Micro-Surface and -Interfacial Tensions Measured Using the Micropipette Technique: Applications in Ultrasound-Microbubbles, Oil-Recovery, Lung-Surfactants, Nanoprecipitation, and Microfluidics


Abstract
:
Graphical Abstract

1. Introduction
1.1. Franklin and Friends at the Royal Society
“At length being at Clapham where there is, on the common, a large pond, which I observed to be one day very rough with the wind, I fetched out a cruet of oil, and dropt a little of it on the water. I law it spread itself with surprizing swiftness upon the surface; but the effect of smoothing the waves was not produced; for I had applied it first on the leeward side of the pond, where the waves were largest and the wind drove my oil back upon the shore. I then went to the windward side, where they began to form; and there the oil, though not more than a tea spoonful, produced an instant calm over a space several yards square, which spread amazingly, and extended itself gradually till it reached the lee side, making all that quarter of the pond, perhaps half an acre, as smooth as a looking-glass.”
1.2. Surfactant-Colleagues at Unilever and Cambridge
1.3. From BLMs in Cambridge (1983) to DIBs in Oxford (2005) and Beyond
2. Basic Micropipette Manipulation Techniques for Surface and Interfacial Tension Measurement
2.1. Principles of Capillary Action and the Micropipette
2.2. Micropipette Manipulation Apparatus
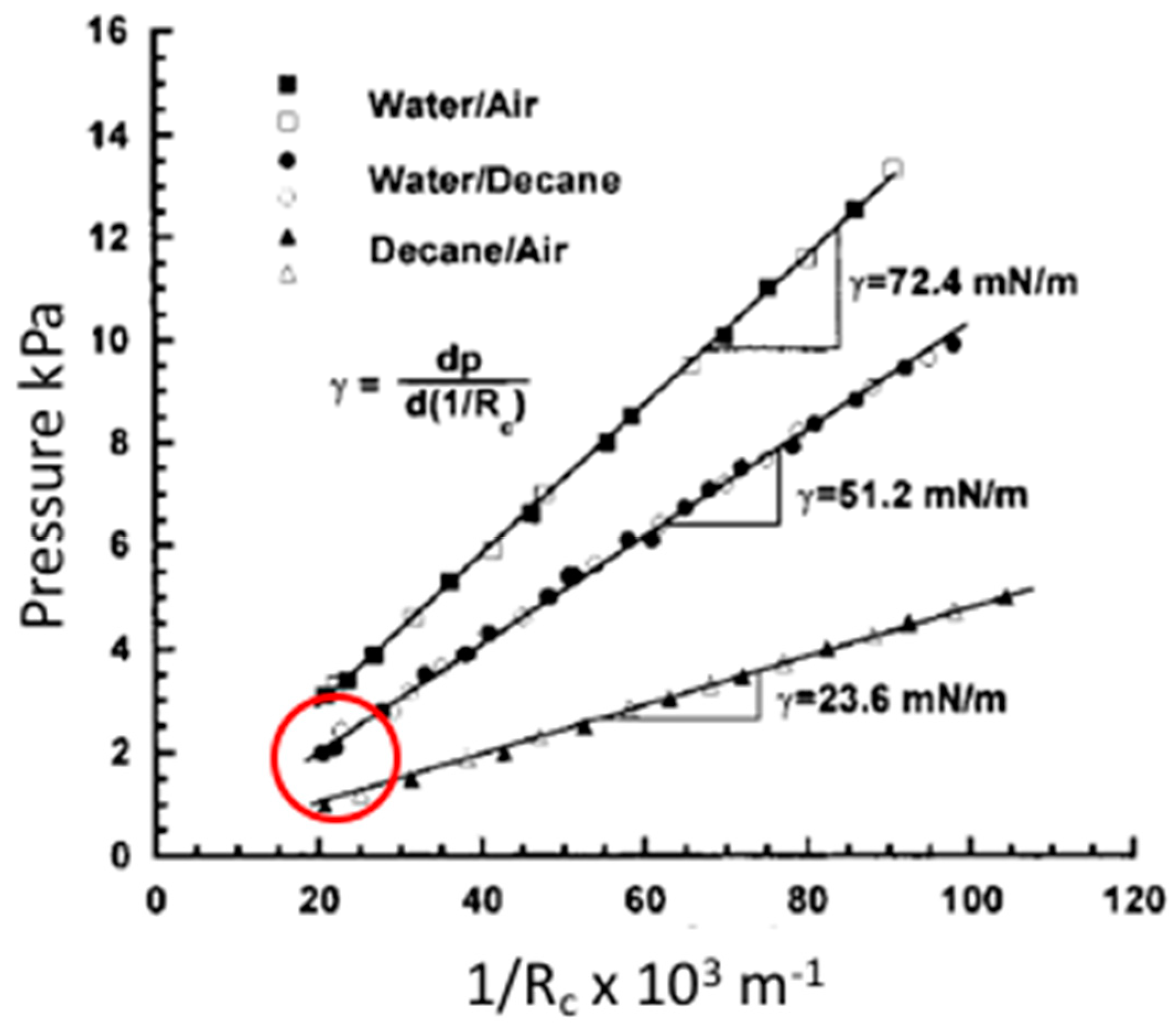
2.3. Gas-Liquid Interfaces
2.4. Liquid-Liquid Interfaces
2.5. More Advanced Techniques: Micropipette Interfacial Area-Expansion Method (MIAM)
3. Entities: Soluble and Insoluble Surfactants as Monolayers, Micelles, Vesicles and Emulsions
3.1. Long-Chain Alcohols form Soluble Monolayers and Emulsions
3.2 Soluble Monolayer: Detergents form Micelles
3.3 Insoluble Monolayer: Lipids form Vesicles
3.4. Surfactants Can Adsorb at Oil-Water Interfaces and the Oil Can Swell Micelles
4. Equilibrium and Dynamic Surface Tension: Adsorption of Soluble Surfactants
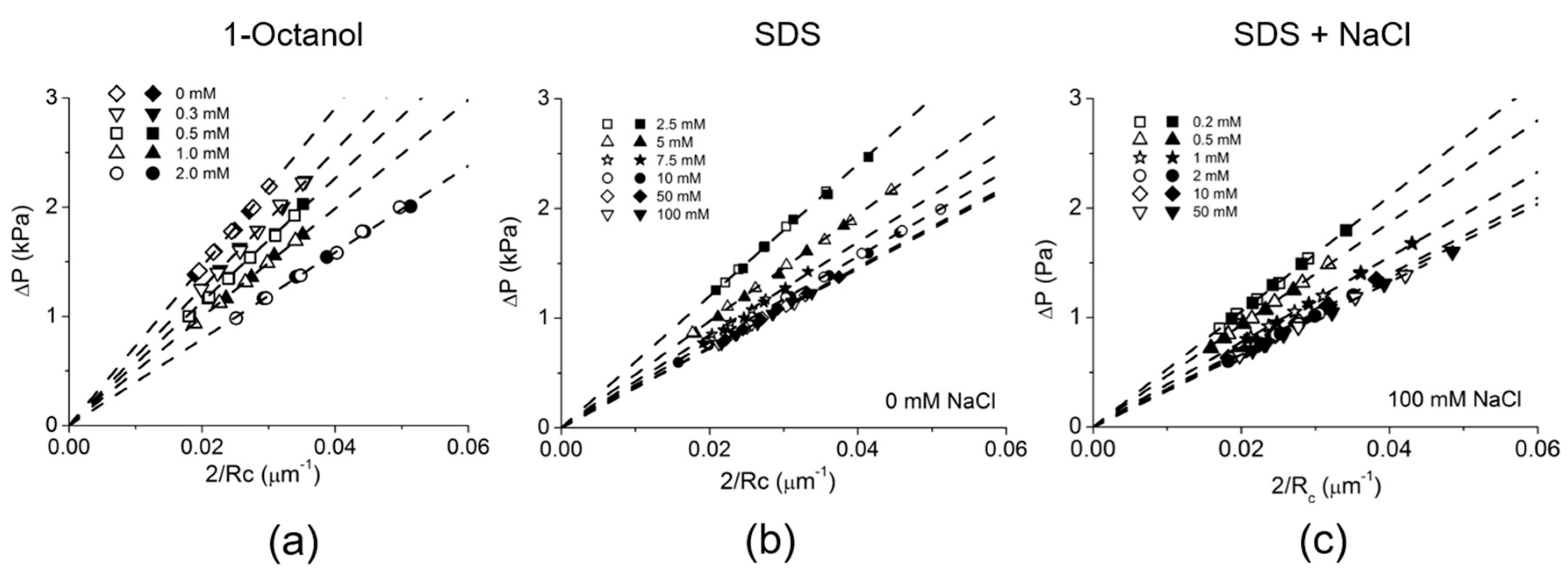
4.1. Equilibrium Surface Tension for Adsorption of Soluble Surfactants: Octanol and Sodium Dodecyl Sulfate
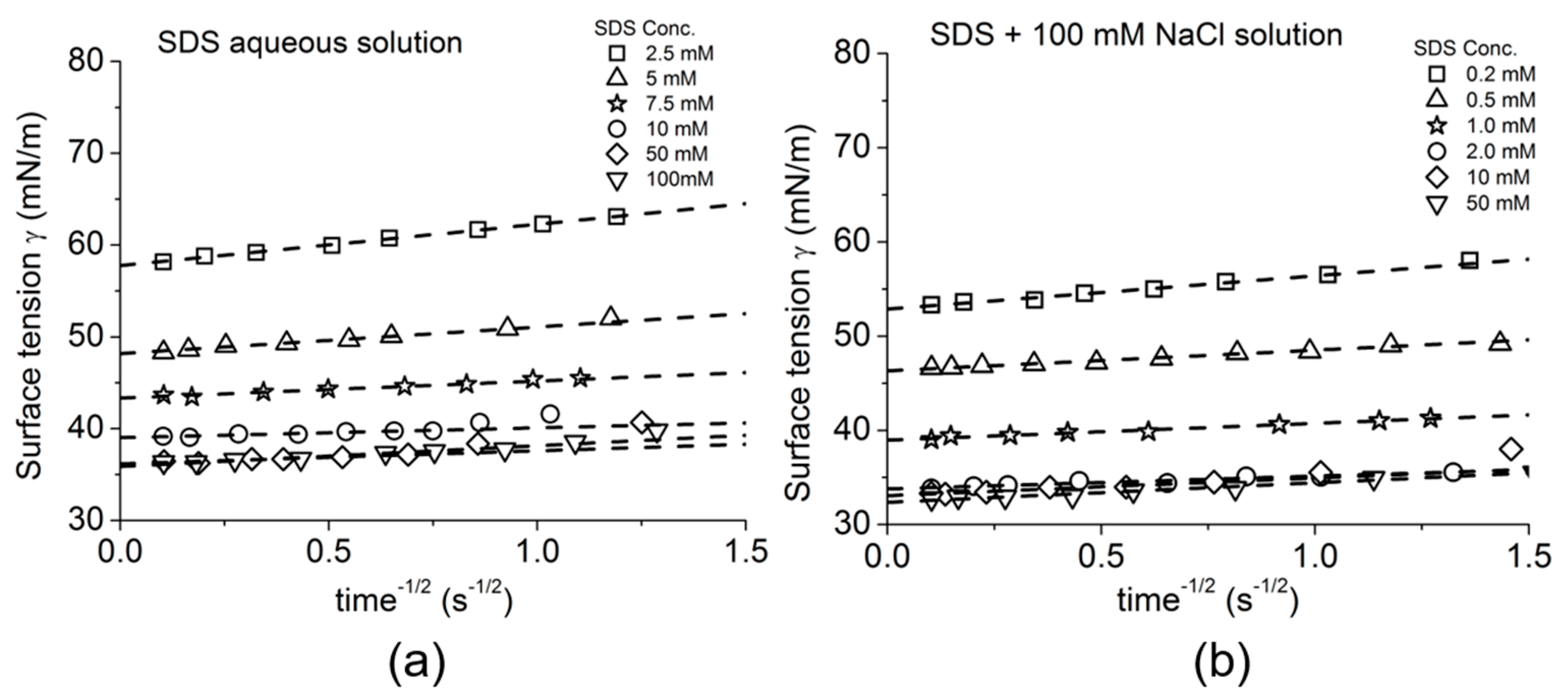
4.2. Dynamic Surface Tension for Adsorption of Soluble Surfactants
4.3. Practical Limits of Theoretical Analysis
4.4. Dynamic Surface Tension: 1-Octanol and SDS Adsorption
4.4.1. 1-Octanol
4.4.2. Sodium Dodecyl Sulfate
4.5. Ward–Tordai Long-Time Adsorption Analyses for 1-Octanol and SDS Adsorption
5. Equilibrium and Dynamic Surface Tension: Adsorption of Insoluble Surfactants
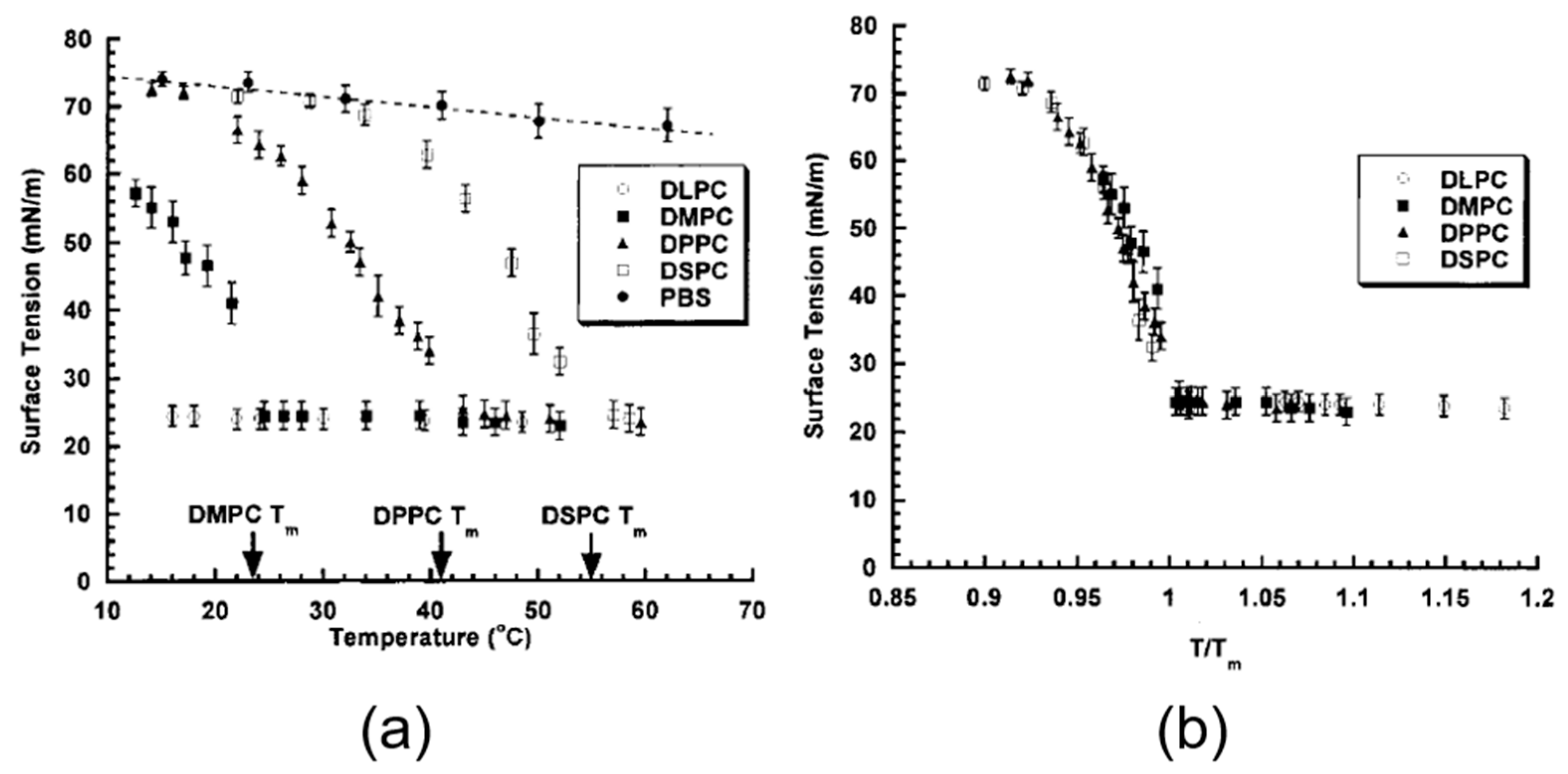
5.1. Equilibrium Surface Tension for Adsorption of Phospholipids vs. Temperature
5.2. Dynamic Surface Tension for Adsorption of Phospholipids
- Pβ’ or ripple phase from 34.45–41.3 °C
- Lβ planar gel phase from 18–34.4 °C
6. Applications in Medical Imaging (Ultrasound), Oil Recovery, Nanoprecipitation, the Biology of the Lung Interface, and Microfluidics
6.1. Gas Micro-Bubbles for Ultrasound Contrast: Surface Tension and their Dissolution into Water at the Scale of the Microbubble
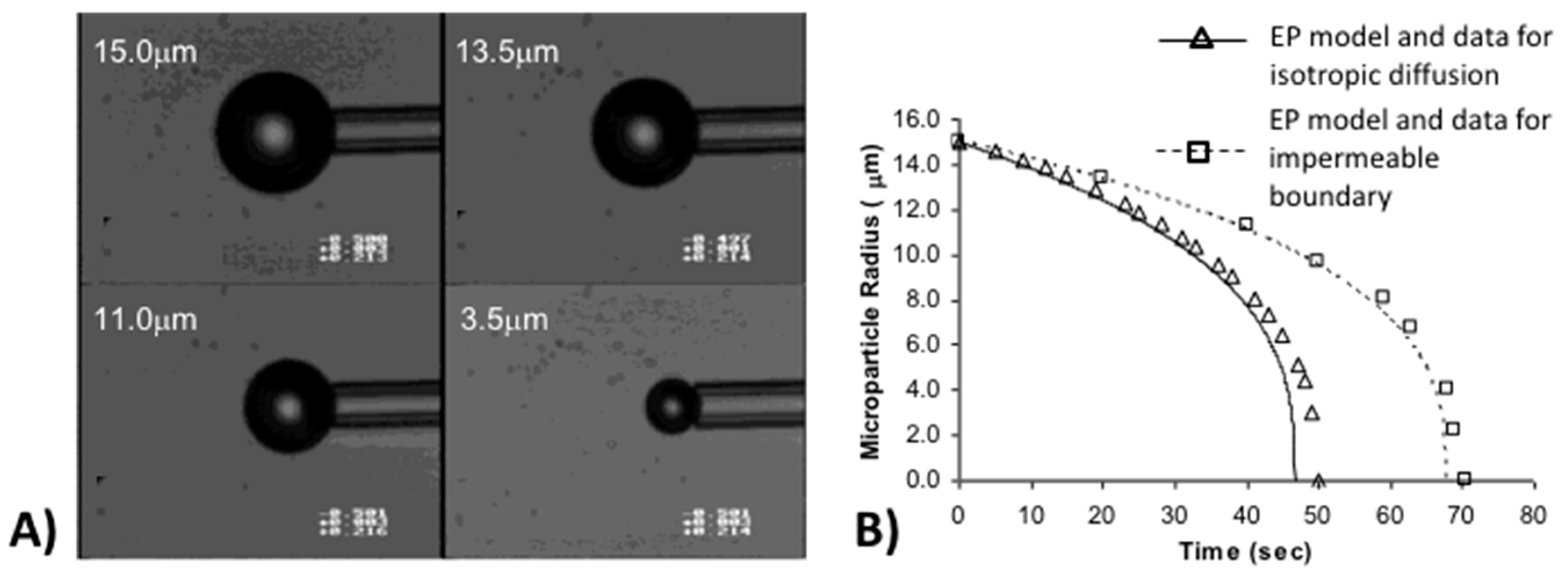
6.1.1. Test of the Epstein–Plesset Model for Gas Microparticle Dissolution in Aqueous Media
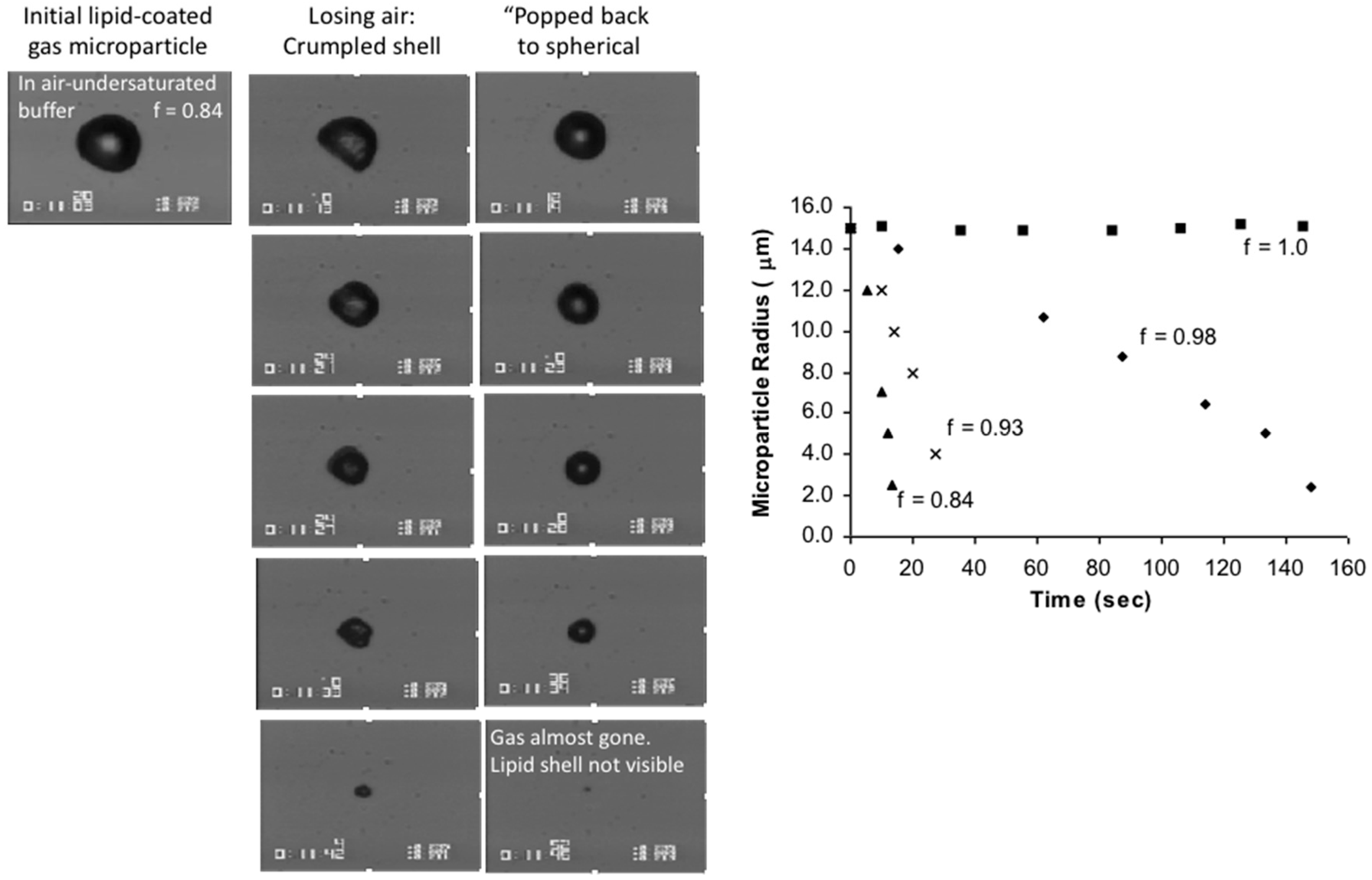
6.1.2. When the Tension-in-the-Surface is Zero: Effect of Gas Saturation in Solution on Microbubble Dissolution
6.2. Surface Tension Measurements of Microdroplets for Oil Recovery
6.3. Surface Tension or “Tension-in-the Surface”: The Biology of the Lung-Air Surface
6.3.1. Lung Surfactant Components
6.3.2. Equilibrium and Dynamic Surface Tensions for the Lung Surfactant Formulations
6.3.3. Morphological Changes of Membranes Formed at the Interface
6.4. Interfacial Tension is a Key Parameter in Nanoprecipitation
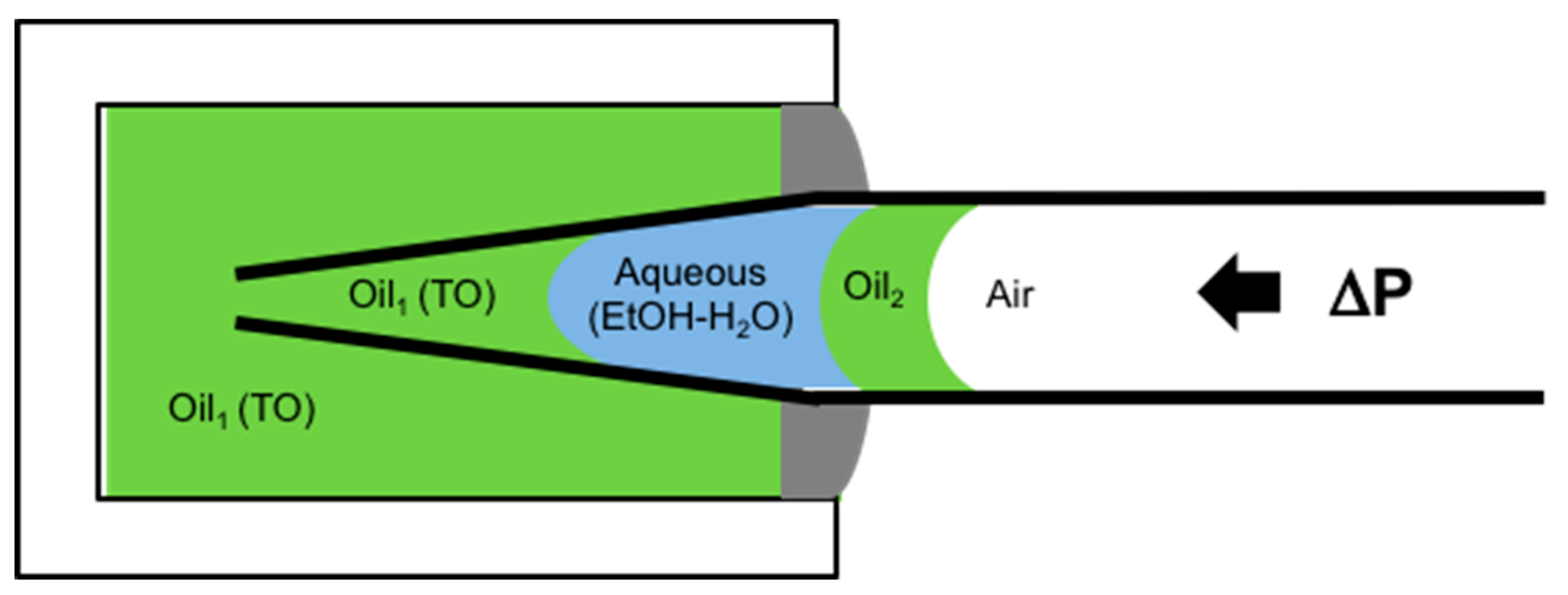
6.4.1. Tapered Micropipette Manipulation Technique for Volatile Oil-Water System (Water-Ethanol Mixtures)
6.4.2. Lipid Adsorption at the Triolein-Ethanol/Water Interface
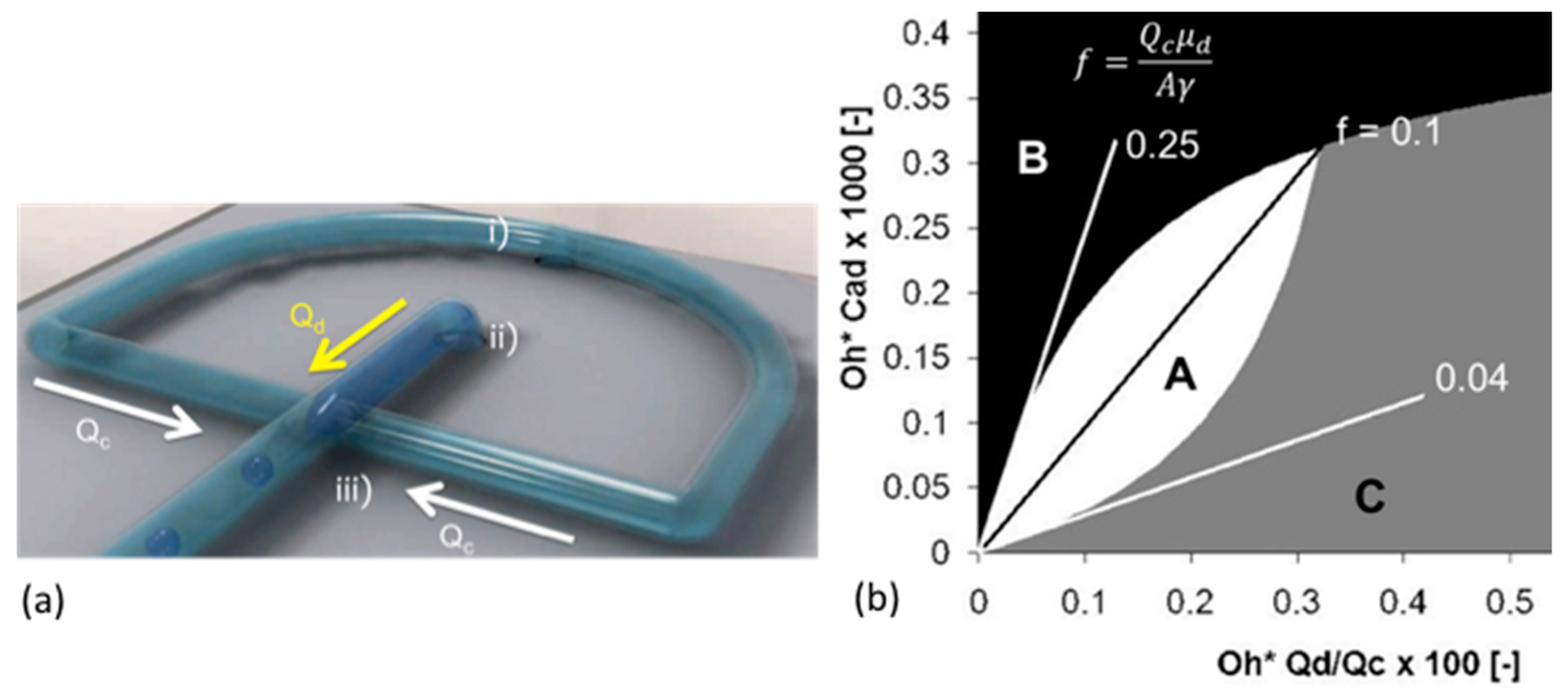
6.5. Microsurface Tensions and Droplet Dissolution for Microfluidics
- An ability to measure all relevant interfacial tensions for each component that tend to be unique to the industrial process and so are not readily obtained from the literature,
- Make individual particles as a function of composition,
- Observe any emerging microstructures inside or on the particles,
- Make measurements of fundamental properties like mechanical deformation of these materials at the single microparticle level and,
- Measure any dissolution (e.g., diffusion coefficients) into the suspending medium.
7. Summary and Conclusions
Acknowledgments
Dedication

Funding
Author Contributions
Conflicts of Interest
References
- Needham, D.; Haydon, D.A. Tensions and free-energies of formation of solventless lipid bilayers—measurement of high contact angles. Biophys. J. 1983, 41, 251–257. [Google Scholar] [CrossRef]
- Honaker, L.W.; Lagerwall, J.P.F.; Jampani, V.S.R. Microfluidic Tensiometry Technique for the Characterization of the Interfacial Tension between Immiscible Liquids. Langmuir 2018, 34, 2403–2409. [Google Scholar] [CrossRef] [PubMed]
- Metz, T.; Paust, N.; Zengerle, R.; Koltay, P. Capillary driven movement of gas bubbles in tapered structures. Microfluid. Nanofluidics 2010, 9, 341–355. [Google Scholar] [CrossRef]
- Renvoisé, P.; Bush, J.W.M.; Prakash, M.; Quéré, D. Drop propulsion in tapered tubes. Epl (Europhys. Lett.) 2009, 86, 64003. [Google Scholar]
- Zhou, H.; Yao, Y.; Chen, Q.; Li, G.; Yao, S. A facile microfluidic strategy for measuring interfacial tension. Appl. Phys. Lett. 2013, 103, 234102. [Google Scholar] [CrossRef]
- Lee, S.; Kim, D.H.; Needham, D. Equilibrium and Dynamic Interfacial Tension Measurements at Microscopic Interfaces Using a Micropipet Technique. 2. Dynamics of Phospholipid Monolayer Formation and Equilibrium Tensions at the Water−Air Interface. Langmuir 2001, 17, 5544–5550. [Google Scholar] [CrossRef]
- Kinoshita, K.; Parra, E.K.; Needham, D. New Sensitive Micro-Measurements of Dynamic Surface Tension and Diffusion Coefficients: Validated and Tested for the Adsorption of 1-Octanol at a Microscopic Air-Water Interface and its Dissolution into Water. J. Colloid Interface Sci. 2017, 488, 166–179. [Google Scholar] [CrossRef]
- Kinoshita, K.; Parra, E.; Needham, D. Adsorption of ionic surfactants at microscopic air-water interfaces using the micropipette interfacial area-expansion method: Measurement of the diffusion coefficient and renormalization of the mean ionic activity for SDS. J. Colloid Interface Sci. 2017, 504, 765–779. [Google Scholar] [CrossRef]
- Lee, S.; Kim, D.H.; Needham, D. Equilibrium and Dynamic Interfacial Tension Measurements at Microscopic Interfaces Using a Micropipet Technique. 1. A New Method for Determination of Interfacial Tension. Langmuir 2001, 17, 5537–5543. [Google Scholar] [CrossRef]
- Parra, E.; Kinoshita, K.; Needham, D. Micropipette Technique Study of Natural and Synthetic Lung Surfactants at the Air-Water Interface: Presence of a SP-B Analog Peptide Promotes Membrane Aggregation, Formation of Tightly Stacked Lamellae, and Growth of Myelin Figures. Langmuir 2016, 32, 10570–10581. [Google Scholar] [CrossRef]
- Yeung, A.; Dabros, T.; Masliyah, J.; Czarnecki, J. Micropipette: A new technique in emulsion research. Colloid Surf. A 2000, 174, 169–181. [Google Scholar] [CrossRef]
- Duncan, P.B.; Needham, D. Test of the Epstein-Plesset model for gas microparticle dissolution in aqueous media: Effect of surface tension and gas undersaturation in solution. Langmuir 2004, 20, 2567–2578. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Costello, M.J.; Duncan, P.B.; Needham, D. Mechanical properties and microstructure of polycrystalline phospholipid monolayer shells: Novel solid microparticles. Langmuir 2003, 19, 8455–8466. [Google Scholar] [CrossRef]
- Kim, D.H.; Klibanov, A.L.; Evans, E.A.; Needham, D. Viscoelastic properties of phospholipid monolayer shells on air microbubbles. Biophys. J. 1998, 74, A313. [Google Scholar]
- Needham, D.; Walke, P.; Utoft, A.; Hervella, P.; Kinoshita, K. Nucleation, Growth and Characterization of Triolein Nanoparticles Prepared in the Absence and Presence of Phospholipids by Rapid Solvent Shifting Technique. 2019; unpubished data, in preparation. [Google Scholar]
- Utoft, A. Micropipette Manipulation Studies: Material Characterization of Multiphase, Multicomponent Systems; University of Southern Denmark: Odense, Denmark, 2017. [Google Scholar]
- Utoft, A.; Kinoshita, K.; Needham, D. Characterization of the ternary triolein-water-ethanol system: Solubility, Interfacial tension and supersaturation. Langmuir 2018, submitted. [Google Scholar]
- Kinoshita, K.; Parra, E.; Hussein, A.; Walke, P.; Bruijn, R.D.; Needham, D. Single Particle Science and Engineering Guiding Scale-Up: Microparticle Formation and Dissolution of Solvent-Emulsion, PLGA, Ibuprofen, and Ibuprofen/PLGA Microspheres. Processes 2016, 4, 49. [Google Scholar] [CrossRef]
- Franklin, B.; Brownrigg, W. Of the stilling of waves by means of oil. Extracted from sundry letters between Benjamin Franklin, LL. D. F. R. S. William Brownrigg, M.D.F.R.S. and the Reverend Mr. Farish. Phil. Trans. 1774, 64, 445–460. [Google Scholar]
- Mertens, J. Oil on troubled waters: Benjamin Franklin and the honor of Dutch Seamen. Phys. Today 2006, 59, 36. [Google Scholar] [CrossRef]
- Tanford, C. The Hydrophobic Effect: Formation of Micelles & Biological Membranes; John Wiley and Sons, Inc.: New York, NY, USA, 1980. [Google Scholar]
- Wikipedia. William Bate Hardy. Available online: https://en.wikipedia.org/wiki/William_Bate_Hardy (accessed on 30 January 2019).
- Hardy, W.B. The tension of composite fluid surfaces and the mechanical stability of films of fluid. P R Soc Lond A-Conta 1912, 86, 610–635. [Google Scholar] [CrossRef]
- Unilever. Our History. Available online: https://www.unilever.co.uk/about/who-we-are/our-history/ (accessed on 30 January 2019).
- Matijevic, E.; Pethica, B.A. The Properties of Ionized Monolayers. 1. Sodium Dodecyl Sulphate at the Air-Water Interface. Trans. Faraday Soc. 1958, 54, 1382–1389. [Google Scholar] [CrossRef]
- Mingins, J.; Zobel, F.G.; Pethica, B.A.; Smart, C. Potential differences due to spread monolayers at the polar oil/water interface. Proc. R. Soc. Lond. A. Math. Phys. Sci. 1971, 324, 99. [Google Scholar] [CrossRef]
- Mingins, J.; Pethica, B.A. Properties of Monolayers at the Air/Water Interface. Part 2? Effect of Temperature on the Double Layer and Water Dipole Contributions to the Surface Potential of Sodium Octadecyl Sulphate. J. Chem. Soc. Faraday Trans. 1 1973, 69, 500–513. [Google Scholar] [CrossRef]
- Taylor, J.A.G.; Mingins, J.; Pethica, B.A.; Tan, B.Y.J.; Jackson, C.M. Phase changes and mosaic formation in single and mixed phospholipid monolayers at the oil-water interface. Biochim. Et Biophys. Acta (Bba) Biomembr. 1973, 323, 157–160. [Google Scholar] [CrossRef]
- Mingins, J.; Owens, N.F.; Taylor, J.A.; Brooks, J.H.; Pethica, B.A. Entropies of Compression of Charged Monolayers at Aqueous Interfaces. In Monolayers; Goddard, E.D., Ed.; ACS: Washington, DC, USA, 1975; Volume 144. [Google Scholar]
- Pethica, B.A.; Standish, M.M.; Mingins, J.; Smart, C.; Iles, D.H.; Feinstein, M.E.; Hossain, S.A.; Pethica, J.B. The Significance of Volta and Compensation States and the Measurement of Surface Potentials of Monolayers. Monolayers 1975, 144, 123–134. [Google Scholar] [CrossRef]
- Pethica, B.A.; Mingins, J.; Taylor, J.A.G. Phospholipid interactions in monolayers. J. Colloid Interface Sci. 1976, 55, 2–8. [Google Scholar] [CrossRef]
- Taylor, J.A.G.; Mingins, J.; Pethica, B.A. Phospholipid monolayers at the n-heptane/water interface. Part 2.—Dilute monolayers of saturated 1,2-diacyl-lecithins and -cephalins. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1976, 72, 2694–2702. [Google Scholar] [CrossRef]
- Yue, B.; Jackson, C.; Taylor, J.A.G.; Mingins, J.; Pethica, B.A. Phospholipid monolayers at nonpolar oil/water interfaces: Phase transition in distearoyl-lecithin films at the n-heptane aqueous sodium chloride interface. J. Chem. Soc. Faraday Trans. 1976, 72, 2685–2693. [Google Scholar] [CrossRef]
- Hall, D.G.; Pethica, B.A. Thermodynamics of the Volta effect for surface films. Proc. R. Soc. Lond. A. Math. Phys. Sci. 1977, 354, 425. [Google Scholar] [CrossRef]
- Pethica, B.A.; Glasser, M.L.; Mingins, J. Intermolecular forces in monolayers at air/water interfaces. J. Colloid Interface Sci. 1981, 81, 41–51. [Google Scholar] [CrossRef]
- Middleton, S.R.; Pallas, N.R.; Mingins, J.; Pethica, B.A. Thermodynamics of Ionized Monolayers: Surface Manometry on Very Low Density Spread Monolayers of Sodium Octadecyl Sulfate at the Air/Water Interface and Analysis of Ionic Double Layer Contributions to the Isotherms. J. Phys. Chem. C 2011, 115, 8056–8063. [Google Scholar] [CrossRef]
- Keynes, R.D. Denis Arthur Haydon, 21 February 1930–29 November 1988. Biogr. Mems Fell. R. Soc. 1990, 36, 199–216. [Google Scholar]
- White, S.H. Formation of "solvent-free" black lipid bilayer membranes from glyceryl monooleate dispersed in squalene. Biophys. J. 1978, 23, 337–347. [Google Scholar] [CrossRef]
- Taylor, J.; Haydon, D.A. Stabilization of thin films of liquid hydrocarbon by alkyl chain interaction. Disc. Faraday Soc. 1966, 42, 51. [Google Scholar] [CrossRef]
- Requena, J.; Billett, D.F.; Haydon, D.A. Van der Waals forces in oil-water systems from the study of thin lipid films. I. Measurement of the contact angle and the estimation of the van der Waals free energy of thinning of a film. Proc. Roy. Soc. Lond. A. Math. Phys. Sci. 1975, 347. [Google Scholar] [CrossRef]
- Gruen, D.W.R. A mean-field model of the alkane-saturated lipid bilayer above its phase transition. I. Development of the model. Biophys. J. 1981, 33, 149–166. [Google Scholar] [CrossRef]
- Gruen, D.W.R.; Haydon, D.A. A mean-field model of the alkane-saturated lipid bilayer above its phase transition. II. Results and comparison with experiment. Biophys. J. 1981, 33, 167–188. [Google Scholar] [CrossRef]
- Bayley, H.; Cronin, B.; Heron, A.; Holden, M.A.; Hwang, W.L.; Syeda, R.; Thompson, J.; Wallace, M. Droplet interface bilayers. Mol. Biosyst. 2008, 4, 1191–1208. [Google Scholar] [CrossRef] [PubMed]
- Holden, M.A.; Needham, D.; Bayley, H. Functional bionetworks from nanoliter water droplets. J. Am. Chem. Soc. 2007, 129, 8650–8655. [Google Scholar] [CrossRef] [PubMed]
- Bayley, H.; Holden, M.; Heron, A.; Needham, D. Formation of Bilayers of Amphpathic Molecules. WO 2008/012552, 2008. [Google Scholar]
- Sarles, S.A.; Leo, D.J. Tailored Current—Voltage Relationships of Droplet-Interface Bilayers Using Biomolecules and External Feedback Control. J. Intell. Mater. Syst. Struct. 2009, 20, 1233–1247. [Google Scholar] [CrossRef]
- Needham, D. Lipid structures: A brief history of multisomes. Nat. Nanotechnol. 2011, 6, 761–792. [Google Scholar] [CrossRef] [PubMed]
- Villar, G.; Graham, A.D.; Bayley, H. A tissue-like printed material. Science 2013, 340, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Villar, G.; Heron, A.J.; Bayley, H. Formation of droplet networks that function in aqueous environments. Nat. Nanotechnol. 2011, 6, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Trantidou, T.; Friddin, M.S.; Salehi-Reyhani, A.; Ces, O.; Elani, Y. Droplet microfluidics for the construction of compartmentalised model membranes. Lab A Chip 2018, 18, 2488–2509. [Google Scholar] [CrossRef] [PubMed]
- Challita, E.J.; Najem, J.S.; Monroe, R.; Leo, D.J.; Freeman, E.C. Encapsulating Networks of Droplet Interface Bilayers in a Thermoreversible Organogel. Sci. Rep. 2018, 8, 6494. [Google Scholar] [CrossRef] [PubMed]
- Hochmuth, R.M.; Mohandas, N. Uniaxial loading of the red-cell membrane. J. Biomech. 1972, 5, 501–509. [Google Scholar] [CrossRef]
- Evans, E.A. A new material concept for the red cell membrane. Biophys. J. 1973, 13, 926. [Google Scholar] [CrossRef]
- Evans, E.A. New Membrane Concept Applied to the Analysis of Fluid Shear- and Micropipette-Deformed Red Blood Cells. Biophys J. 1973, 13, 941–954. [Google Scholar] [CrossRef]
- Skalak, R.; Tozeren, A.; Zarda, R.P.; Chien, S. Strain Energy Function of Red Blood Cell Membranes. Biophys. J. 1973, 13, 245–264. [Google Scholar] [CrossRef]
- Evans, E.; Hochmuth, R.M. A solid-liquid composite model of the red blood cell membrane. J. Membr. Biol. 1977, 30, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.; Sung, K.L.; Skalak, R.; Usami, S.; Tözeren, A. Theoretical and experimental studies on viscoelastic properties of erythrocyte membrane. Biophys. J. 1978, 24, 463–487. [Google Scholar] [CrossRef]
- Evans, E.; Hochmuth, R.M. Mechanochemical Properties of Membranes. In Current Topics in Membranes and Transport; Bonner, F., Kleinzeller, A., Eds.; Academic Press: New York, NY, USA, 1978; Volume 10, p. 1. [Google Scholar]
- Evans, E.; Skalak, R. Mechanics and Thermodynamics of Biomembranes; CRC: Boca Raton, FL, USA, 1980. [Google Scholar]
- Tözeren, H.; Chien, S.; Tözeren, A. Estimation of viscous dissipation inside an erythrocyte during aspirational entry into a micropipette. Biophys. J. 1984, 45, 1179–1184. [Google Scholar] [CrossRef]
- Meiselman, H.J.; Lichtman, M.A.; LaCelle, P.L. White cell mechanics: basic science and clinical aspects. In Proceedings of the a Symposium Held at the Kroc Foundation, Santa Ynez Valley, CA, USA, 2–6 May 1984. [Google Scholar]
- Dong, C.; Skalak, R.; Sung, K.L.P.; Schmid-Schonbein, G.W.; Chien, S. Passive deformation analysis of human leukocytes. J. Biomech. Eng. 1988, 110, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Evans, E. Micromethods for measurement of deformability and adhesivity properties of blood cells and synthetic membrane vesicles. In Physical Basis of Cell-Cell Adhesion; Bongrand, P., Ed.; CRC: Boca Raton, FL, USA, 1988. [Google Scholar]
- Sung, K.; Dong, C.; Schmid-Schoenbein, G.W.; Chien, S.; Skalak, R. Leukocyte relaxation properties. Biophys. J. 1988, 54, 331. [Google Scholar] [CrossRef]
- Needham, D.; Armstrong, M.; Hatchell, D.L.; Nunn, R.S. Rapid deformation of passive polymorphonuclear leukocytes—The effects of pentoxifylline. J. Cell. Physiol. 1989, 140, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Hochmuth, R.M.; Needham, D. The viscosity of neutrophils and their transit times through small pores. Biorheology 1990, 27, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Needham, D.; Hochmuth, R.M. Rapid flow of passive neutrophils into a 4 mu-m pipette and measurement of cytoplasmic viscosity. J. Biomech. Eng. Trans. Asme 1990, 112, 269–276. [Google Scholar] [CrossRef]
- Needham, D. Possible role of cell cycle-dependent morphology, geometry, and mechanical properties in tumor cell metastasis. Cell Biophys. 1991, 18, 99–121. [Google Scholar] [CrossRef]
- Transontay, R.; Needham, D.; Yeung, A.; Hochmuth, R.M. Time-dependent recovery of passive neutrophils after large deformation. Biophys. J. 1991, 60, 856–866. [Google Scholar] [CrossRef]
- Sonkin, P.L.; Freedman, S.F.; Needham, D.; Rao, K.M.K.; Hatchell, D.L. Pentoxifylline modulates deformability, f-actin content, and superoxide anion production of polymorphonuclear leukocytes from diabetic cats. Exp. Eye Res. 1992, 55, 831–838. [Google Scholar] [CrossRef]
- Hochmuth, R.M.; Tingbeall, H.P.; Beaty, B.B.; Needham, D.; Transontay, R. Viscosity of passive human neutrophils undergoing small deformations. Biophys. J. 1993, 64, 1596–1601. [Google Scholar] [CrossRef]
- Evans, E.; Kwok, R. Mechanical calorimetry of large dimyristoylphosphatidylcholine vesicles in the phase transition region. Biochemistry 1982, 21, 4874–4879. [Google Scholar] [CrossRef]
- Evans, E.; Needham, D. Physical-properties of surfactant bilayer-membranes—Thermal transitions, elasticity, rigidity, cohesion, and colloidal interactions. J. Phys. Chem-Us 1987, 91, 4219–4228. [Google Scholar] [CrossRef]
- Evans, E.; Rawicz, W. Entropy-driven tension and bending elasticity in condensed-fluid membranes. Phys. Rev. Lett. 1990, 64, 2094–2097. [Google Scholar] [CrossRef]
- Evans, E.; Rawicz, W.; Hofmann, A.F. Lipid bilayer expansion and mechanical disruption in solutions of water soluble bile acid. In Proceedings of the XIII International Bile Salt Meeting. Bile Acids in Gastroenterology: Basic & Clinical AdvancesFalk Symposium, San Diego, CA, USA, 30 September–2 October 1994. [Google Scholar]
- Needham, D.; Evans, E. Structure and Mechanical-Properties of Giant Lipid (DMPC) Vesicle Bilayers from 20 °C below to 10 °C above the liquid crystal-crystalline phase transition at 24° C. Biochemistry 1988, 27, 8261–8269. [Google Scholar] [CrossRef]
- Needham, D.; McIntosh, T.J.; Evans, E.A. Thermomechanical and transition properties of dimyristoylphosphatidylcholine/cholesterol bilayers. Biochemistry 1988, 27, 4668–4673. [Google Scholar] [CrossRef]
- Needham, D.; Nunn, R.S. Elastic deformation and failure of lipid bilayer membranes containing cholesterol. Biophys. J. 1990, 58, 997–1009. [Google Scholar] [CrossRef]
- Needham, D.; Stoicheva, N.; Zhelev, D.V. Exchange of monooleoylphosphatidylcholine as monomer and micelle with membranes containing poly(ethylene glycol)-lipid. Biophys. J. 1997, 73, 2615–2629. [Google Scholar] [CrossRef]
- Needham, D.; Zhelev, D. The mechanochemistry of lipid vesicles examined by micropipet manipulation techniques. In Vesicles; Rosoff, M., Ed.; Marcell Dekker: New York, NY, USA, 1996; Volume 62, p. 373. [Google Scholar]
- Noppl-Simson, D.; Needham, D. Avidin-Biotin interactions at vesicle surfaces: surface binding, cross-bridge formation and lateral interactions. Biophys. J. 1996, 70, 1391–1401. [Google Scholar] [CrossRef]
- Olbrich, K.C. Water Permeability and Mechanical Properties of Unsaturated Lipid Membranes and Sarcolemmal Vesicles; Duke University: Durham, UK, 1997. [Google Scholar]
- Rawicz, W.; Olbrich, K.C.; McIntosh, T.; Needham, D.; Evans, E. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys. J. 2000, 79, 328–339. [Google Scholar] [CrossRef]
- Zhelev, D.V.; Needham, D.; Hochmuth, R.M. A novel micropipet method for measuring the bending modulus of vesicle membranes. Biophys. J. 1994, 67, 720–727. [Google Scholar] [CrossRef]
- Walton, A. Surface tension and capillary rise. Phys. Educ. 1972, 7, 491. [Google Scholar] [CrossRef]
- Birdi, K.S.; Vu, D.T.; Winter, A.; Norregard, A. Capillary Rise of Liquids in Rectangular Tubings. Colloid Polym. Sci. 1988, 266, 470–474. [Google Scholar] [CrossRef]
- Adamson, A.W.; Gast, A.P. Physical Chemistry of Surface; John Wiley and Sons: New York, NY, USA, 1997. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.R.; Ferris, L.A.; Thalladi, V.R. Selective growth of a stable drug polymorph by suppressing the nucleation of corresponding metastable polymorphs. Angew. Chem. Int. Ed. 2007, 46, 4333–4336. [Google Scholar] [CrossRef] [PubMed]
- Vargaftik, N.B.V.; Voljak, L.D. International Table of the Surface Tension of Water. J. Phys. Chem. Ref. Data 1983, 12, 817–820. [Google Scholar] [CrossRef]
- Demond, A.H.; Lindner, A.S. Estimation of Interfacial-Tension between Organic Liquids and Water. Environ. Sci. Technol. 1993, 27, 2318–2331. [Google Scholar] [CrossRef]
- U.S. Coast Guard. CHRIS—Hazardous Chemical Data; U.S. Government Printing Office: Washington, DC, USA, 1978.
- Israelachvili, J.N. Intermolecular and Surface Force, 3rd ed.; Academic Press: New York, NY, USA, 2011. [Google Scholar]
- Vazquez, G.; Alvarez, E.; Navaza, J.M. Surface-Tension of Alcohol Plus Water from 20-Degrees-C to 50-Degrees-C. J. Chem. Eng. Data 1995, 40, 611–614. [Google Scholar] [CrossRef]
- Su, J.T.; Needham, D. Mass Transfer in the Dissolution of a Multicomponent Liquid Droplet in an Immiscible Liquid Environment. Langmuir 2013, 29, 13339–13345. [Google Scholar] [CrossRef] [PubMed]
- Epstein, P.S.; Plesset, M.S. On the Stability of Gas Bubbles in Liquid-Gas Solutions. J. Chem. Phys. 1950, 18, 1409–1505. [Google Scholar] [CrossRef]
- Einstein, A. On the Motion of Small Particles Suspended in Liquids at Rest Required by the Molecular-Kinetic Theory of Heat. Ann. Der Phys. 1905, 17, 549–560. [Google Scholar] [CrossRef]
- Phillips, J.N. The Energetics of Micelle Formation. T Faraday Soc 1955, 51, 561–569. [Google Scholar] [CrossRef]
- Pisárčik, M.; Devínsky, F.; Pupák, M. Determination of micelle aggregation numbers of alkyltrimethylammonium bromide and sodium dodecyl sulfate surfactants using time-resolved fluorescence quenching. Open Chem. 2015, 13, 922–931. [Google Scholar] [CrossRef]
- Tausk, R.J.; Karmiggelt, J.; Oudshoorn, C.; Overbeek, J.T. Physical chemical studies of short-chain lecithin homologues. I. Influence of the chain length of the fatty acid ester and of electrolytes on the critical micelle concentration. Biophys. Chem. 1974, 1, 175–183. [Google Scholar] [CrossRef]
- Mohwald, H. Phospholipid Monolayers. In Handbook of Biological Physics; Lipowsky, R., Sackmann, E., Eds.; Elsevier Science B.V.: Amsterdam, The Netherlands, 1995; Volume 1. [Google Scholar]
- Albrecht, O.; Gruler, H.; Sackmann, E. Polymorphism of phospholipid monolayers. J. Phys. 1978, 39, 301–313. [Google Scholar] [CrossRef]
- Smith, R.; Tanford, C. The critical micelle concentration of l-dipalmitoylphosphatidylcholine in water and water-methanol solutions. J. Mol. Biol. 1972, 67, 75–83. [Google Scholar] [CrossRef]
- Kinoshita, K.; Yamazaki, M. Organic solvents induce interdigitated gel structures in multilamellar vesicles of dipalmitoylphosphatidylcholine. Bba-Biomembranes 1996, 1284, 233–239. [Google Scholar] [CrossRef]
- Kinoshita, K.; Yamazaki, M. Phase transition between hexagonal II (H-II) and liquid-crystalline phase induced by interaction between solvents and segments of the membrane surface of dioleoylphosphatidylethanolamine. Bba-Biomembranes 1997, 1330, 199–206. [Google Scholar] [CrossRef]
- Kinoshita, K.; Furuike, S.; Yamazaki, M. Intermembrane distance in multilamellar vesicles of phosphatidylcholine depends on the interaction free energy between solvents and the hydrophilic segments of the membrane surface. Biophys. Chem. 1998, 74, 237–249. [Google Scholar] [CrossRef]
- Cullis, P.R.; Mayer, L.D.; Bally, M.B.; Madden, T.D.; Hope, M.J. Generating and loading of liposomal systems for drug-delivery applications. Adv. Drug Deliv. Rev. 1989, 3, 267–282. [Google Scholar] [CrossRef]
- Needham, D.; Zhelev, D. Chapter 9. Use of Micropipet Manipulation Techniques to Measure the Properties of Giant Lipid Vesicles. In Giant Vesicles; Luisi, P.L., Walde, P., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 2000; pp. 103–147. [Google Scholar]
- Parra, E.; Needham, D. Mechanic assays based on micropipette aspiration. In The Giant Vesicle Book; Marques, C., Dimova, R., Eds.; CRC Press—Taylor & Francis: Boca Raton, FL, USA, 2018; in press. [Google Scholar]
- Mingins, J.; Taylor, J.A.G.; Pethica, B.A.; Jackson, C.M.; Yue, B.Y.T. Phospholipid monolayers at non-polar oil/water interfaces. Part 3.—Effect of chain length on phase transitions in saturated di-acyl lecithins at the n-heptane/aqueous sodium chloride interface. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1982, 78, 323–339. [Google Scholar] [CrossRef]
- Haydon, D.A.; Elliott, J.R. Surface potential changes in lipid monolayers and the ‘cut-off’ in anaesthetic effects of N-alkanols. Biochim. Et Biophys. Acta (Bba)-Biomembr. 1986, 863, 337–340. [Google Scholar] [CrossRef]
- Mueller, P.; Rudin, D.O.; Ti Tien, H.; Wescott, W.C. Reconstitution of Cell Membrane Structure in vitro and its Transformation into an Excitable System. Nature 1962, 194, 979. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.R.; Needham, D.; Dilger, J.P.; Haydon, D.A. The effects of bilayer thickness and tension on gramicidin single-channel lifetime. Biochim. Et Biophys. Acta (Bba)-Biomembr. 1983, 735, 95–103. [Google Scholar] [CrossRef]
- Hladky, S.B.; Haydon, D.A. Ion transfer across lipid membranes in the presence of gramicidin A: I. Studies of the unit conductance channel. Biochim. Et Biophys. Acta (Bba)-Biomembr. 1972, 274, 294–312. [Google Scholar] [CrossRef]
- Baoukina, S.; Monticelli, L.; Risselada, H.J.; Marrink, S.J.; Tieleman, D.P. The molecular mechanism of lipid monolayer collapse. Proc. Natl. Acad. Sci. USA 2008, 105, 10803–10808. [Google Scholar] [CrossRef] [PubMed]
- Girard-Egrot, A.; Blum, L. Langmuir-Blodgett Technique for Synthesis of Biomimetic Lipid Membranes. In Nanobiotechnology of Biomimetic Membranes, Martin, D., Ed.; Springer: Basel, Switzerland, 2007; pp. 23–74. [Google Scholar]
- MacDonald, R.C.; Simon, S.A. Lipid monolayer states and their relationships to bilayers. Proc. Natl. Acad. Sci. USA 1987, 84, 4089–4093. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.; Needham, D. Giant vesicle bilayers composed of mixtures of lipids, cholesterol and polypeptides—Thermomechanical and (mutual) adherence properties. Faraday Discuss. 1986, 81, 267–280. [Google Scholar] [CrossRef]
- Needham, D. Measurement of interbilayer adhesion energies. Methods Enzymol. 1993, 220, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.; Rawicz, W.; Smith, B.A. Concluding remarks back to the future: Mechanics and thermodynamics of lipid biomembranes. Faraday Discuss. 2013, 161, 591–611. [Google Scholar] [CrossRef]
- Casals, C.; Canadas, O. Role of lipid ordered/disordered phase coexistence in pulmonary surfactant function. Bba-Biomembranes 2012, 1818, 2550–2562. [Google Scholar] [CrossRef]
- Zuo, Y.Y.; Veldhuizen, R.A.; Neumann, A.W.; Petersen, N.O.; Possmayer, F. Current perspectives in pulmonary surfactant--inhibition, enhancement and evaluation. Biochim. Et Biophys. Acta 2008, 1778, 1947–1977. [Google Scholar] [CrossRef] [PubMed]
- Keating, E.; Zuo, Y.Y.; Tadayyon, S.M.; Petersen, N.O.; Possmayer, F.; Veldhuizen, R.A. A modified squeeze-out mechanism for generating high surface pressures with pulmonary surfactant. Biochim. Et Biophys. Acta 2012, 1818, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.G.; Perez-Gil, J. Protein-lipid interactions and surface activity in the pulmonary surfactant system. Chem. Phys. Lipids 2006, 141, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Taeusch, H.W.; Bernardino de la Serna, J.; Perez-Gil, J.; Alonso, C.; Zasadzinski, J.A. Inactivation of pulmonary surfactant due to serum-inhibited adsorption and reversal by hydrophilic polymers: Experimental. Biophys. J. 2005, 89, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Hauser, H. Short-chain phospholipids as detergents. Biochim. Et Biophys. Acta (Bba)-Biomembr. 2000, 1508, 164–181. [Google Scholar] [CrossRef]
- Israelachvili, J. The Science and Applications of Emulsions—An Overview. Colloid Surf. A 1994, 91, 1–8. [Google Scholar] [CrossRef]
- Lee, J.; Needham, D. Swollen Micelles: When does Bulk Matter Appear? Duke University: Durham, NC, USA, 2000. [Google Scholar]
- Haynes, W.M. CRC Handbook of Chemistry and Physics; Haynes, W.M., Ed.; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Hommelen, J.R. The elimination of errors due to evaporation of the solute in the determination of surface tensions. J. Colloid Sci. 1959, 14, 385–400. [Google Scholar] [CrossRef]
- Chang, C.H.; Wang, N.H.L.; Franses, E.I. Adsorption Dynamics of Single and Binary Surfactants at the Air-Water-Interface. Colloid Surf. 1992, 62, 321–332. [Google Scholar] [CrossRef]
- Elworthy, P.H.A.K.J.M. The surface tension of sodium dodecylsulfate solutions and the phase separation model of micelle formation. J. Colloid Interface Sci. 1966, 21, 331–347. [Google Scholar] [CrossRef]
- Christov, N.C.; Danov, K.D.; Kralchevsky, P.A.; Ananthapadmanabhan, K.P.; Lips, A. Maximum bubble pressure method: Universal surface age and transport mechanisms in surfactant solutions. Langmuir 2006, 22, 7528–7542. [Google Scholar] [CrossRef] [PubMed]
- Casandra, A.; Ismadji, S.; Noskov, B.A.; Liggieri, L.; Lin, S.Y. A study on the method of short-time approximation—Criteria for applicability. Int. J. Heat Mass. Trans. 2015, 90, 752–760. [Google Scholar] [CrossRef]
- Eastoe, J.; Dalton, J.S. Dynamic surface tension and adsorption mechanisms of surfactants at the air-water interface. Adv. Colloid Interface Sci. 2000, 85, 103–144. [Google Scholar] [CrossRef]
- Fainerman, V.B.; Miller, R. Dynamic Surface-Tension Measurements in the Submillisecond Range. J. Colloid Interface Sci. 1995, 175, 118–121. [Google Scholar] [CrossRef]
- Fainerman, V.B.; Miller, R. Adsorption kinetics of short-chain alcohols at the water/air interface: Diffusion-controlled adsorption under the conditions of a nonequilibrium surface layer. J. Colloid Interface Sci. 1996, 178, 168–175. [Google Scholar] [CrossRef]
- Fainerman, V.B.; Mys, V.D.; Makievski, A.V.; Petkov, J.T.; Miller, R. Dynamic surface tension of micellar solutions in the millisecond and submillisecond time range. J. Colloid Interface Sci. 2006, 302, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Wang, W.J.; Hsu, C.T. Adsorption kinetics of 1-octanol at the air-water interface. Langmuir 1997, 13, 6211–6218. [Google Scholar] [CrossRef]
- Ozdemir, O.; Karaguzel, C.; Nguyen, A.V.; Celik, M.S.; Miller, J.D. Contact angle and bubble attachment studies in the flotation of trona and other soluble carbonate salts. Miner. Eng. 2009, 22, 168–175. [Google Scholar] [CrossRef]
- Macleod, C.A.; Radke, C.J. A Growing Drop Technique for Measuring Dynamic Interfacial-Tension. J. Colloid Interface Sci. 1993, 160, 435–448. [Google Scholar] [CrossRef]
- Zhang, X.G.; Harris, M.T.; Basaran, O.A. Measurement of Dynamic Surface-Tension by a Growing Drop Technique. J. Colloid Interface Sci. 1994, 168, 47–60. [Google Scholar] [CrossRef]
- Bleys, G.; Joos, P. Adsorption-Kinetics of Bolaform Surfactants at the Air Water Interface. J Phys Chem-Us 1985, 89, 1027–1032. [Google Scholar] [CrossRef]
- Defay, R.; Hommelen, J.R.I. Measurement of Dynamic Surface Tensions of Aqueous Solutions by the Oscilating Jet Method. J. Colloid Sci. 1958, 13, 553–564. [Google Scholar] [CrossRef]
- Horozov, T.S.; Dushkin, C.D.; Danov, K.D.; Arnaudov, L.N.; Velev, O.D.; Mehreteab, A.; Broze, G. Effect of the surface expansion and wettability of the capillary on the dynamic surface tension measured by the maximum bubble pressure method. Colloids Surf. A Physicochem. Eng. Asp. 1996, 113, 117–126. [Google Scholar] [CrossRef]
- Ma, G.; Allen, H.C. DPPC Langmuir monolayer at the air-water interface: Probing the tail and head groups by vibrational sum frequency generation spectroscopy. Langmuir 2006, 22, 5341–5349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Fan, Q.H.; Wang, Y.E.; Neal, C.R.; Zuo, Y.Y. Comparative study of clinical pulmonary surfactants using atomic force microscopy. Bba-Biomembranes 2011, 1808, 1832–1842. [Google Scholar] [CrossRef] [PubMed]
- Putz, G.; Walch, M.; Van Eijk, M.; Haagsman, H.P. A spreading technique for forming film in a captive bubble. Biophys. J. 1998, 75, 2229–2239. [Google Scholar] [CrossRef]
- Schurch, S.; Bachofen, H.; Goerke, J.; Possmayer, F. A Captive Bubble Method Reproduces the Insitu Behavior of Lung Surfactant Monolayers. J. Appl. Physiol. 1989, 67, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Franses, E.I. Dynamic Tension Behavior of Aqueous Octanol Solutions under Constant-Area and Pulsating-Area Conditions. Chem. Eng. Sci. 1994, 49, 313–325. [Google Scholar] [CrossRef]
- Ward, A.F.H.; Tordai, L. Time-Dependence of Boundary Tensions of Solutions. 1. The Role of Diffusion in Time-Effects. J. Chem. Phys. 1946, 14, 453–461. [Google Scholar] [CrossRef]
- Fainerman, V.B.; Makievski, A.V.; Miller, R. The Analysis of Dynamic Surface-Tension of Sodium Alkyl Sulfate-Solutions, Based on Asymptotic Equations of Adsorption Kinetic-Theory. Colloid Surf. A 1994, 87, 61–75. [Google Scholar] [CrossRef]
- Li, X.; Shaw, R.; Evans, G.M.; Stevenson, P. A simple numerical solution to the Ward–Tordai equation for the adsorption of non-ionic surfactants. Comput. Chem. Eng. 2010, 34, 146–153. [Google Scholar] [CrossRef]
- Chien-Hsiang Chang, E.I.F. Adsorption dynamics of surfactants at the air/water interface: A critical review of mathematical models, data, and mechanisms. Colloid Surf. 1995, 100, 1–45. [Google Scholar] [CrossRef]
- Lu, J.R.; Purcell, I.P.; Lee, E.M.; Simister, E.A.; Thomas, R.K.; Rennie, A.R.; Penfold, J. The Composition and Structure of Sodium Dodecyl-Sulfate Dodecanol Mixtures Adsorbed at the Air-Water-Interface—A Neutron Reflection Study. J. Colloid Interface Sci. 1995, 174, 441–455. [Google Scholar] [CrossRef]
- Li, P.X.; Li, Z.X.; Shen, H.H.; Thomas, R.K.; Penfold, J.; Lu, J.R. Application of the Gibbs Equation to the Adsorption of Nonionic Surfactants and Polymers at the Air-Water Interface: Comparison with Surface Excesses Determined Directly using Neutron Reflectivity. Langmuir 2013, 29, 9324–9334. [Google Scholar] [CrossRef]
- Xu, H.; Li, P.X.; Ma, K.; Thomas, R.K.; Penfold, J.; Lu, J.R. Limitations in the Application of the Gibbs Equation to Anionic Surfactants at the Air/Water Surface: Sodium Dodecylsulfate and Sodium Dodecylmonooxyethylenesulfate Above and Below the CMC. Langmuir 2013, 29, 9335–9351. [Google Scholar] [CrossRef] [PubMed]
- Firooz, A.; Chen, P. Surface tension and adsorption kinetics of amphiphiles in aqueous solutions: The role of carbon chain length and temperature. J. Colloid Interface Sci. 2012, 370, 183–191. [Google Scholar] [CrossRef]
- Fainerman, V.B.; Lucassen-Reynders, E.H. Adsorption of single and mixed ionic surfactants at fluid interfaces. Adv. Colloid Interface Sci. 2002, 96, 295–323. [Google Scholar] [CrossRef]
- Koynova, R.; Caffrey, M. Phases and phase transitions of the phosphatidylcholines. Biochim. Et Biophys. Acta (Bba)-Rev. Biomembr. 1998, 1376, 91–145. [Google Scholar] [CrossRef]
- Goebel, A.; Lunkenheimer, K. Interfacial Tension of the Water/n-Alkane Interface. Langmuir 1997, 13, 369–372. [Google Scholar] [CrossRef]
- Cooper, V.G.; Yedgar, S.; Barenholz, Y. Diffusion coefficients of mixed micelles of Triton X-100 and sphingomyelin and of sonicated sphingomyelin liposomes, measured by autocorrelation spectroscopy of Rayleigh scattered light. Biochim. Et Biophys. Acta (Bba)-Biomembr. 1974, 363, 86–97. [Google Scholar] [CrossRef]
- Keller, M.W.; Glasheen, W.; Kaul, S. Albunex: A Safe and Effective Commercially Produced Agent for Myocardial Contrast Echocardiography. J. Am. Soc. Echocardiogr. 1989, 2, 48–52. [Google Scholar] [CrossRef]
- Duncan, P.B.; Needham, D. Microdroplet dissolution into a second-phase solvent using a micropipet technique: Test of the Epstein-Plesset model for an aniline-water system. Langmuir 2006, 22, 4190–4197. [Google Scholar] [CrossRef] [PubMed]
- Su, J.T.; Duncan, P.B.; Momaya, A.; Jutila, A.; Needham, D. The Effects of Hydrogen Bonding on the Diffusion of Water in n-Alkanes and n-Alcohols Measured with a Novel Single Microdroplet Method. J. Chem. Phys. 2010, 132, 044506. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.L.; Houghton, G. Effect of an impermeable wall on bubble collapse in diffusion coefficient measurements. Chem. Eng. Sci. 1968, 23, 1502–1503. [Google Scholar] [CrossRef]
- Ram, A.; Narkis, M. Melt Viscosity of Polyethylene: Shear Dependence. J. Polym. Sci. 1966, 10, 481–489. [Google Scholar] [CrossRef]
- Rickard, D.L.; Duncan, P.B.; Needham, D. Hydration Potential of Lysozyme: Protein Dehydration Using a Single Microparticle Technique. Biophys. J. 2010, 98, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Kwok, R.; Evans, E. Thermoelasticity of Large Lecithin Bilayer Vesicles. Biophys. J. 1981, 35, 637–652. [Google Scholar] [CrossRef]
- Waugh, R.; Evans, E.A. Thermoelasticity of red blood cell membrane. Biophys. J. 1979, 26, 115–131. [Google Scholar] [CrossRef]
- Needham, D.; Zhelev, D.V. Lysolipid exchange with lipid vesicle membranes. Ann. Biomed. Eng. 1995, 23, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Raghavendran, K.; Willson, D.; Notter, R.N. Surfactant Therapy for Acute Lung Injury and Acute Respiratory Distress Syndrome. Crit. Care Clin. 2011, 27, 525–529. [Google Scholar] [CrossRef]
- Ainsworth, S.B. Pathophysiology of neonatal respiratory distress syndrome: Implications for early treatment strategies. Treat. Respir. Med. 2005, 4, 423–437. [Google Scholar] [CrossRef]
- Moya, F.R.; Gadzinowski, J.; Bancalari, E.; Salinas, V.; Kopelman, B.; Bancalari, A.; Kornacka, M.K.; Merritt, T.A.; Segal, R.; Schaber, C.J.; et al. A multicenter, randomized, masked, comparison trial of lucinactant, colfosceril palmitate, and beractant for the prevention of respiratory distress syndrome among very preterm infants. Pediatrics 2005, 115, 1018–1029. [Google Scholar] [CrossRef]
- Agassandian, M.; Mallampalli, R.K. Surfactant phospholipid metabolism. Biochim. Et Biophys. Acta 2013, 1831, 612–625. [Google Scholar] [CrossRef]
- Olmeda, B.; García-Álvarez, B.; Pérez-Gil, J. Structure–function correlations of pulmonary surfactant protein SP-B and the saposin-like family of proteins. Eur. Biophys. J. 2013, 42, 209–222. [Google Scholar] [CrossRef]
- Merrill, J.D.; Ballard, R.A. Pulmonary surfactant for neonatal respiratory disorders. Curr. Opin. Pediatrics 2003, 15, 149–154. [Google Scholar] [CrossRef]
- Waring, A.J.; Walther, F.J.; Gordon, L.M.; Hernandez-Juviel, J.M.; Hong, T.; Sherman, M.A.; Alonso, C.; Alig, T.; Braun, A.; Bacon, D. The role of charged amphipathic helices in the structure and function of surfactant protein B. J. Pept. Res. 2005, 66, 364–374. [Google Scholar] [CrossRef]
- Walther, F.J.; Waring, A.J.; Hernandez-Juviel, J.M.; Gordon, L.M.; Wang, Z.D.; Jung, C.L.; Ruchala, P.; Clark, A.P.; Smith, W.M.; Sharma, S.; et al. Critical Structural and Functional Roles for the N-Terminal Insertion Sequence in Surfactant Protein B Analogs. PLoS ONE 2010, 5, e8672. [Google Scholar] [CrossRef]
- Walther, F.J.; Hernández-Juviel, J.M.; Gordon, L.M.; Waring, A.J. Synthetic surfactant containing SP-B and SP-C mimics is superior to single-peptide formulations in rabbits with chemical acute lung injury. PeerJ 2014, 2, e393. [Google Scholar] [CrossRef]
- Alonso, C.; Bringezu, F.; Brezesinski, G.; Waring, A.J.; Zasadzinski, J.A. Modifying calf lung surfactant by hexadecanol. Langmuir 2005, 21, 1028–1035. [Google Scholar] [CrossRef]
- Parra, E.; Moleiro, L.H.; Lopez-Montero, I.; Cruz, A.; Monroy, F.; Perez-Gil, J. A combined action of pulmonary surfactant proteins SP-B and SP-C modulates permeability and dynamics of phospholipid membranes. Biochem. J. 2011, 438, 555–564. [Google Scholar] [CrossRef]
- Parra, E.; Kinoshita, K.; Needham, D. Supporting Information: Micropipette Technique Study of Natural and Synthetic Lung Surfactants at the Air-Water Interface: Presence of a SP-B Analog Peptide Promotes Membrane Aggregation, Formation of Tightly Stacked Lamellae, and Growth of Myelin Figures. Langmuir 2016, 32, 10570–10581. [Google Scholar] [CrossRef]
- Parra, E.; Kinoshita, K. Needham. (4.85 MB). Available online: https://pubs.acs.org/doi/suppl/10.1021/acs.langmuir.6b01420/suppl_file/la6b01420_si_002.mpg (accessed on 30 January 2019).
- Parra, E.; Kinoshita, K.; Needham, D. (3.69 MB). Available online: https://pubs.acs.org/doi/suppl/10.1021/acs.langmuir.6b01420/suppl_file/la6b01420_si_003.mpg (accessed on 30 January 2019).
- Needham, D.; Arslanagic, A.; Glud, K.; Hervella, P.; Karimi, L.; Høeilund-Carlsen, P.-F.; Kinoshita, K.; Mollenhauer, J.; Parra, E.; Utoft, A.; et al. Bottom up design of nanoparticles for anti-cancer diapeutics: “put the drug in the cancer’s food”. J. Drug. Target. 2016, 24, 836–856. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Firestone, R.A. Low-Density Lipoprotein as a Vehicle for Targeting Antitumor Compounds to Cancer Cells. Bioconjugate Chem. 1994, 5, 105–113. [Google Scholar] [CrossRef]
- Merriel, S.W.D.; Carroll, R.; Hamilton, F.; Hamilton, W. Association between unexplained hypoalbuminaemia and new cancer diagnoses in UK primary care patients. Fam. Pract. 2016, 33, 449–452. [Google Scholar] [CrossRef]
- Yue, S.; Li, J.; Lee, S.-Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl Ester Accumulation Induced by PTEN Loss and PI3K/AKT Activation Underlies Human Prostate Cancer Aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef]
- Rodrigues dos Santos, C.; Domingues, G.; Matias, I.; Matos, J.; Fonseca, I.; de Almeida, J.M.; Dias, S. LDL-cholesterol signaling induces breast cancer proliferation and invasion. Lipids Health Dis. 2014, 13, 16. [Google Scholar] [CrossRef]
- Gallagher, E.J.; Zelenko, Z.; Neel, B.A.; Antoniou, I.M.; Rajan, L.; Kase, N.; LeRoith, D. Elevated tumor LDLR expression accelerates LDL cholesterol-mediated breast cancer growth in mouse models of hyperlipidemia. Oncogene 2017, 36, 6462–6471. [Google Scholar] [CrossRef]
- Karthika, S.; Radhakrishnan, T.K.; Kalaichelvi, P. A Review of Classical and Nonclassical Nucleation Theories. Cryst. Growth Des. 2016, 16, 6663–6681. [Google Scholar] [CrossRef]
- Horn, D.; Rieger, J. Organic Nanoparticles in the Aqueous Phase–Theory, Experiment, and Use. Angew. Chem. Int. Ed. 2001, 40, 4330–4361. [Google Scholar] [CrossRef]
- Aarts, P.A.; Gijeman, O.L.; Kremer, J.M.; Wiersema, P.H. Dynamics of phospholipid aggregation in ethanol--water solutions. Chem. Phys. Lipids 1977, 19, 267–274. [Google Scholar] [CrossRef]
- Dunn, R.O.; Schwab, A.W.; Bagby, M.O. Physical Property and Phase Studies of Nonaqueous Triglyceride Unsaturated Long-Chain Fatty Alcohol Methanol Systems. J. Disper. Sci. Technol. 1992, 13, 77–93. [Google Scholar] [CrossRef]
- Bitterfield, D.L.; Utoft, A.; Needham, D. An Activity-Based Dissolution Model for Solute-Containing Microdroplets. Langmuir 2016, 32, 12749–12759. [Google Scholar] [CrossRef]
- Parra, E.; Hervella, P.; Needham, D. Real-Time Visualization of the Precipitation and Phase Behavior of Octaethylporphyrin in Lipid Microparticles. J. Pharm. Sci. 2017, 106, 1025–1041. [Google Scholar] [CrossRef]
- Utoft, A.; Kinoshita, K.; Bitterfield, D.; David, N. Manipulating Single Microdroplets of NaCl Solutions: Solvent Dissolution, Microcrystallization, and Crystal Morphology. Langmuir 2018, 34, 3626–3641. [Google Scholar] [CrossRef]
- De Bruijn, R.; Schaaf, J.V.D.; Patil, N.; Schouten, J. Process for Preparing Monodispersed Emulsions. WO 2010/031709 A1, 2010. [Google Scholar]






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alkane | Alkane-Water γOW (mN/m) | Alkane-Air γOA (mN/m) |
| Hexane (C6) | 51.1 | 18.4 |
| Octane (C8) | 50.8 | 21.6 |
| Decane (C10) | 51.2 (52.0) | 23.6 (23.8) |
| Alkanol | Alkanol-Water γOW (mN/m) | Alkanol-Air γOA (mN/m) |
| 1-Hexanol (C6) | 6.8 | 25.8 |
| 1-Octanol (C8) | 8.52 | 27.5 |
| 1-decanol (C10) | 8.97 | 28.9 |
| Techniques | Short-Time Adsorption (s) | Interfacial Area (µm2) | Functions and Drawbacks | Ref. |
|---|---|---|---|---|
| MIAM | <1 | 103–105 | Interfacial area expansion 150–200 times No requirement of contact angle information Leakage for high viscous and sticky materials for glass surface | [6,9,7,8] |
| PBT | 10−1 | 107 | Reliable data in the middle time range (0.1 s to mins) Limit of long-time adsorption (bubble detachment) Bubble expansion rate increasing cause error | [139,140] |
| Growing-drop | 10−2 | 107 | Wide range (milliseconds to more than hours) dynamic surface tension measurement Need to keep constant flow, material loss from tip climbing | [141,142] |
| Oscillating Jet | 10−3–10−2 | 105 | Short time adsorption measurement Not suitable for the long-time adsorption range of (>10 s), no equilibrium tension | [87,143,144] |
| MBPM | <10−3 | 105 | Wide range, short-time and long-time, adsorption measurement Leakage of material, requirement of hydrophobic treatment inner wall and hydrophilic tip | [133,136,145] |
| Langmuir–Wilhelmy | >20 | ~102 cm2–m2 | Easily assembles with microscopy and spectroscopy methods Not suitable for fast area exchange because of leakage of material Contact angle requirement | [146,147] |
| CBS | ~10−1 | 107 | Leakage proof, mimic alveolar environment Limited interfacial area expansion (two times) range for adsorption dynamic | [148,149] |
| PBS | 1 | 106 | Mimic breath control, easy to operate Material leakage, lacks operational flexibility | [122,150] |
| % SMB | # Analysed Tubes | Thickness (μm) | Linear Growth Rate (μm/s) | Volume Growth Rate (μm3/s) |
|---|---|---|---|---|
| 0.1 | 5 | 4.46 ± 1.85 | 1.76 ± 1.24 | 28.37 ± 28.37 |
| 1 | 15 | 3.41 ± 1.07 | 2.68 ± 3.84 | 22.38 ± 25.11 |
| 2 | 20 | 4.73 ± 2.19 | 2.23 ± 2.34 | 26.65 ± 14.10 |
| 4 | 21 | 3.48 ± 1.24 | 2.73 ± 1.70 | 22.14 ± 12.95 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Needham, D.; Kinoshita, K.; Utoft, A. Micro-Surface and -Interfacial Tensions Measured Using the Micropipette Technique: Applications in Ultrasound-Microbubbles, Oil-Recovery, Lung-Surfactants, Nanoprecipitation, and Microfluidics. Micromachines 2019, 10, 105. https://doi.org/10.3390/mi10020105
Needham D, Kinoshita K, Utoft A. Micro-Surface and -Interfacial Tensions Measured Using the Micropipette Technique: Applications in Ultrasound-Microbubbles, Oil-Recovery, Lung-Surfactants, Nanoprecipitation, and Microfluidics. Micromachines. 2019; 10(2):105. https://doi.org/10.3390/mi10020105
Chicago/Turabian StyleNeedham, David, Koji Kinoshita, and Anders Utoft. 2019. "Micro-Surface and -Interfacial Tensions Measured Using the Micropipette Technique: Applications in Ultrasound-Microbubbles, Oil-Recovery, Lung-Surfactants, Nanoprecipitation, and Microfluidics" Micromachines 10, no. 2: 105. https://doi.org/10.3390/mi10020105