
The Middle Fragment of Helicobacter pylori CagA Induces Actin Rearrangement and Triggers Its Own Uptake into Gastric Epithelial Cells


Abstract
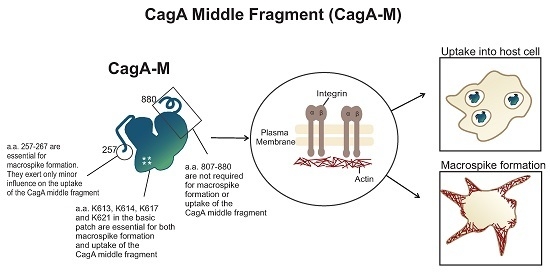
:
1. Introduction
2. Results
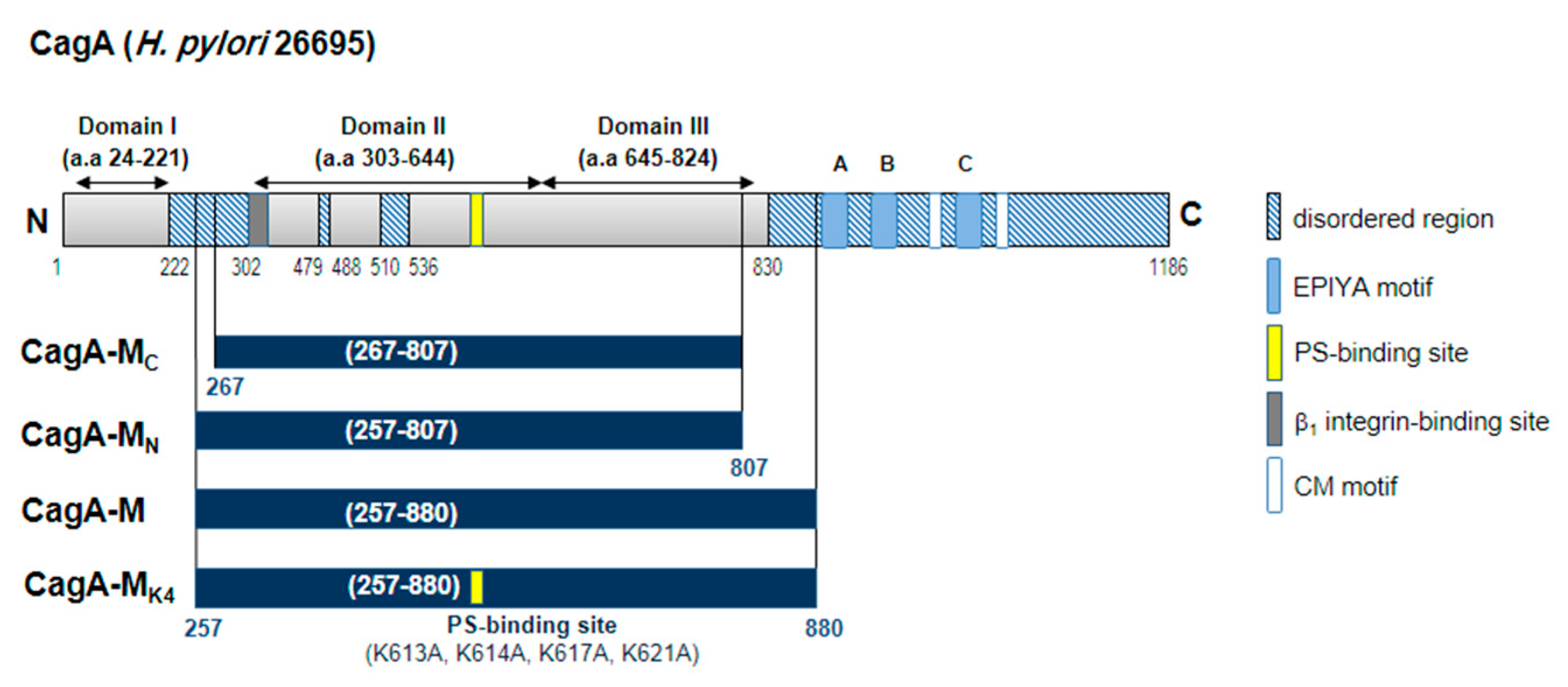
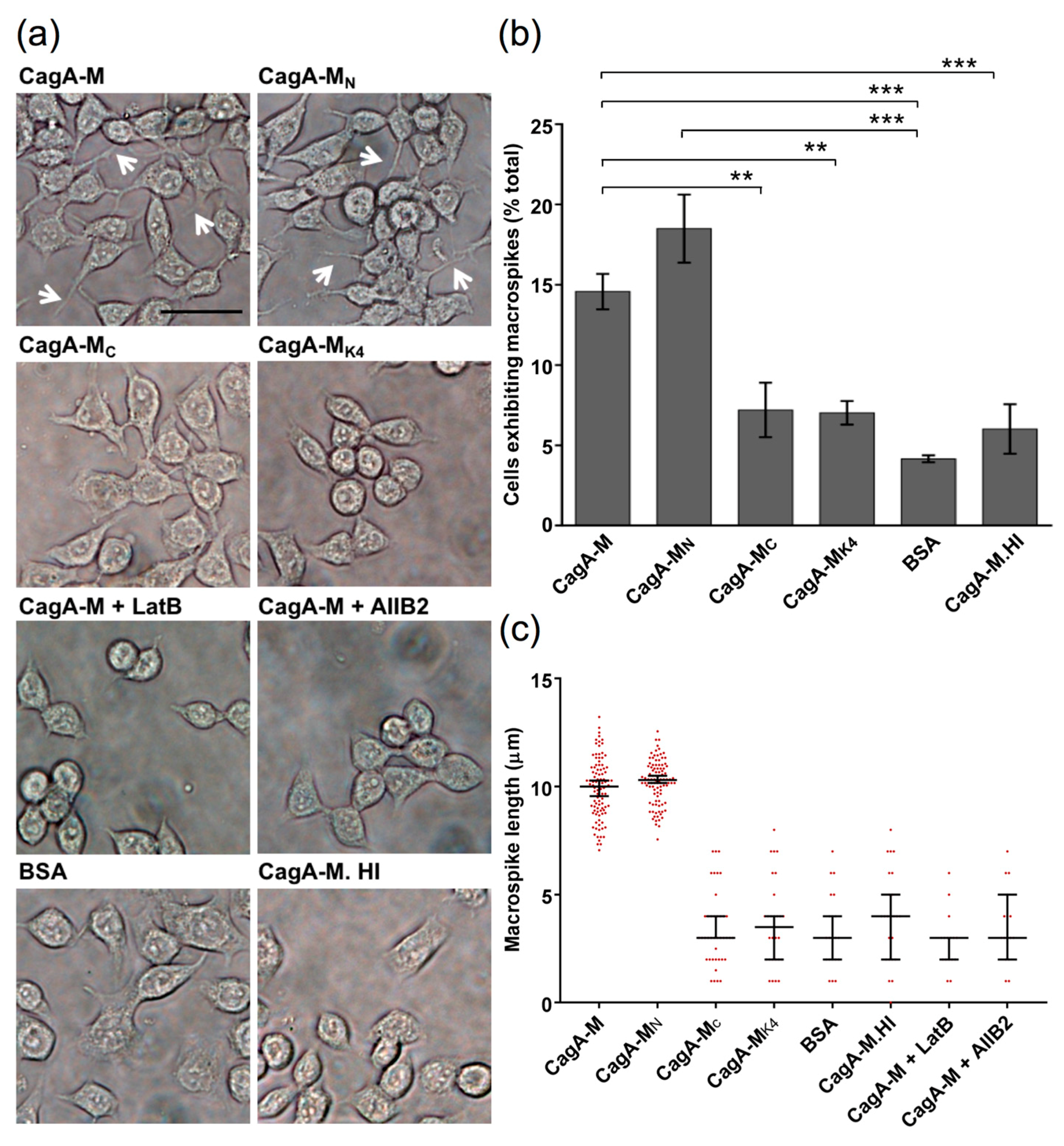
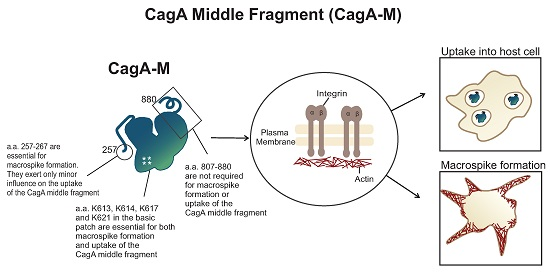
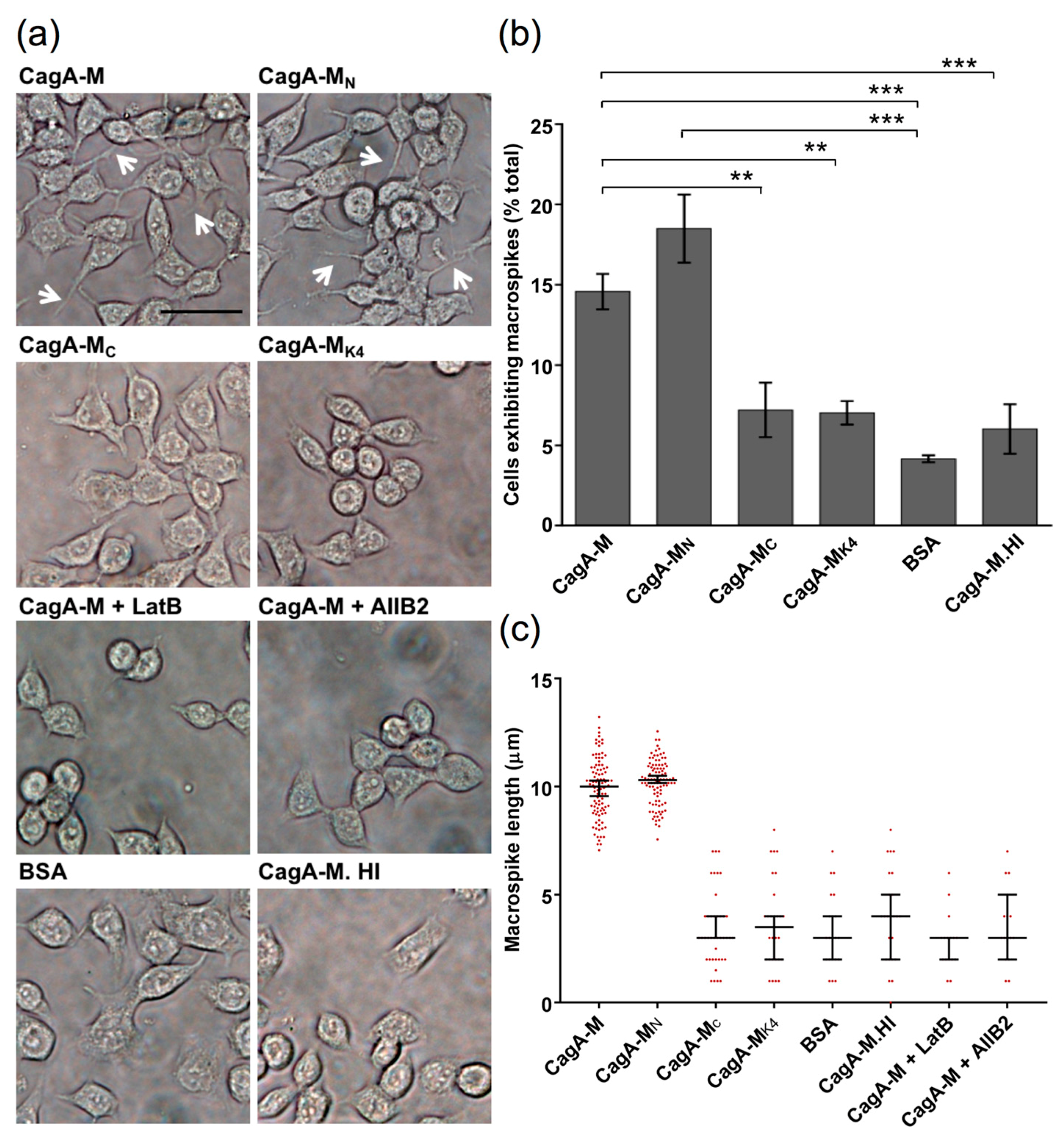
2.1. The Middle Fragment of CagA (CagA-M, aa 257–880) Alone Is Sufficient for Altering Host Cell Morphology
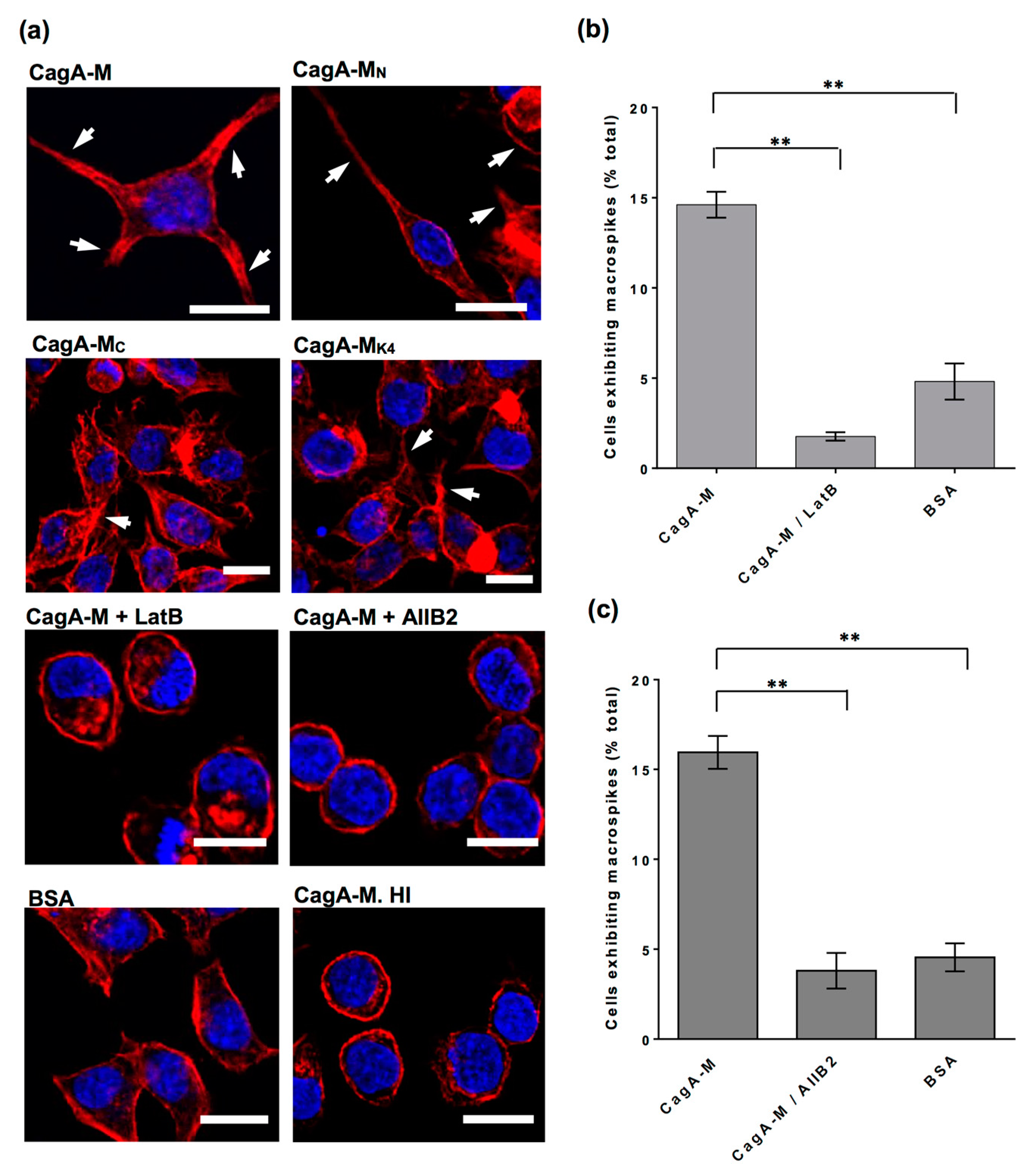
2.2. β1 Integrin Activation Is Required for the Morphological Changes Induced by the Middle Fragment of CagA
2.3. Amino Acid Residues 257 to 266 and the Lipid-Binding Site within the Middle Fragment of CagA Are Essential for Induction of Host Cell Morphological Changes
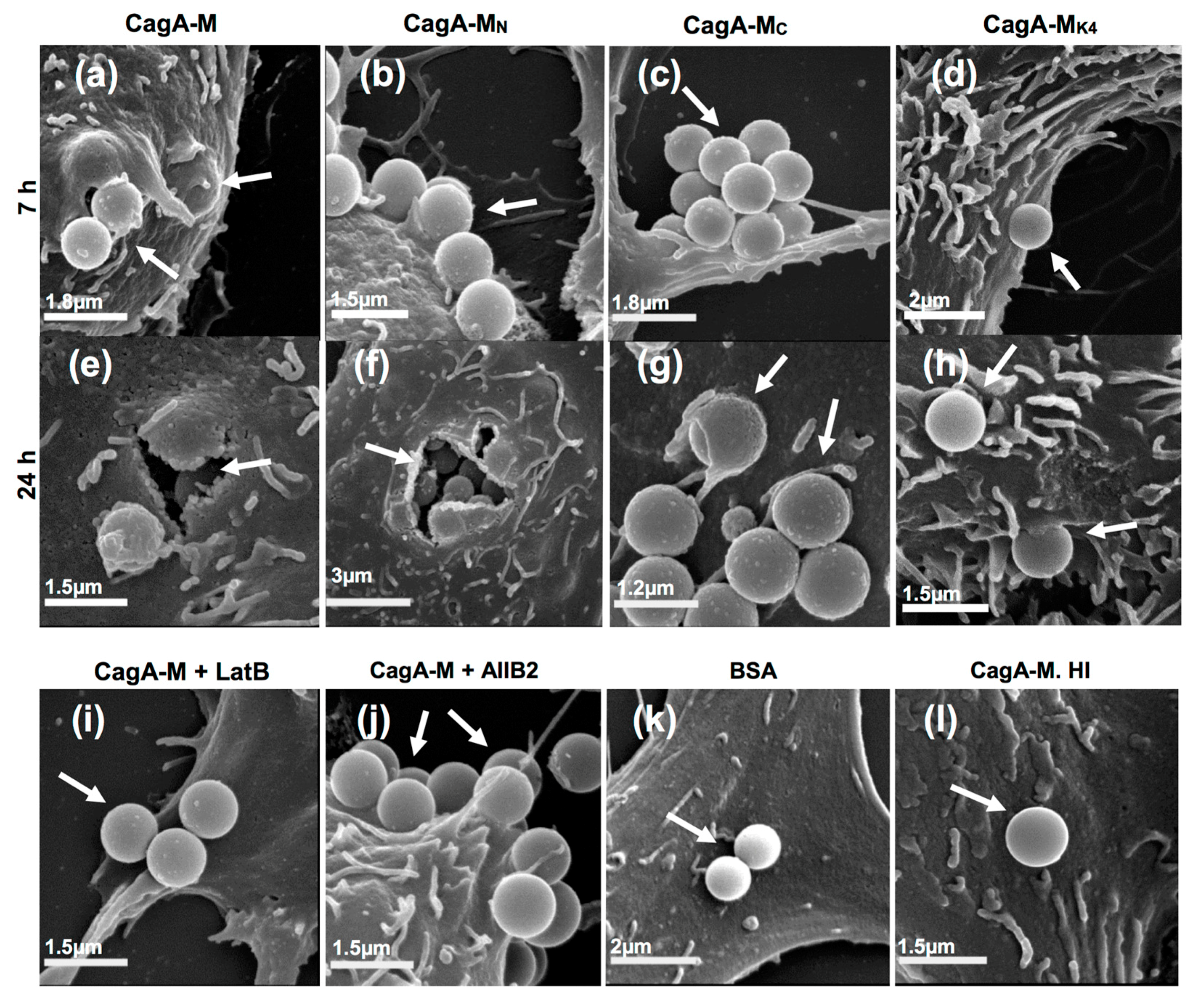
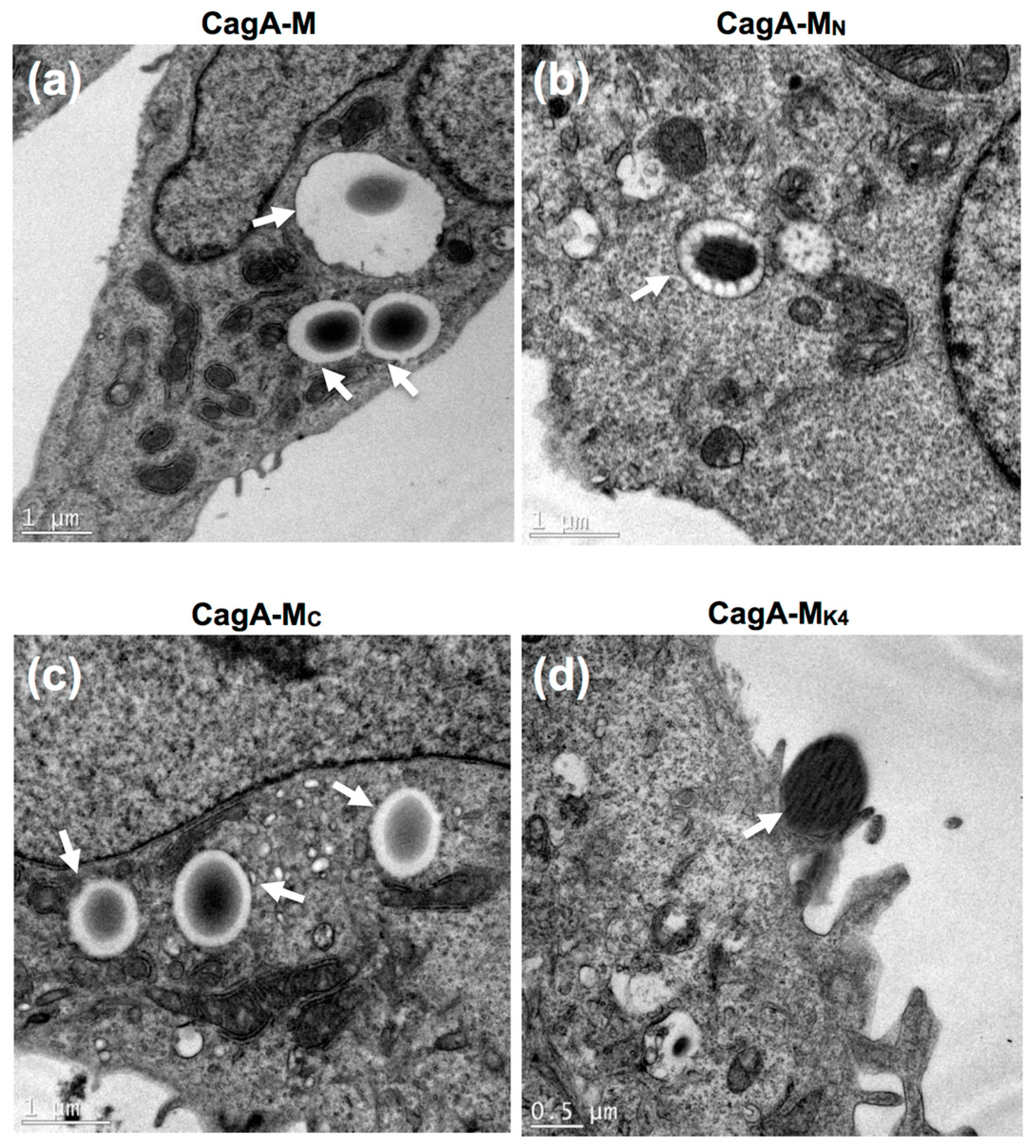
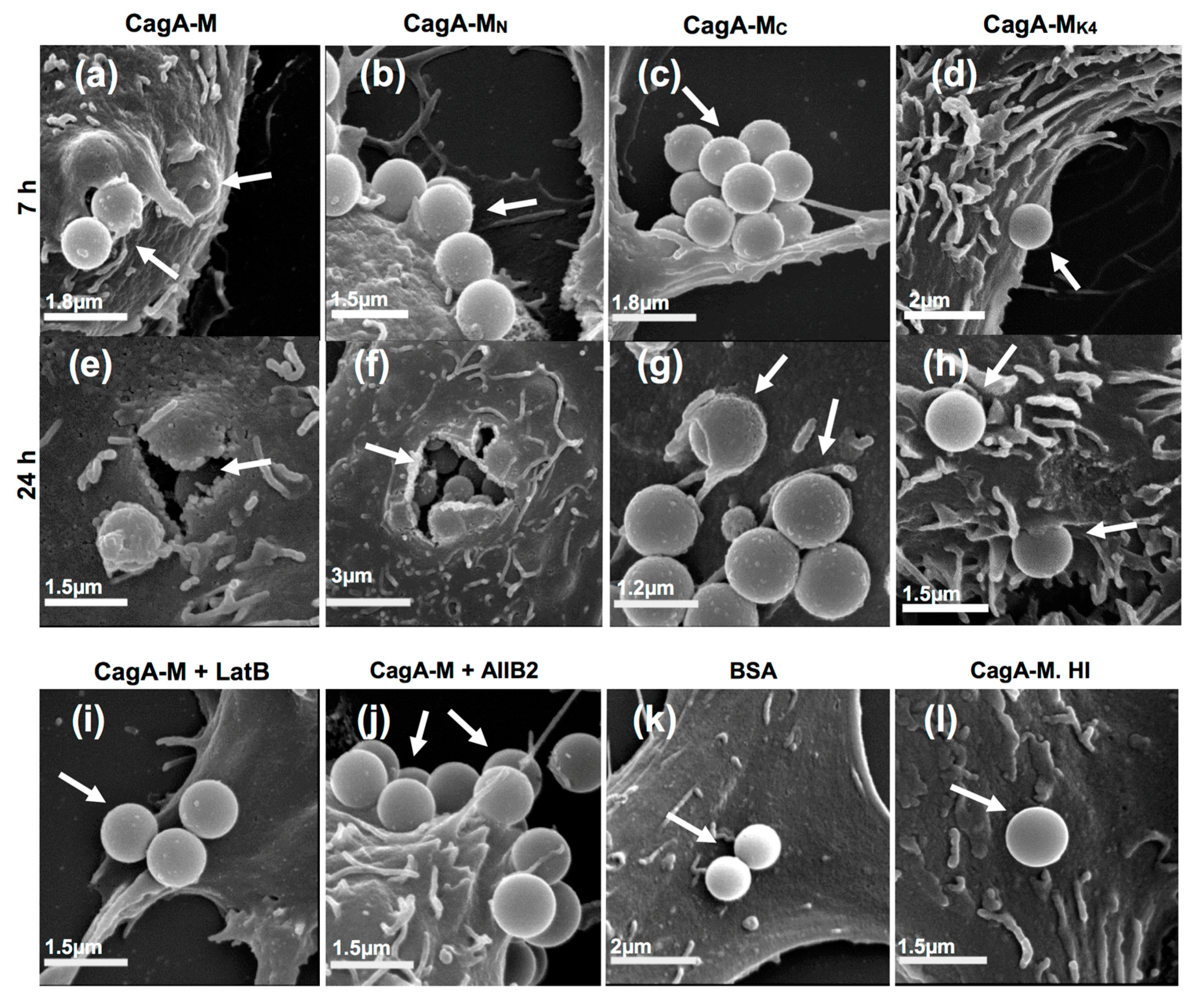
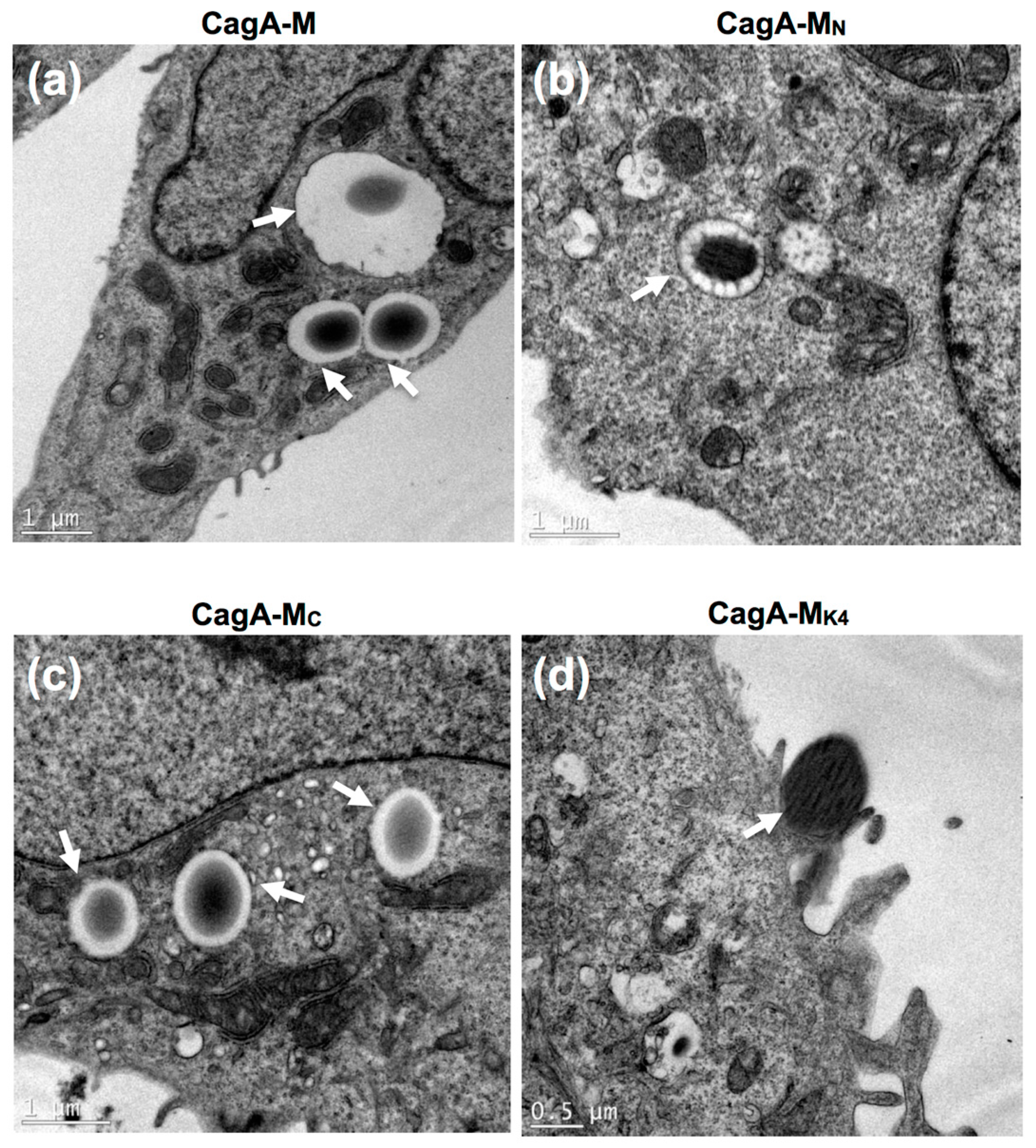
2.4. The Middle Fragment of CagA Immobilized on Latex Beads Is Capable of Binding and Entering AGS Cells
2.5. Actin Cytoskeletal Rearragnement and β1 Integrin Activation Are Essential for the Uptake of CagA Middle Fragment-Coated Beads into AGS Cells
3. Discussion
4. Materials and Methods
4.1. Gene Synthesis, Cloning, Protein Expression, and Purification
4.2. Tissue Culture and Treatment with CagA Fragments
4.3. Inhibition of Actin Cytoskeleton Polymerization and β1 Integrin Activity
4.4. Latex Bead Binding Assay
4.5. Staining of Actin Microfilaments and Confocal Laser Scanning Microscopy
4.6. Scanning Electron Microscopy (SEM)
4.7. Transmission Electron Microscopy (TEM)
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wroblewski, L.E.; Peek, R.M., Jr.; Wilson, K.T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef] [PubMed]
- Peek, R.M., Jr.; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Covacci, A.; Censini, S.; Bugnoli, M.; Petracca, R.; Burroni, D.; Macchia, G.; Massone, A.; Papini, E.; Xiang, Z.; Figura, N.; et al. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. USA 1993, 90, 5791–5795. [Google Scholar] [CrossRef] [PubMed]
- Tummuru, M.K.; Cover, T.L.; Blaser, M.J. Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: Evidence of linkage to cytotoxin production. Infect. Immun. 1993, 61, 1799–1809. [Google Scholar] [PubMed]
- Hatakeyama, M. Helicobacter pylori and gastric carcinogenesis. J. Gastroenterol. 2009, 44, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.R.; Vogelmann, R.; Covacci, A.; Tompkins, L.S.; Nelson, W.J.; Falkow, S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 2003, 300, 1430–1434. [Google Scholar] [CrossRef] [PubMed]
- Conlin, V.S.; Curtis, S.B.; Zhao, Y.; Moore, E.D.; Smith, V.C.; Meloche, R.M.; Finlay, B.B.; Buchan, A.M. Helicobacter pylori infection targets adherens junction regulatory proteins and results in increased rates of migration in human gastric epithelial cells. Infect. Immun. 2004, 72, 5181–5192. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.T.; Israel, D.A.; Washington, M.K.; Krishna, U.; Fox, J.G.; Rogers, A.B.; Neish, A.S.; Collier-Hyams, L.; Perez-Perez, G.I.; Hatakeyama, M.; et al. Activation of β-catenin by carcinogenic Helicobacter pylori. Proc. Natl. Acad. Sci. USA 2005, 102, 10646–10651. [Google Scholar] [CrossRef] [PubMed]
- Murata-Kamiya, N.; Kurashima, Y.; Teishikata, Y.; Yamahashi, Y.; Saito, Y.; Higashi, H.; Aburatani, H.; Akiyama, T.; Peek, R.M., Jr.; Azuma, T.; et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the β-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 2007, 26, 4617–4626. [Google Scholar] [CrossRef] [PubMed]
- Peek, R.M., Jr.; Moss, S.F.; Tham, K.T.; Perez-Perez, G.I.; Wang, S.; Miller, G.G.; Atherton, J.C.; Holt, P.R.; Blaser, M.J. Helicobacter pylori cagA+ strains and dissociation of gastric epithelial cell proliferation from apoptosis. J. Natl. Cancer Inst. 1997, 89, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Jurik, A.; Hausser, E.; Kutter, S.; Pattis, I.; Prassl, S.; Weiss, E.; Fischer, W. The coupling protein Cagβ and its interaction partner CagZ are required for type IV secretion of the Helicobacter pylori CagA protein. Infect. Immun. 2010, 78, 5244–5251. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Mimuro, H.; Suzuki, T.; Tanaka, J.; Asahi, M.; Haas, R.; Sasakawa, C. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol. Cell 2002, 10, 745–755. [Google Scholar] [CrossRef]
- Suzuki, M.; Mimuro, H.; Suzuki, T.; Park, M.; Yamamoto, T.; Sasakawa, C. Interaction of CagA with Crk plays an important role in Helicobacter pylori-induced loss of gastric epithelial cell adhesion. J. Exp. Med. 2005, 202, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Nesic, D.; Miller, M.C.; Quinkert, Z.T.; Stein, M.; Chait, B.T.; Stebbins, C.E. Helicobacter pylori CagA inhibits PAR1-MARK family kinases by mimicking host substrates. Nat. Struct. Mol. Biol. 2010, 17, 130–132. [Google Scholar] [PubMed]
- Hatakeyama, M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat. Rev. Cancer 2004, 4, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Mentis, A.F. Pathogenesis of Helicobacter pylori infection. Helicobacter 2007, 12 (Suppl. 1), 10–14. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Helicobacter pylori CagA—A bacterial intruder conspiring gastric carcinogenesis. Int. J. Cancer 2006, 119, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Tsutsumi, R.; Fujita, A.; Yamazaki, S.; Asaka, M.; Azuma, T.; Hatakeyama, M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc. Natl. Acad. Sci. USA 2002, 99, 14428–14433. [Google Scholar] [CrossRef] [PubMed]
- Murata-Kamiya, N.; Kikuchi, K.; Hayashi, T.; Higashi, H.; Hatakeyama, M. Helicobacter pylori exploits host membrane phosphatidylserine for delivery, localization, and pathophysiological action of the CagA oncoprotein. Cell Host Microbe 2010, 7, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Senda, M.; Morohashi, H.; Higashi, H.; Horio, M.; Kashiba, Y.; Nagase, L.; Sasaya, D.; Shimizu, T.; Venugopalan, N.; et al. Tertiary structure-function analysis reveals the pathogenic signaling potentiation mechanism of Helicobacter pylori oncogenic effector CagA. Cell Host Microbe 2012, 12, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Soto, L.F.; Kutter, S.; Sewald, X.; Ertl, C.; Weiss, E.; Kapp, U.; Rohde, M.; Pirch, T.; Jung, K.; Retta, S.F.; et al. Helicobacter pylori type IV secretion apparatus exploits β1 integrin in a novel RGD-independent manner. PLoS Pathog. 2009, 5, e1000684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, T.; Zabler, D.; Urman, S.; Rohde, M.; Hartig, R.; Wessler, S.; Misselwitz, R.; Berger, J.; Sewald, N.; Konig, W.; et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 2007, 449, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Terradot, L.; Waksman, G. Architecture of the Helicobacter pylori Cag-type IV secretion system. FEBS J. 2011, 278, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Kaplan-Turkoz, B.; Jimenez-Soto, L.F.; Dian, C.; Ertl, C.; Remaut, H.; Louche, A.; Tosi, T.; Haas, R.; Terradot, L. Structural insights into Helicobacter pylori oncoprotein CagA interaction with β1 integrin. Proc. Natl. Acad. Sci. USA 2012, 109, 14640–14645. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Higashi, H.; Lu, H.; Azuma, T.; Hatakeyama, M. Structural basis and functional consequence of Helicobacter pylori CagA multimerization in cells. J. Biol. Chem. 2006, 281, 32344–32352. [Google Scholar] [CrossRef] [PubMed]
- Woon, A.P.; Tohidpour, A.; Alonso, H.; Saijo-Hamano, Y.; Kwok, T.; Roujeinikova, A. Conformational analysis of isolated domains of Helicobacter pylori CagA. PLoS ONE 2013, 8, e79367. [Google Scholar] [CrossRef] [PubMed]
- Segal, E.D.; Cha, J.; Lo, J.; Falkow, S.; Tompkins, L.S. Altered states: Involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc. Natl. Acad. Sci. USA 1999, 96, 14559–14564. [Google Scholar] [CrossRef] [PubMed]
- Spector, I.; Shochet, N.R.; Kashman, Y.; Groweiss, A. Latrunculins: Novel marine toxins that disrupt microfilament organization in cultured cells. Science 1983, 219, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Wakatsuki, T.; Schwab, B.; Thompson, N.C.; Elson, E.L. Effects of cytochalasin D and latrunculin B on mechanical properties of cells. J. Cell Sci. 2001, 114, 1025–1036. [Google Scholar] [PubMed]
- Calderwood, D.A.; Shattil, S.J.; Ginsberg, M.H. Integrins and actin filaments: Reciprocal regulation of cell adhesion and signaling. J. Biol. Chem. 2000, 275, 22607–22610. [Google Scholar] [CrossRef] [PubMed]
- Swanson, J.A. Shaping cups into phagosomes and macropinosomes. Nat. Rev. Mol. Cell Biol. 2008, 9, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhu, X.; Jin, Y.; Shan, W.; Huang, Y. Mechanism study of cellular uptake and tight junction opening mediated by goblet cell-specific trimethyl chitosan nanoparticles. Mol. Pharm. 2014, 11, 1520–1532. [Google Scholar] [CrossRef] [PubMed]
- Tahara, K.; Yamamoto, H.; Kawashima, Y. Cellular uptake mechanisms and intracellular distributions of polysorbate 80-modified poly (d,l-lactide-co-glycolide) nanospheres for gene delivery. Eur. J. Pharm. Biopharm. 2010, 75, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pan, S.Q. Agrobacterium delivers VirE2 protein into host cells via clathrin-mediated endocytosis. Sci. Adv. 2017, 3, e1601528. [Google Scholar] [CrossRef] [PubMed]
- Voss, B.J.; Gaddy, J.A.; McDonald, W.H.; Cover, T.L. Analysis of surface-exposed outer membrane proteins in Helicobacter pylori. J. Bacteriol. 2014, 196, 2455–2471. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Gorrell, R.J.; Guan, J.; Xin, Y.; Tafreshi, M.A.; Hutton, M.L.; McGuckin, M.A.; Ferrero, R.L.; Kwok, T. A novel NOD1- and CagA-independent pathway of interleukin-8 induction mediated by the Helicobacter pylori type IV secretion system. Cell Microbiol. 2013, 15, 554–570. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Average Number of Beads Per Field a (Mean ± S.D.) | |
|---|---|---|
| Engulfed Beads b | Extracellular Beads | |
| CagA-M | 4.1 ± 1.4 | 4.2 ± 1.1 |
| CagA-MN | 4.5 ± 1.7 | 4.9 ± 1.5 |
| CagA-MC | 2.4 ± 1.1 | 2.5 ± 1.2 |
| CagA-MK4 | 1.2 ± 0.4 | 1.8 ± 0.7 |
| BSA | 0 ± 0 | 0.2 ± 0.4 |
| CagA-M. HI | 0 ± 0 | 0.2 ± 0.5 |
| CagA-M + LatB | 0 ± 0 | 0.2 ± 0.4 |
| CagA-M + AIIB2 | 0 ± 0 | 0.2 ± 0.5 |
| CagA-M | CagA-MN | CagA-MC | CagA-MK4 | |
|---|---|---|---|---|
| Average number of beads per field | 3.2 | 3.4 | 2.2 | 1.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tohidpour, A.; Gorrell, R.J.; Roujeinikova, A.; Kwok, T. The Middle Fragment of Helicobacter pylori CagA Induces Actin Rearrangement and Triggers Its Own Uptake into Gastric Epithelial Cells. Toxins 2017, 9, 237. https://doi.org/10.3390/toxins9080237
Tohidpour A, Gorrell RJ, Roujeinikova A, Kwok T. The Middle Fragment of Helicobacter pylori CagA Induces Actin Rearrangement and Triggers Its Own Uptake into Gastric Epithelial Cells. Toxins. 2017; 9(8):237. https://doi.org/10.3390/toxins9080237
Chicago/Turabian StyleTohidpour, Abolghasem, Rebecca J. Gorrell, Anna Roujeinikova, and Terry Kwok. 2017. "The Middle Fragment of Helicobacter pylori CagA Induces Actin Rearrangement and Triggers Its Own Uptake into Gastric Epithelial Cells" Toxins 9, no. 8: 237. https://doi.org/10.3390/toxins9080237