
An Integrated Centrifugal Degassed PDMS-Based Microfluidic Device for Serial Dilution

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Theory and Working Principle
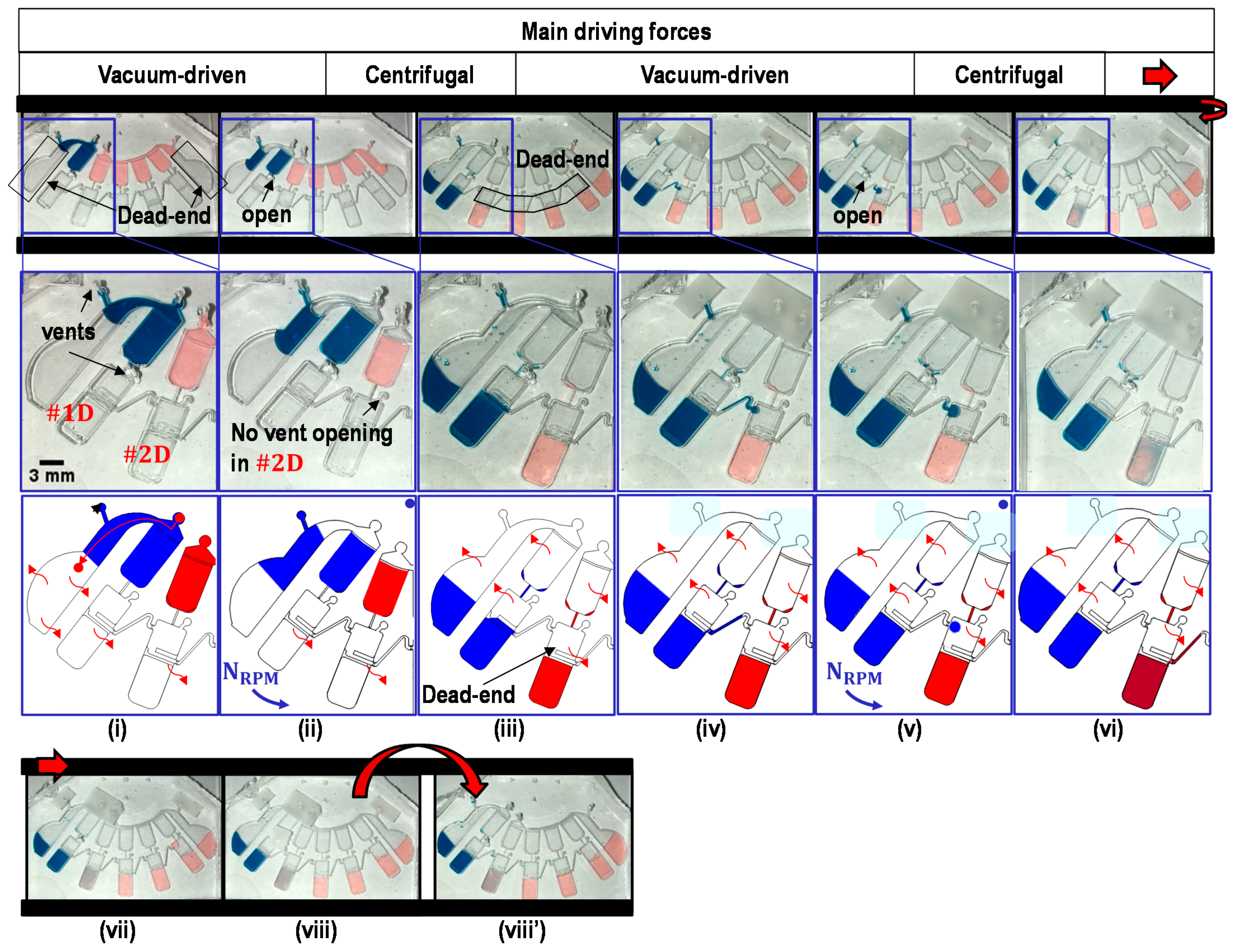
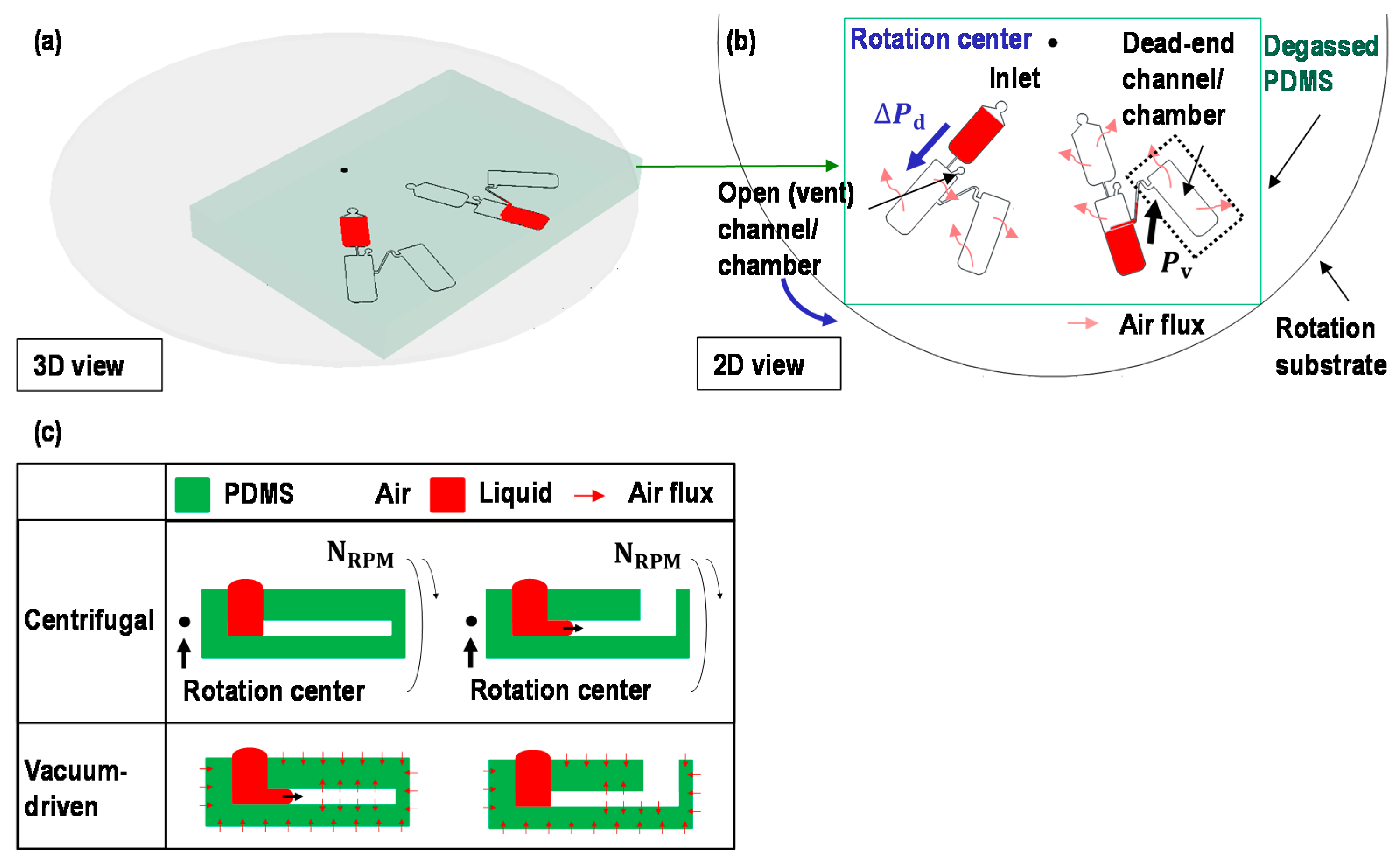
2.1. Switch a Dominant Force between Centrifugal Force and Vacuum-Driven Force
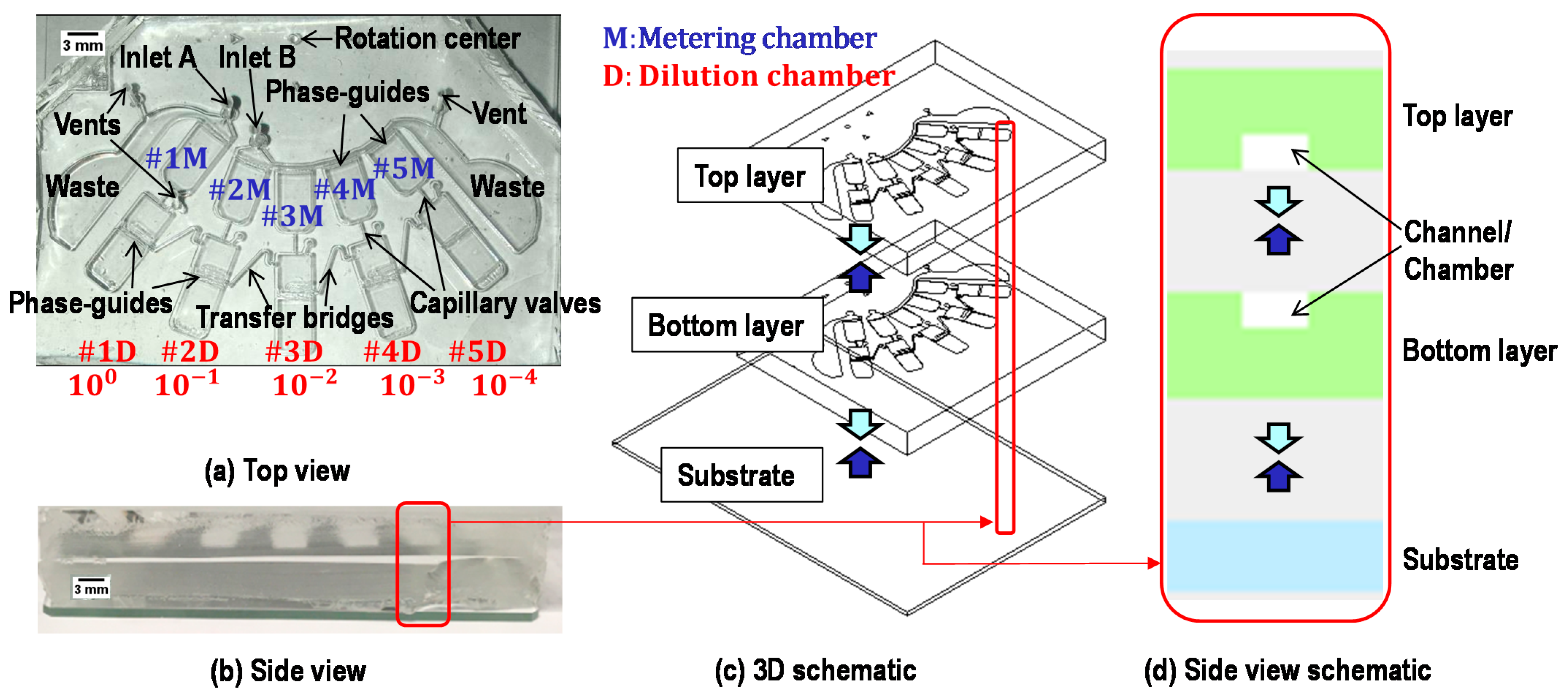
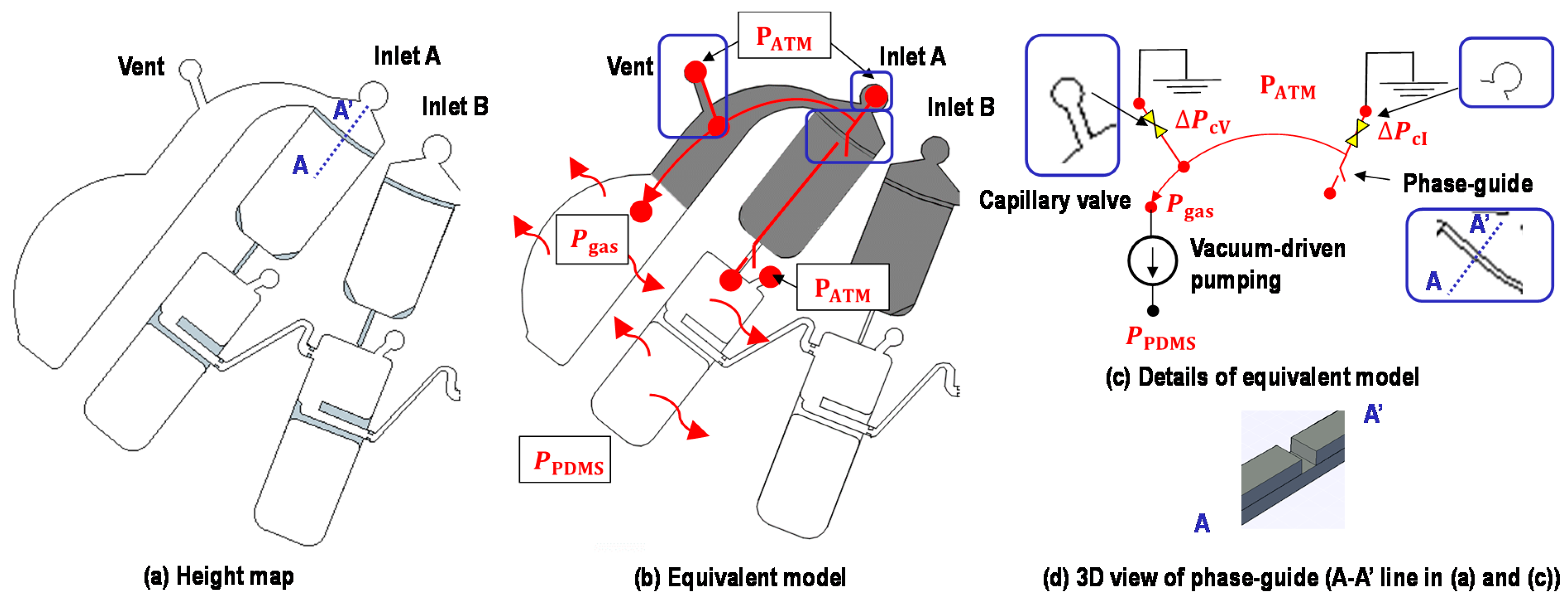
2.2. Device Structure
2.3. Working Principle
2.4. Metering of Liquids
2.5. Transfer of Metered Liquids into Dilution Chambers
2.6. Partial Transfer of Liquids across the Transfer Bridge
2.7. Sample Mixing
3. Materials and Methods
3.1. Device Fabrication
3.2. Sample Preparation for qPCR
3.3. Performance of qPCR
4. Results
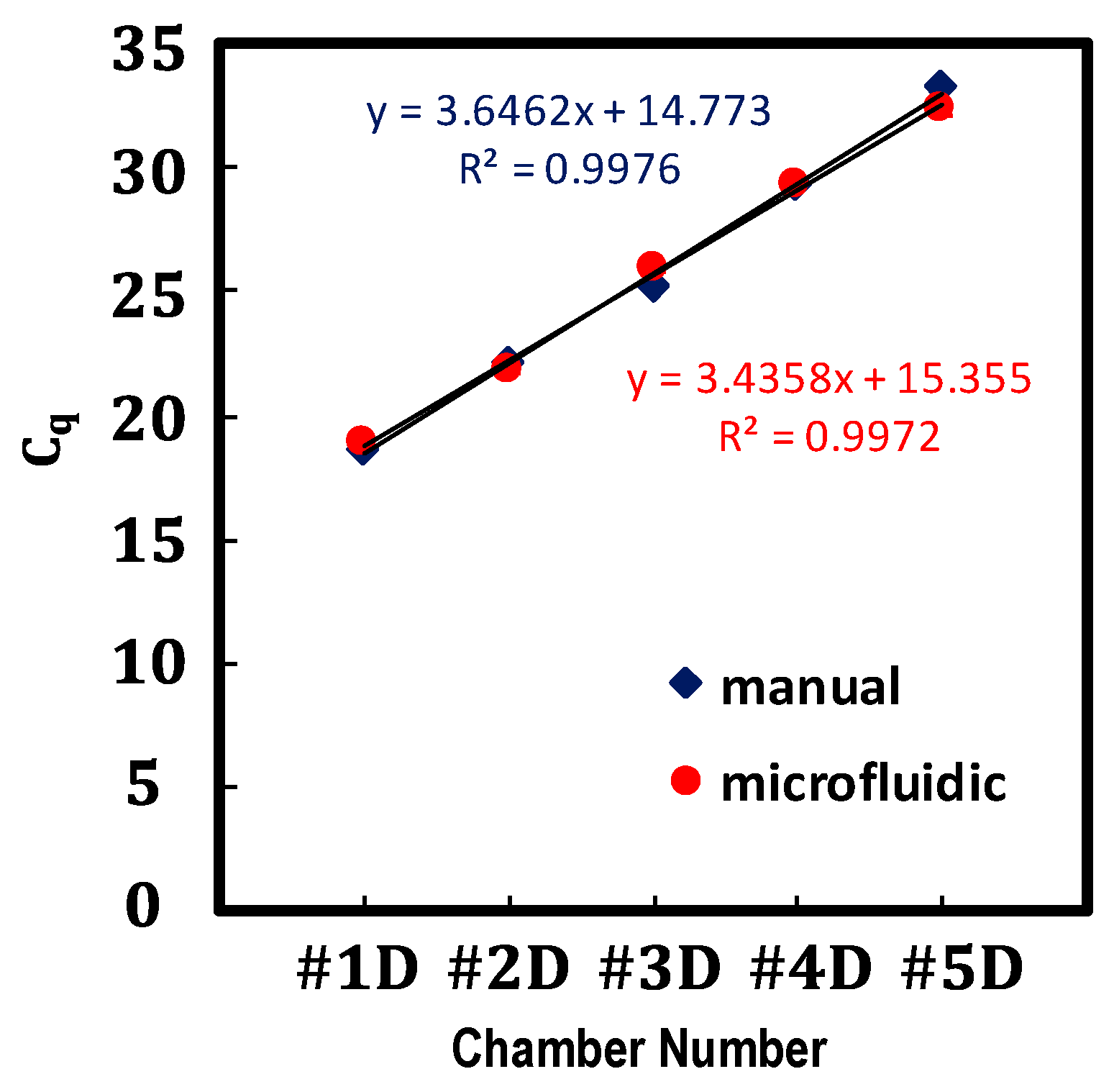
4.1. Volume Analysis
4.2. Proof-of-Concept: Verification of the Performance of the Proposed Serial Dilution by qPCR
4.3. Durability, Limitations and Future Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shamloo, A.; Ma, N.; Poo, M.-M.; Sohn, L.L.; Heilshorn, S.C. Endothelial cell polarization and chemotaxis in a microfluidic device. Lab Chip 2008, 8, 1292–1299. [Google Scholar] [CrossRef]
- Juelg, P.; Specht, M.; Kipf, E.; Lehnert, M.; Eckert, C.; Keller, M.; Hutzenlaub, T.; von Stetten, F.; Zengerle, R.; Paust, N. Automated serial dilutions for high-dynamic-range assays enabled by fill-level-coupled valving in centrifugal microfluidics. Lab Chip 2019, 19, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Strohmeier, O.; Rombach, M.; Mark, D.; Zengerle, R.; Roth, G.; von Stetten, F. Fully integrated dilution series generation on a laboratory centrifuge. In Proceedings of the 2011 16th International Solid-State Sensors, Actuators and Microsystems Conference, Beijing, China, 5–9 June 2011; pp. 2952–2955. [Google Scholar]
- Wang, T.; Zhang, M.; Dreher, D.D.; Zeng, Y. Ultrasensitive microfluidic solid-phase ELISA using an actuatable microwell-patterned PDMS chip. Lab Chip 2013, 13, 4190–4197. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, G. A power-free, parallel loading microfluidic reactor array for biochemical screening. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Yuan, L.; Zheng, Y.F.; Chen, W. Automatic liquid handling for life science: A critical review of the current state of the art. J. Lab. Autom. 2012, 17, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Felton, M.J. Product review: Liquid handling: Dispensing reliability. Anal. Chem. 2003, 75, 397 A–399 A. [Google Scholar] [CrossRef] [Green Version]
- Tecan. Fluent® Laboratory Automation Workstation for Cell-Based Assays. Available online: https://pdf.medicalexpo.com/pdf/tecan/fluent-laboratory-automation-workstation-cell-based-assays/80772-194104.html (accessed on 21 April 2021).
- Xu, L.; Lee, H.; Brasil Pinheiro, M.V.; Schneider, P.; Jetta, D.; Oh, K.W. Phaseguide-assisted blood separation microfluidic device for point-of-care applications. Biomicrofluidics 2015, 9. [Google Scholar] [CrossRef] [Green Version]
- Ji, T.; Liu, Z.; Wang, G.; Guo, X.; Lai, C.; Chen, H.; Huang, S.; Xia, S.; Chen, B.; Jia, H. Detection of COVID-19: A review of the current literature and future perspectives. Biosens. Bioelectron. 2020, 166. [Google Scholar] [CrossRef]
- Xu, L.; Lee, H.; Jetta, D.; Oh, K.W. Vacuum-driven power-free microfluidics utilizing the gas solubility or permeability of polydimethylsiloxane (PDMS). Lab Chip 2015, 15, 3962–3979. [Google Scholar] [CrossRef]
- Xu, L.; Lee, H.; Oh, K.W. Syringe-assisted point-of-care micropumping utilizing the gas permeability of polydimethylsiloxane. Microfluid. Nanofluid. 2014, 17, 745–750. [Google Scholar] [CrossRef]
- Xu, L.; Wang, A.; Li, X.; Oh, K.W. Passive micropumping in microfluidics for point-of-care testing. Biomicrofluidics 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Olanrewaju, A.; Beaugrand, M.; Yafia, M.; Juncker, D. Capillary microfluidics in microchannels: From microfluidic networks to capillaric circuits. Lab Chip 2018, 18, 2323–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Kim, C.; Jung, G.; Kim, T.S.; Kang, J.Y.; Oh, K.W. Microfluidic network-based combinatorial dilution device for high throughput screening and optimization. Microfluid. Nanofluid. 2010, 8, 677–685. [Google Scholar] [CrossRef]
- Lee, M.; Lee, K.; Kim, K.H.; Oh, K.W.; Choo, J. SERS-based immunoassay using a gold array-embedded gradient microfluidic chip. Lab Chip 2012, 12, 3720–3727. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.W.; Lee, K.; Ahn, B.; Furlani, E.P. Design of pressure-driven microfluidic networks using electric circuit analogy. Lab Chip 2012, 12, 515–545. [Google Scholar] [CrossRef]
- Choi, N.; Lee, K.; Lim, D.W.; Lee, E.K.; Chang, S.-I.; Oh, K.W.; Choo, J. Simultaneous detection of duplex DNA oligonucleotides using a SERS-based micro-network gradient chip. Lab Chip 2012, 12, 5160–5167. [Google Scholar] [CrossRef]
- Lee, K.; Kim, C.; Kim, Y.; Ahn, B.; Bang, J.; Kim, J.; Panchapakesan, R.; Yoon, Y.-K.; Kang, J.Y.; Oh, K.W. Microfluidic concentration-on-demand combinatorial dilutions. Microfluid. Nanofluid. 2011, 11, 75–86. [Google Scholar] [CrossRef]
- Gao, Y.; Sun, J.; Lin, W.-H.; Webb, D.J.; Li, D. A compact microfluidic gradient generator using passive pumping. Microfluid. Nanofluid. 2012, 12, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Shangguan, J.-W.; Xu, B.-Y.; Yu, X.-D.; Xu, J.-J.; Chen, H.-Y. Abnormal Liquid Chasing Effect in Paper Capillary Enables Versatile Gradient Generation on Microfluidic Paper Analytical Devices. Anal. Chem. 2020, 92, 2722–2730. [Google Scholar] [CrossRef]
- Lee, Y.; Choi, M.; Kim, S.-J. Method to prevent backflow in a capillarity network for bioassays: Exploiting time constant ratios. Sens. Actuators B Chem. 2018, 255, 3630–3635. [Google Scholar] [CrossRef]
- Lee, Y.; Seder, I.; Kim, S.-J. Influence of surface tension-driven network parameters on backflow strength. RSC Adv. 2019, 9, 10345–10351. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Wang, A.; Koh, D.; Schneider, P.; Oh, K.W. A robust, portable and backflow-free micromixing device based on both capillary-and vacuum-driven flows. Lab Chip 2018, 18, 276–284. [Google Scholar] [CrossRef]
- Kim, S.-J.; Lim, Y.T.; Yang, H.; Shin, Y.B.; Kim, K.; Lee, D.-S.; Park, S.H.; Kim, Y.T. Passive microfluidic control of two merging streams by capillarity and relative flow resistance. Anal. Chem. 2005, 77, 6494–6499. [Google Scholar] [CrossRef]
- Wu, J.; Kumar-Kanojia, A.; Hombach-Klonisch, S.; Klonisch, T.; Lin, F. A radial microfluidic platform for higher throughput chemotaxis studies with individual gradient control. Lab Chip 2018, 18, 3855–3864. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.; Zehnle, S.; Juelg, P.; Hutzenlaub, T.; Zengerle, R.; Paust, N. Review on pneumatic operations in centrifugal microfluidics. Lab Chip 2019, 19, 3745–3770. [Google Scholar] [CrossRef] [PubMed]
- Clime, L.; Brassard, D.; Geissler, M.; Veres, T. Active pneumatic control of centrifugal microfluidic flows for lab-on-a-chip applications. Lab Chip 2015, 15, 2400–2411. [Google Scholar] [CrossRef]
- Wang, A.; Koh, D.; Schneider, P.; Breloff, E.; Oh, K.W. A compact, syringe-assisted, vacuum-driven micropumping device. Micromachines 2019, 10, 543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Tanaka, N.; Yamazoe, H.; Furutani, S.; Nagai, H.; Kawai, T.; Tanaka, Y. Flow analysis on microcasting with degassed polydimethylsiloxane micro-channels for cell patterning with cross-linked albumin. PLoS ONE 2020, 15, e0232518. [Google Scholar] [CrossRef]
- Hosokawa, K.; Omata, M.; Maeda, M. Immunoassay on a power-free microchip with laminar flow-assisted dendritic amplification. Anal. Chem. 2007, 79, 6000–6004. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, K.; Omata, M.; Sato, K.; Maeda, M. Power-free sequential injection for microchip immunoassay toward point-of-care testing. Lab Chip 2006, 6, 236–241. [Google Scholar] [CrossRef]
- Hosokawa, K.; Sato, K.; Ichikawa, N.; Maeda, M. Power-free poly (dimethylsiloxane) microfluidic devices for gold nanoparticle-based DNA analysis. Lab Chip 2004, 4, 181–185. [Google Scholar] [CrossRef]
- Juncker, D.; Schmid, H.; Drechsler, U.; Wolf, H.; Wolf, M.; Michel, B.; de Rooij, N.; Delamarche, E. Autonomous microfluidic capillary system. Anal. Chem. 2002, 74, 6139–6144. [Google Scholar] [CrossRef]
- Safavieh, R.; Juncker, D. Capillarics: Pre-programmed, self-powered microfluidic circuits built from capillary elements. Lab Chip 2013, 13, 4180–4189. [Google Scholar] [CrossRef] [PubMed]
- Vulto, P.; Podszun, S.; Meyer, P.; Hermann, C.; Manz, A.; Urban, G.A. Phaseguides: A paradigm shift in microfluidic priming and emptying. Lab Chip 2011, 11, 1596–1602. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, K.; Ahn, B.; Xu, J.; Xu, L.; Oh, K.W. A new fabrication process for uniform SU-8 thick photoresist structures by simultaneously removing edge bead and air bubbles. J. Micromech. Microeng. 2011, 21, 125006. [Google Scholar] [CrossRef]
- Wang, A.; Koh, D.; Schneider, P.; Zhai, Y.; Bosinski, B.; Oh, W.K. Backflow-assisted sequential injection in vacuum-driven power-free microfluidic device. In Proceedings of the MicroTAS 2017, Savannah, GA, USA, 22–26 October 2017; pp. 752–753. [Google Scholar]
- Bauer, M.; Ataei, M.; Caicedo, M.; Jackson, K.; Madou, M.; Bousse, L. Burst valves for commercial microfluidics: A critical analysis. Microfluid. Nanofluid. 2019, 23, 86. [Google Scholar] [CrossRef]
- Cho, H.; Kim, H.-Y.; Kang, J.Y.; Kim, T.S. How the capillary burst microvalve works. J. Colloid Interface Sci. 2007, 306, 379–385. [Google Scholar] [CrossRef]
- Nolte, D.D. Invited Review Article: Review of centrifugal microfluidic and bio-optical disks. Rev. Sci. Instrum. 2009, 80. [Google Scholar] [CrossRef] [Green Version]
- Grumann, M.; Geipel, A.; Riegger, L.; Zengerle, R.; Ducrée, J. Batch-mode mixing on centrifugal microfluidic platforms. Lab Chip 2005, 5, 560–565. [Google Scholar] [CrossRef]
- Ren, Y.; Leung, W.W.-F. Vortical flow and mixing in rotating milli-and micro-chambers. Comput. Fluids 2013, 79, 150–166. [Google Scholar] [CrossRef]
- Ren, Y.; Leung, W.W.-F. Numerical and experimental investigation on flow and mixing in batch-mode centrifugal microfluidics. Int. J. Heat Mass Transf. 2013, 60, 95–104. [Google Scholar] [CrossRef]
- TaKaRa PCR Amplification Kit; Cat. #R011; Available online: http://www.takara.co.kr/file/manual/pdf/R011_e.v1906Da.pdf (accessed on 21 April 2021).
- Lifetechnologies. Real-Time PCR Handbook; Available online: https://www.thermofisher.com/content/dam/LifeTech/global/Forms/PDF/real-time-pcr-handbook.pdf (accessed on 21 April 2021).
- Kim, T.-H.; Kim, C.-J.; Kim, Y.; Cho, Y.-K. Centrifugal microfluidic system for a fully automated N-fold serial dilution. Sens. Actuators B Chem. 2018, 256, 310–317. [Google Scholar] [CrossRef]
- Liu, B.; Koh, D.; Wang, A.; Schneider, P.; Oh, K.W. Hermetic encapsulation of negative-pressure-driven PDMS microfluidic devices using paraffin wax and glass. Microsyst. Technol. 2018, 24, 2035–2043. [Google Scholar] [CrossRef]
- Song, J.S.; Lee, S.; Jung, S.H.; Cha, G.C.; Mun, M.S. Improved biocompatibility of parylene—C films prepared by chemical vapor deposition and the subsequent plasma treatment. J. Appl. Polym. Sci. 2009, 112, 3677–3685. [Google Scholar] [CrossRef]
- Lamberti, A.; Marasso, S.; Cocuzza, M. PDMS membranes with tunable gas permeability for microfluidic applications. RSC Adv. 2014, 4, 61415–61419. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, A.; Boroujeni, S.M.; Schneider, P.J.; Christie, L.B.; Mancuso, K.A.; Andreadis, S.T.; Oh, K.W. An Integrated Centrifugal Degassed PDMS-Based Microfluidic Device for Serial Dilution. Micromachines 2021, 12, 482. https://doi.org/10.3390/mi12050482
Wang A, Boroujeni SM, Schneider PJ, Christie LB, Mancuso KA, Andreadis ST, Oh KW. An Integrated Centrifugal Degassed PDMS-Based Microfluidic Device for Serial Dilution. Micromachines. 2021; 12(5):482. https://doi.org/10.3390/mi12050482
Chicago/Turabian StyleWang, Anyang, Samaneh Moghadasi Boroujeni, Philip J. Schneider, Liam B. Christie, Kyle A. Mancuso, Stelios T. Andreadis, and Kwang W. Oh. 2021. "An Integrated Centrifugal Degassed PDMS-Based Microfluidic Device for Serial Dilution" Micromachines 12, no. 5: 482. https://doi.org/10.3390/mi12050482