium mycotoxins deoxynivalenol and T-2 toxin promotes transepithelial passage of doxycycline and paromomycin. BMC Vet. Res. 2012, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nossol, C.; Diesing, A.K.; Walk, N.; Faber-Zuschratter, H.; Hartig, R.; Post, A.; Kluess, J.; Rothkotter, H.J.; Kahlert, S. Air-liquid interface cultures enhance the oxygen supply and trigger the structural and functional differentiation of intestinal porcine epithelial cells (IPEC). Histochem. Cell Biol. 2011, 136, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Karam, S.M. Lineage commitment and maturation of epithelial cells in the gut. Front. Biosci. 1999, 4, D286–D298. [Google Scholar] [CrossRef] [PubMed]
- Geens, M.M.; Niewold, T.A. Optimizing culture conditions of a porcine epithelial cell line IPEC-J2 through a histological and physiological characterization. Cytotechnology 2011, 63, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Fromter, E.; Diamond, J. Route of passive ion permeation in epithelia. Nat. New Biol. 1972, 235, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Feldman, G.; Kiely, B.; Martin, N.; Ryan, G.; McMorrow, T.; Ryan, M.P. Role for TGF-beta in cyclosporine-induced modulation of renal epithelial barrier function. J. Am. Soc. Nephrol. 2007, 18, 1662–1671. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Reardon, C.; Wang, A.; Nazli, A.; McKay, D.M. Transforming growth factor-beta regulation of epithelial tight junction proteins enhances barrier function and blocks enterohemorrhagic Escherichia coli O157:H7-induced increased permeability. Am. J. Pathol. 2005, 167, 1587–1597. [Google Scholar] [CrossRef]
- Basuroy, S.; Seth, A.; Elias, B.; Naren, A.P.; Rao, R. MAPK interacts with occludin and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochem. J. 2006, 393, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Lipschutz, J.H.; Li, S.; Arisco, A.; Balkovetz, D.F. Extracellular signal-regulated kinases 1/2 control claudin-2 expression in Madin-Darby canine kidney strain I and II cells. J. Biol. Chem. 2005, 280, 3780–3788. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, T.; Sakaguchi, T.; Gu, X.; Reinecker, H.C. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology 2000, 118, 1001–1011. [Google Scholar] [CrossRef]
- Campbell, M.; Collery, R.; McEvoy, A.; Gardiner, T.A.; Stitt, A.W.; Brankin, B. Involvement of MAPKs in endostatin-mediated regulation of blood-retinal barrier function. Curr. Eye Res. 2006, 31, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.L.; Ching, D.; Guan, Y.; Firestone, G.L. Requirement for Ras and phosphatidylinositol 3-kinase signaling uncouples the glucocorticoid-induced junctional organization and transepithelial electrical resistance in mammary tumor cells. J. Biol. Chem. 1999, 274, 32818–32828. [Google Scholar] [CrossRef] [PubMed]
- Sumanasekera, W.K.; Sumanasekera, G.U.; Mattingly, K.A.; Dougherty, S.M.; Keynton, R.S.; Klinge, C.M. Estradiol and dihydrotestosterone regulate endothelial cell barrier function after hypergravity-induced alterations in MAPK activity. Am. J. Physiol. Cell Physiol. 2007, 293, C566–C573. [Google Scholar] [CrossRef] [PubMed]
- Sumanasekera, W.K.; Zhao, L.; Ivanova, M.; Morgan, D.D.; Noisin, E.L.; Keynton, R.S.; Klinge, C.M. Effect of estradiol and dihydrotestosterone on hypergravity-induced MAPK signaling and occludin expression in human umbilical vein endothelial cells. Cell Tissue Res. 2006, 324, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Harada, T.; Li, J.; Uchiyama, T.; Han, Y.; Englert, J.A.; Fink, M.P. Bile modulates intestinal epithelial barrier function via an extracellular signal related kinase 1/2 dependent mechanism. Intensive Care Med. 2005, 31, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lu, Q.; Schneeberger, E.E.; Goodenough, D.A. Restoration of tight junction structure and barrier function by down-regulation of the mitogen-activated protein kinase pathway in ras-transformed Madin-Darby canine kidney cells. Mol. Biol. Cell 2000, 11, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Mullin, J.M.; Leatherman, J.M.; Valenzano, M.C.; Huerta, E.R.; Verrechio, J.; Smith, D.M.; Snetselaar, K.; Liu, M.; Francis, M.K.; Sell, C. Ras mutation impairs epithelial barrier function to a wide range of nonelectrolytes. Mol. Biol. Cell 2005, 16, 5538–5550. [Google Scholar] [CrossRef] [PubMed]
- Krizbai, I.A.; Bauer, H.; Bresgen, N.; Eckl, P.M.; Farkas, A.; Szatmari, E.; Traweger, A.; Wejksza, K.; Bauer, H.C. Effect of oxidative stress on the junctional proteins of cultured cerebral endothelial cells. Cell Mol. Neurobiol. 2005, 25, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Wiesnet, M.; Renz, D.; Schaper, W. H2O2 induces paracellular permeability of porcine brain-derived microvascular endothelial cells by activation of the p44/42 MAP kinase pathway. Eur. J. Cell Biol. 2005, 84, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Kevil, C.G.; Oshima, T.; Alexander, B.; Coe, L.L.; Alexander, J.S. H(2)O(2)-mediated permeability: Role of MAPK and occludin. Am. J. Physiol. Cell Physiol. 2000, 279, C21–C30. [Google Scholar] [PubMed]
- Tan, X.; Egami, H.; Ishikawa, S.; Kurizaki, T.; Hirota, M.; Ogawa, M. Zonula occludens-1 (ZO-1) redistribution is involved in the regulation of cell dissociation in pancreatic cancer cells. Dig. Dis. Sci. 2005, 50, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Tamori, Y.; Egami, H.; Ishikawa, S.; Kurizaki, T.; Takai, E.; Hirota, M.; Ogawa, M. Analysis of invasion-metastasis mechanism in pancreatic cancer: Involvement of tight junction transmembrane protein occludin and MEK/ERK signal transduction pathway in cancer cell dissociation. Oncol. Rep. 2004, 11, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.; Franzen, A.; Karlsson, J.O.; Ericson, L.E.; Heldin, N.E.; Nilsson, M. Transforming growth factor-beta and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. J. Cell Sci. 2002, 115, 4227–4236. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, N.; de Kozak, Y.; Jeanny, J.C.; Glotin, A.; Mascarelli, F.; Massin, P.; BenEzra, D.; Behar-Cohen, F. Placental growth factor-1 and epithelial haemato-retinal barrier breakdown: Potential implication in the pathogenesis of diabetic retinopathy. Diabetologia 2007, 50, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.C.; Chan, L.Y.; Li, F.F.; Tang, S.C.; Chan, K.W.; Chan, T.M.; Lam, M.F.; Wieslander, A.; Lai, K.N. Glucose degradation products downregulate ZO-1 expression in human peritoneal mesothelial cells: The role of VEGF. Nephrol. Dial. Transplant. 2005, 20, 1336–1349. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Yang, P.C.; Darmoul, D.; Amadesi, S.; Saito, T.; Cottrell, G.S.; Coelho, A.M.; Singh, P.; Grady, E.F.; Perdue, M.; et al. Mast cell tryptase controls paracellular permeability of the intestine. Role of protease-activated receptor 2 and beta-arrestins. J. Biol. Chem. 2005, 280, 31936–31948. [Google Scholar] [CrossRef] [PubMed]
- Pu, H.; Tian, J.; Andras, I.E.; Hayashi, K.; Flora, G.; Hennig, B.; Toborek, M. HIV-1 Tat protein-induced alterations of ZO-1 expression are mediated by redox-regulated ERK 1/2 activation. J. Cereb. Blood Flow Metab. 2005, 25, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Andras, I.E.; Pu, H.; Tian, J.; Deli, M.A.; Nath, A.; Hennig, B.; Toborek, M. Signaling mechanisms of HIV-1 Tat-induced alterations of claudin-5 expression in brain endothelial cells. J. Cereb Blood. Flow Metab. 2005, 25, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lu, X.H.; Yan, S.; Chai, H.; Yao, Q. HIV protease inhibitor ritonavir increases endothelial monolayer permeability. Biochem. Biophys. Res. Commun. 2005, 335, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Egami, H.; Abe, M.; Nozawa, F.; Hirota, M.; Ogawa, M. Involvement of MMP-7 in invasion of pancreatic cancer cells through activation of the EGFR mediated MEK-ERK signal transduction pathway. J. Clin. Pathol. 2005, 58, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Wang, X.; Aoki, T.; Lo, E.H. Downregulation of matrix metalloproteinase-9 and attenuation of edema via inhibition of ERK mitogen activated protein kinase in traumatic brain injury. J. Neurotrauma 2002, 19, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Czerucka, D.; Dahan, S.; Mograbi, B.; Rossi, B.; Rampal, P. Saccharomyces boulardii preserves the barrier function and modulates the signal transduction pathway induced in enteropathogenic Escherichia coli-infected T84 cells. Infect. Immun. 2000, 68, 5998–6004. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Matter, K. Tight junctions as regulators of tissue remodelling. Curr. Opin. Cell Biol. 2016, 42, 94–101. [Google Scholar]
- Zhou, H.R.; Islam, Z.; Pestka, J.J. Rapid, sequential activation of mitogen-activated protein kinases and transcription factors precedes proinflammatory cytokine mRNA expression in spleens of mice exposed to the trichothecene vomitoxin. Toxicol. Sci. 2003, 72, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Favata, M.F.; Horiuchi, K.Y.; Manos, E.J.; Daulerio, A.J.; Stradley, D.A.; Feeser, W.S.; Van Dyk, D.E.; Pitts, W.J.; Earl, R.A.; Hobbs, F.; et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998, 273, 18623–18632. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; Pasmans, F.; De Backer, P.; Croubels, S. An in vitro model using the IPEC-J2 cell line for efficacy and drug interaction testing of mycotoxin detoxifying agents. Toxicol. in Vitro 2013, 27, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Diesing, A.K.; Nossol, C.; Panther, P.; Walk, N.; Post, A.; Kluess, J.; Kreutzmann, P.; Danicke, S.; Rothkotter, H.J.; Kahlert, S. Mycotoxin deoxynivalenol (DON) mediates biphasic cellular response in intestinal porcine epithelial cell lines IPEC-1 and IPEC-J2. Toxicol. Lett. 2011, 200, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.J.; Song, S.K.; Lee, I.K.; Ko, S.; Han, S.E.; Bae, S.; Ji, S.Y.; Park, B.C.; Song, K.D.; Lee, H.K.; et al. Barrier protection via Toll-like receptor 2 signaling in porcine intestinal epithelial cells damaged by deoxynivalnol. Vet. Res. 2016, 47. [Google Scholar] [CrossRef] [PubMed]
- Danicke, S.; Hegewald, A.K.; Kahlert, S.; Kluess, J.; Rothkotter, H.J.; Breves, G.; Doll, S. Studies on the toxicity of deoxynivalenol (DON), sodium metabisulfite, DON-sulfonate (DONS) and de-epoxy-DON for porcine peripheral blood mononuclear cells and the Intestinal Porcine Epithelial Cell lines IPEC-1 and IPEC-J2, and on effects of DON and DONS on piglets. Food Chem. Toxicol. 2010, 48, 2154–2162. [Google Scholar] [PubMed]
- Daenicke, S.; Keese, C.; Goyarts, T.; Doll, S. Effects of deoxynivalenol (DON) and related compounds on bovine peripheral blood mononuclear cells (PBMC) in vitro and in vivo. Mycotoxin Res. 2011, 27, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Sundstol Eriksen, G.; Pettersson, H.; Lundh, T. Comparative cytotoxicity of deoxynivalenol, nivalenol, their acetylated derivatives and de-epoxy metabolites. Food Chem. Toxicol. 2004, 42, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Nasri, T.; Bosch, R.R.; Voorde, S.; Fink-Gremmels, J. Differential induction of apoptosis by type A and B trichothecenes in Jurkat T-lymphocytes. Toxicol. in Vitro 2006, 20, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Pierron, A.; Mimoun, S.; Murate, L.S.; Loiseau, N.; Lippi, Y.; Bracarense, A.P.; Schatzmayr, G.; He, J.W.; Zhou, T.; Moll, W.D.; et al. Microbial biotransformation of DON: molecular basis for reduced toxicity. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Broekaert, N.; Devreese, M.; Demeyere, K.; Berthiller, F.; Michlmayr, H.; Varga, E.; Adam, G.; Meyer, E.; Croubels, S. Comparative in vitro cytotoxicity of modified deoxynivalenol on porcine intestinal epithelial cells. Food Chem. Toxicol. 2016, 95, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Juan-Garcia, A.; Juan, C.; Konig, S.; Ruiz, M.J. Cytotoxic effects and degradation products of three mycotoxins: alternariol, 3-acetyl-deoxynivalenol and 15-acetyl-deoxynivalenol in liver hepatocellular carcinoma cells. Toxicol. Lett. 2015, 235, 8–16. [Google Scholar] [CrossRef] [PubMed]






© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Springler, A.; Hessenberger, S.; Schatzmayr, G.; Mayer, E. Early Activation of MAPK p44/42 Is Partially Involved in DON-Induced Disruption of the Intestinal Barrier Function and Tight Junction Network. Toxins 2016, 8, 264. https://doi.org/10.3390/toxins8090264
Springler A, Hessenberger S, Schatzmayr G, Mayer E. Early Activation of MAPK p44/42 Is Partially Involved in DON-Induced Disruption of the Intestinal Barrier Function and Tight Junction Network. Toxins. 2016; 8(9):264. https://doi.org/10.3390/toxins8090264
Chicago/Turabian StyleSpringler, Alexandra, Sabine Hessenberger, Gerd Schatzmayr, and Elisabeth Mayer. 2016. "Early Activation of MAPK p44/42 Is Partially Involved in DON-Induced Disruption of the Intestinal Barrier Function and Tight Junction Network" Toxins 8, no. 9: 264. https://doi.org/10.3390/toxins8090264