
Microfluidic Fabrication of Hydrocortisone Nanocrystals Coated with Polymeric Stabilisers



Abstract
:1. Introduction
2. Materials and Methods
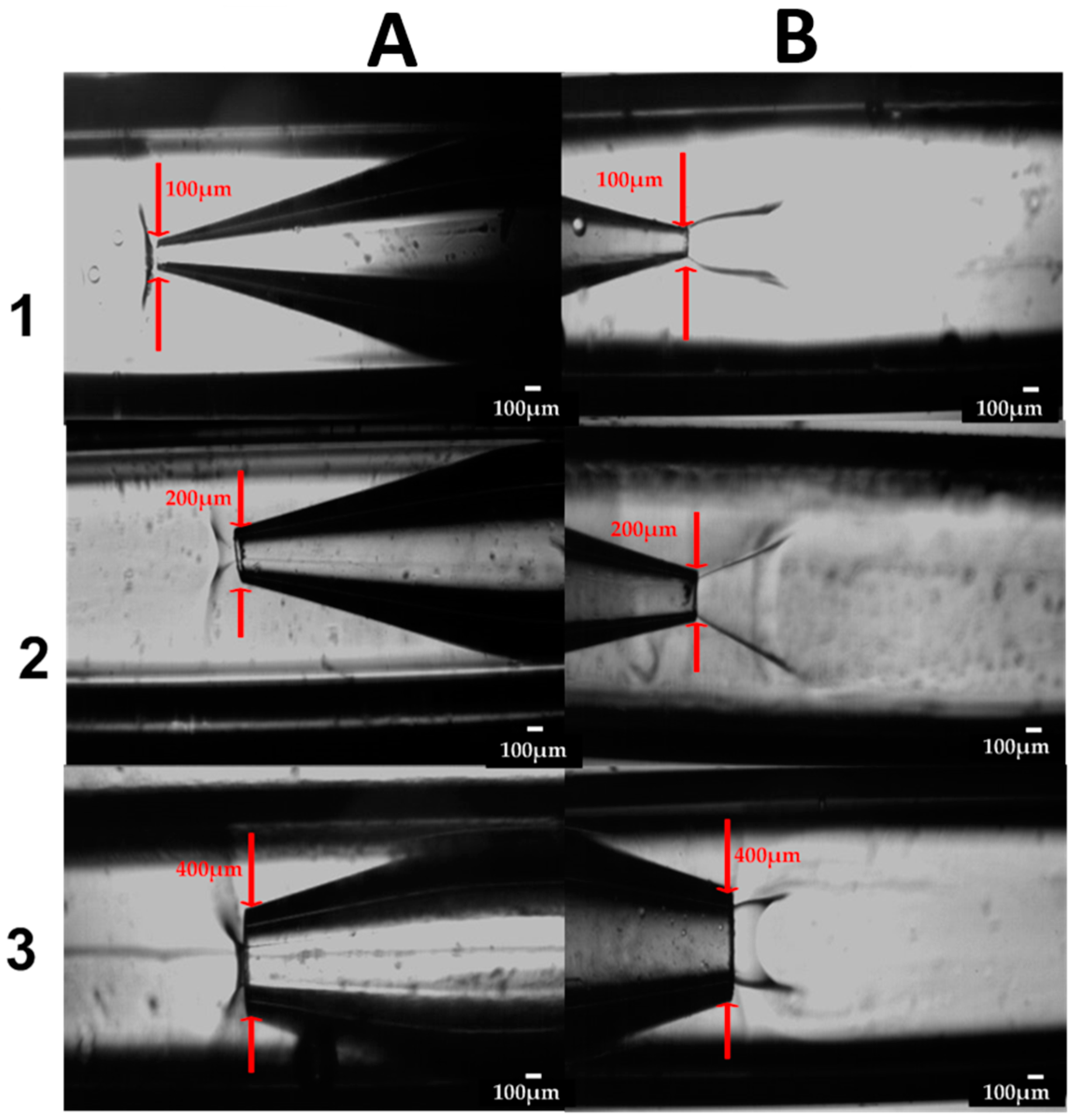
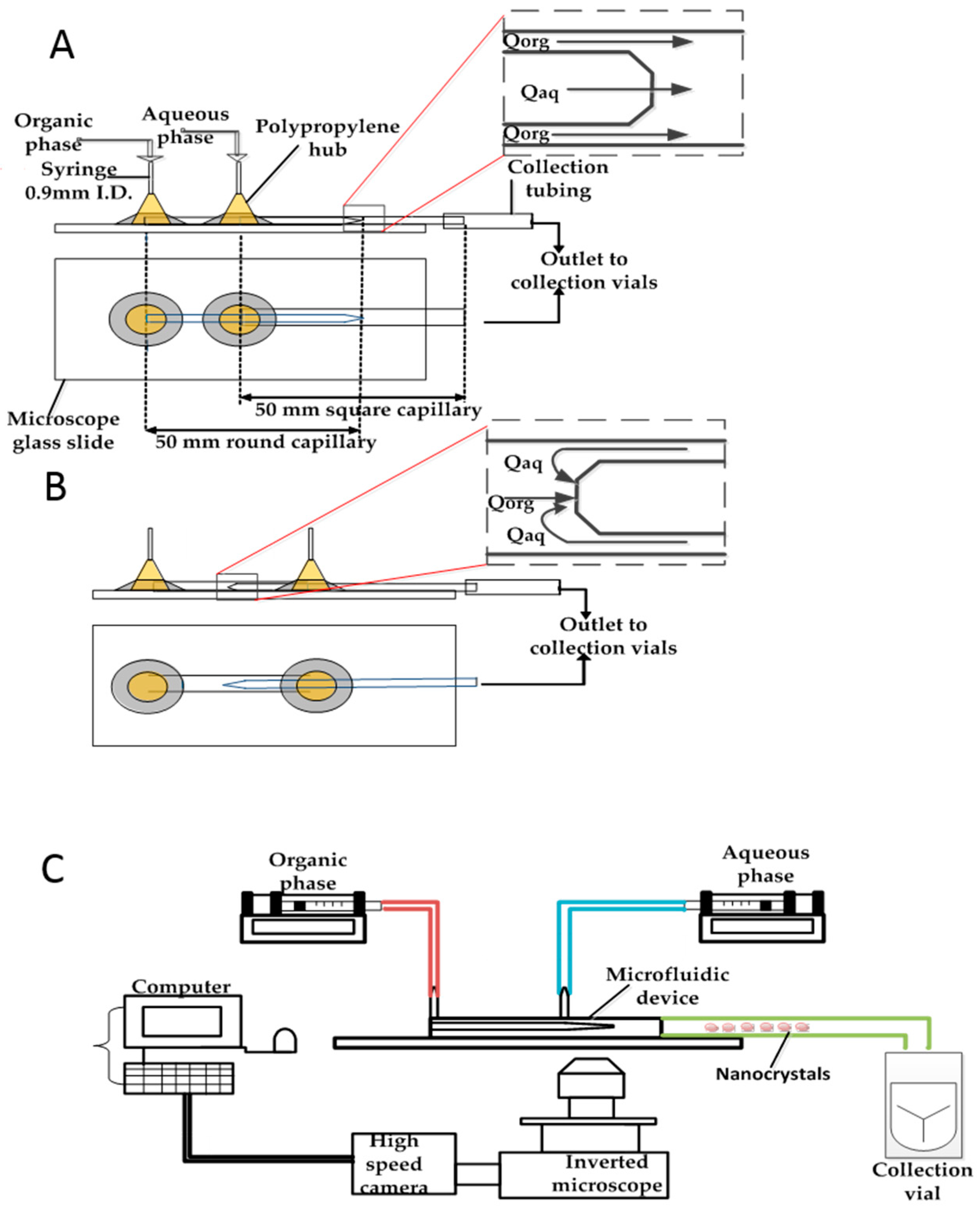
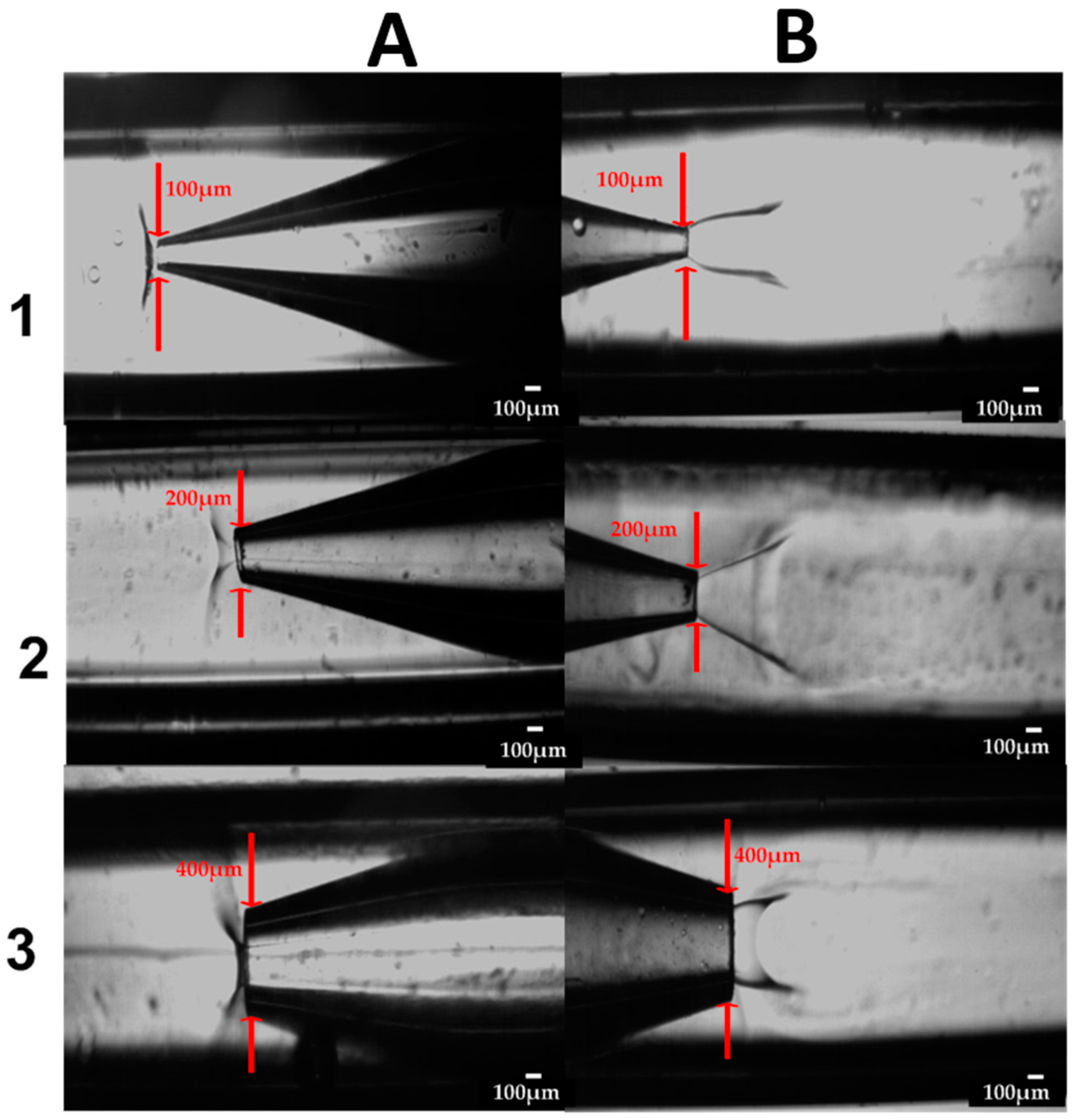
2.1. Fabrication of the Glass Capillary Devices
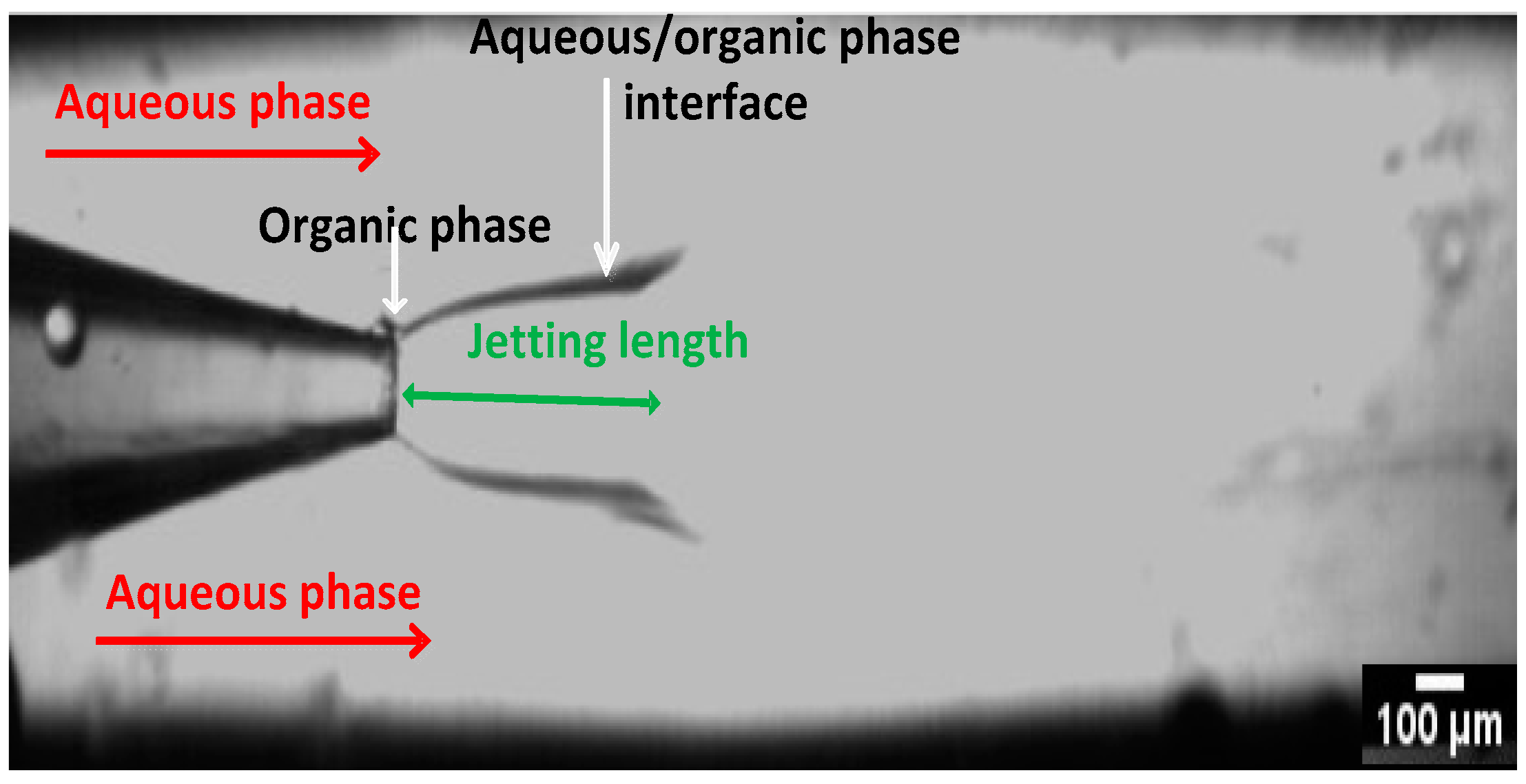
2.2. Set up and Preparation of Nanocrystals
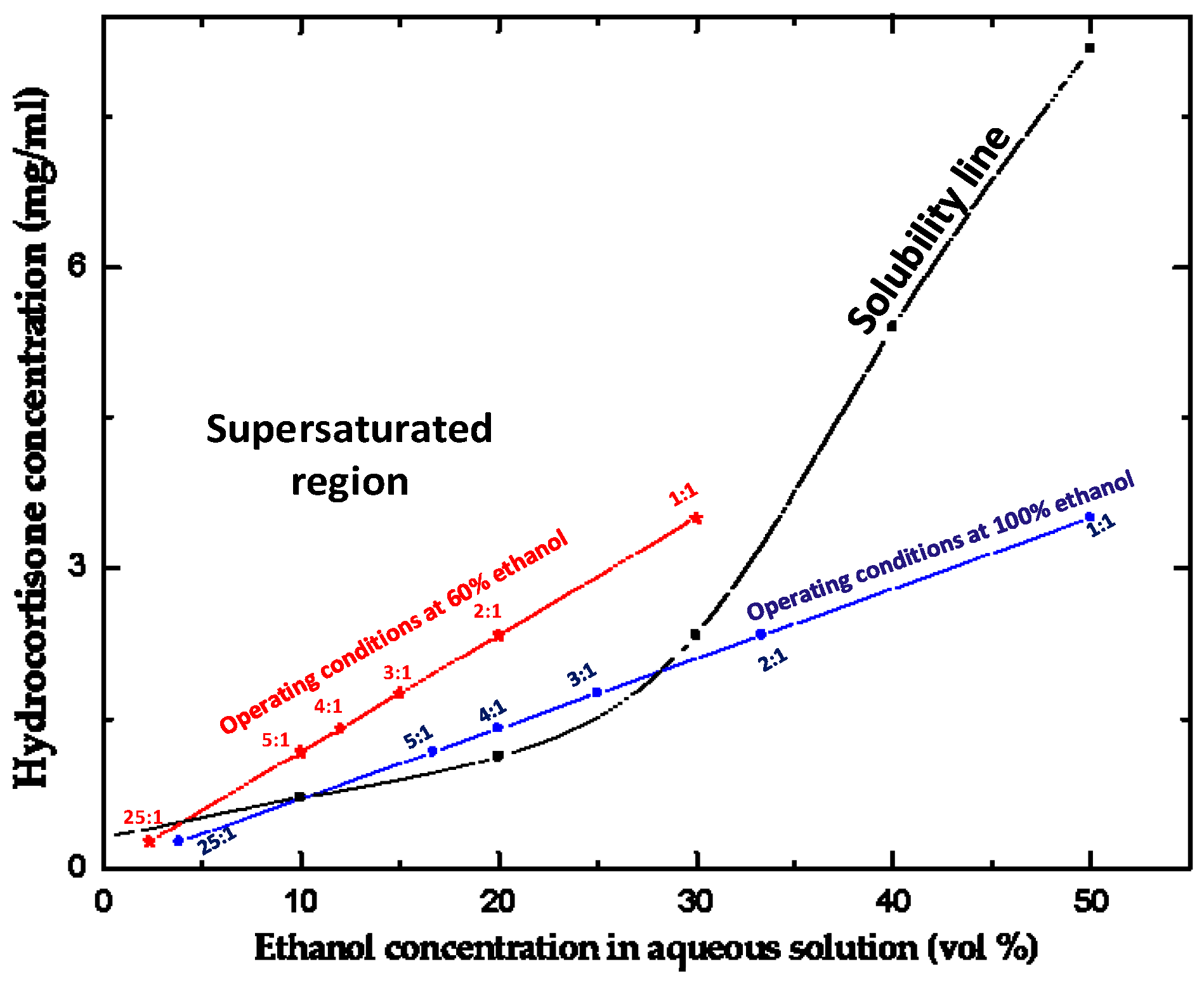
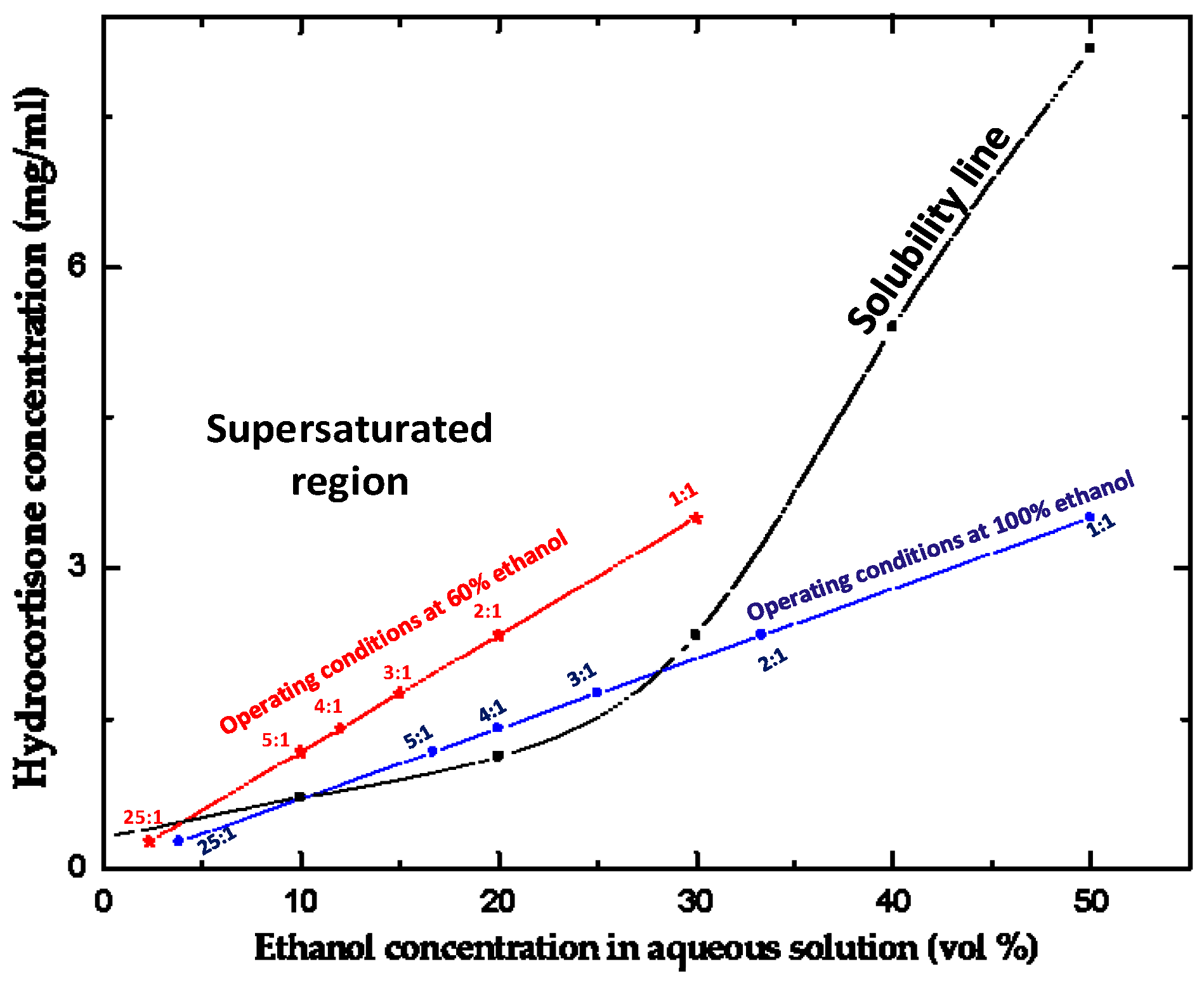
2.3. Solubility of Hydrocortisone in Ethanol/Water Mixtures
2.4. Characterisation of Produced Nanocrystals (Quantitative and Qualitative Analyses)
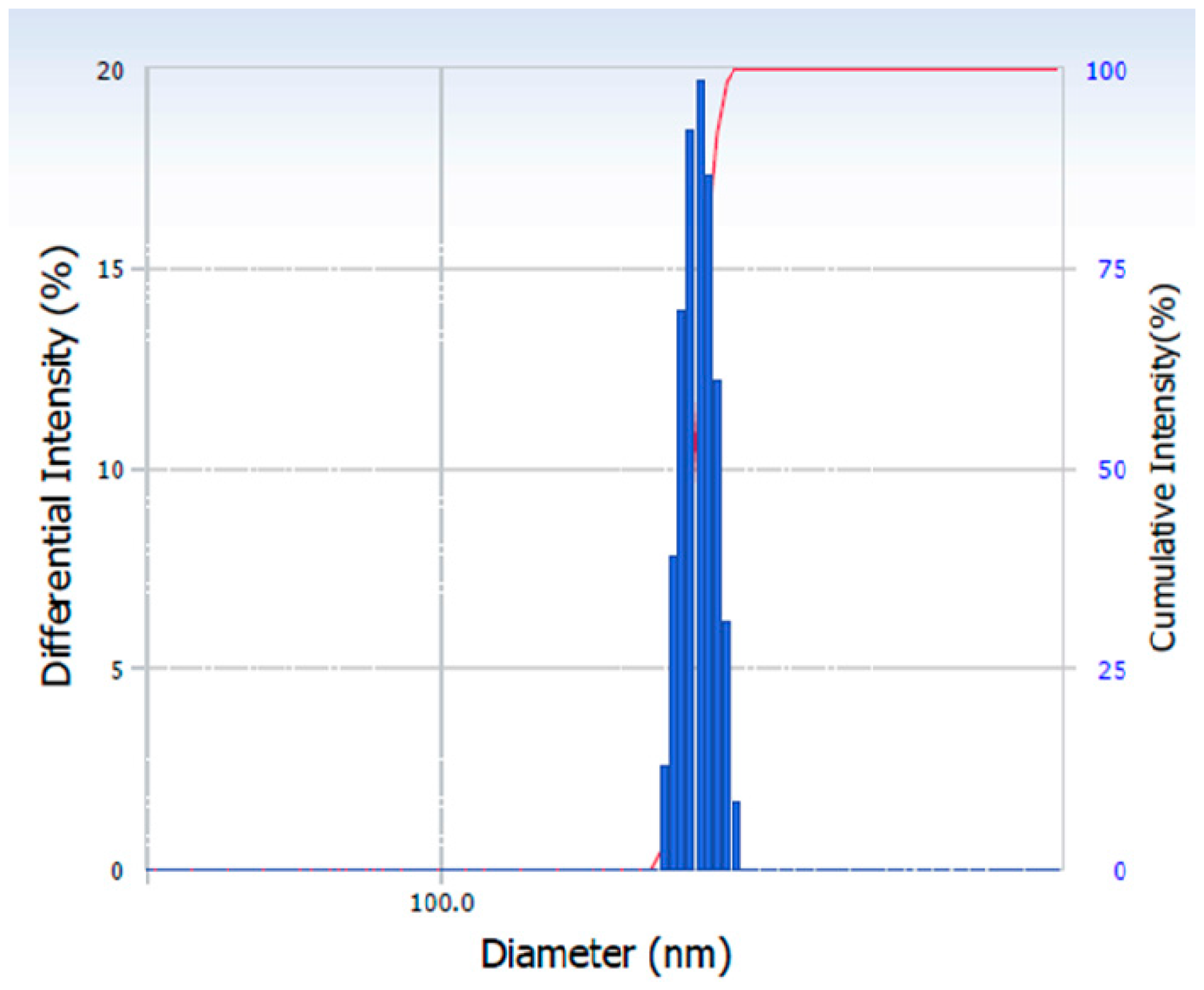
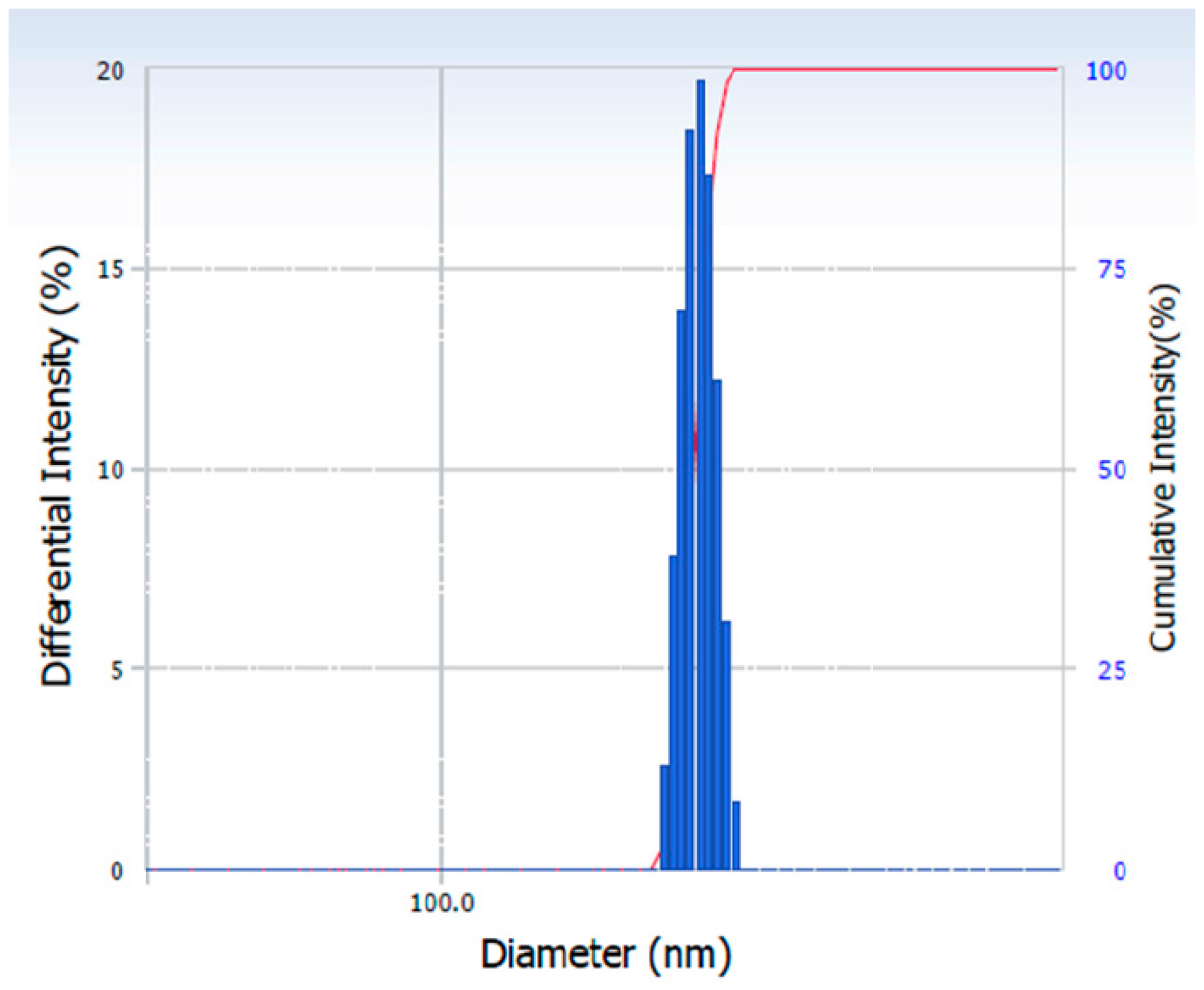
2.4.1. Particle Size Analysis
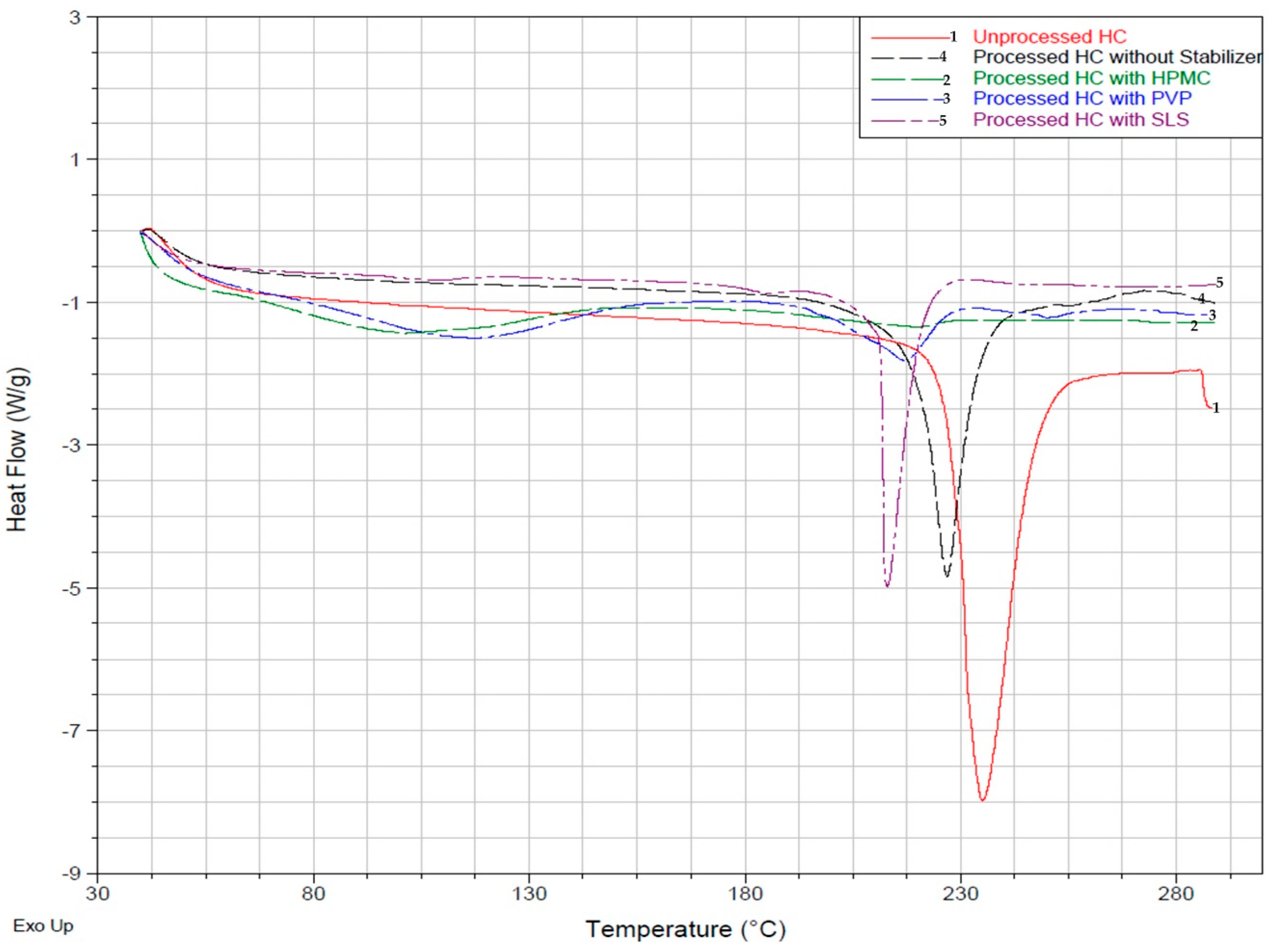
2.4.2. Differential Scanning Calorimetry (DSC) Analysis
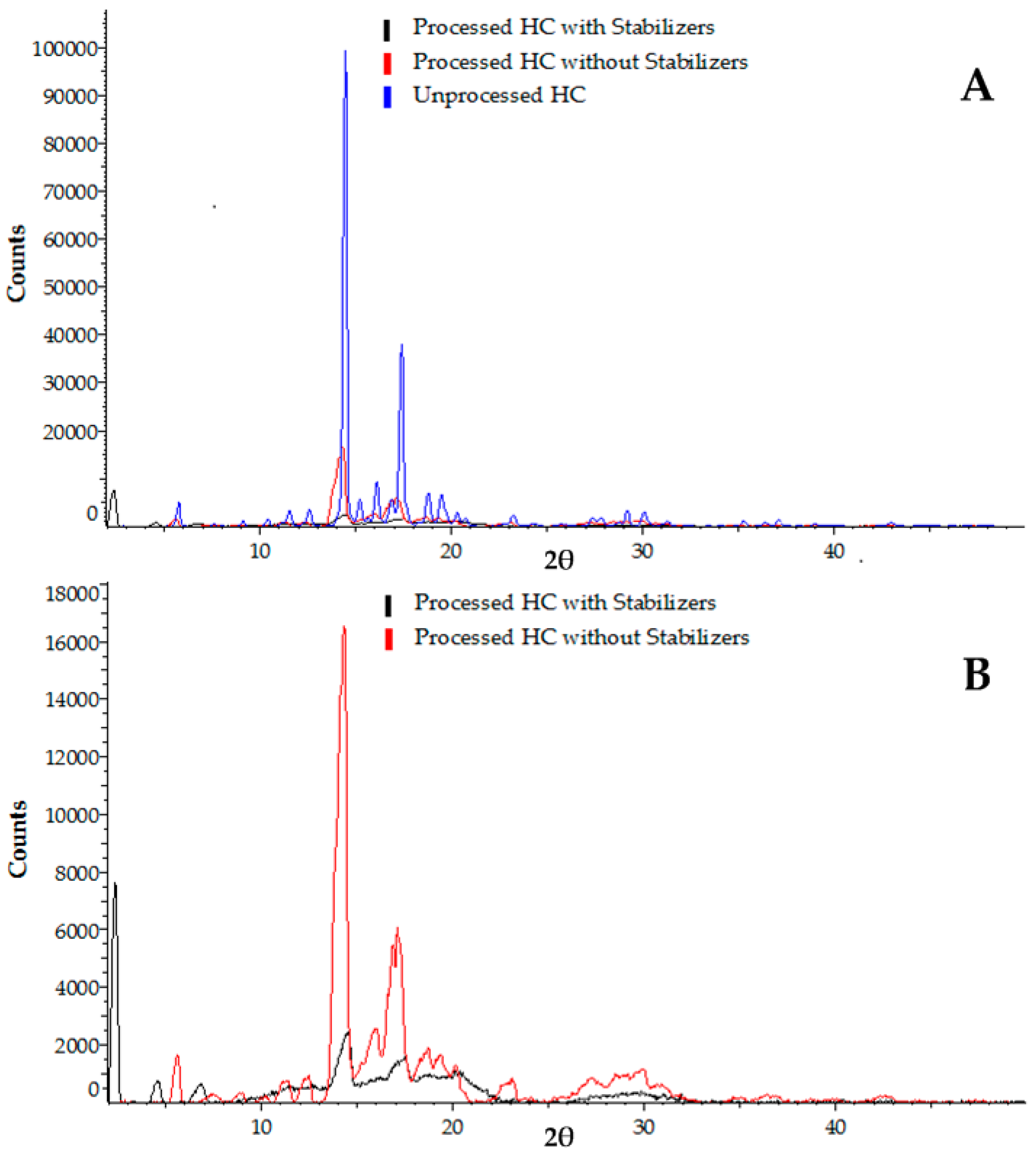
2.4.3. X-ray Powder Diffraction (XRPD) Analysis
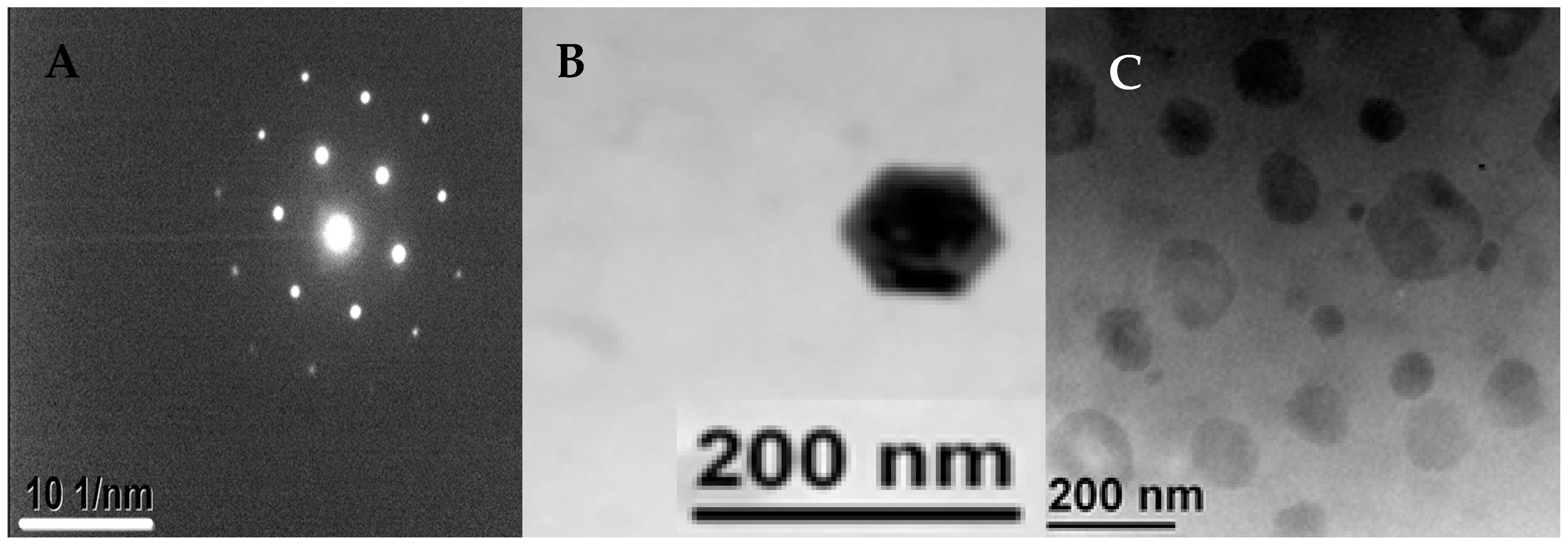
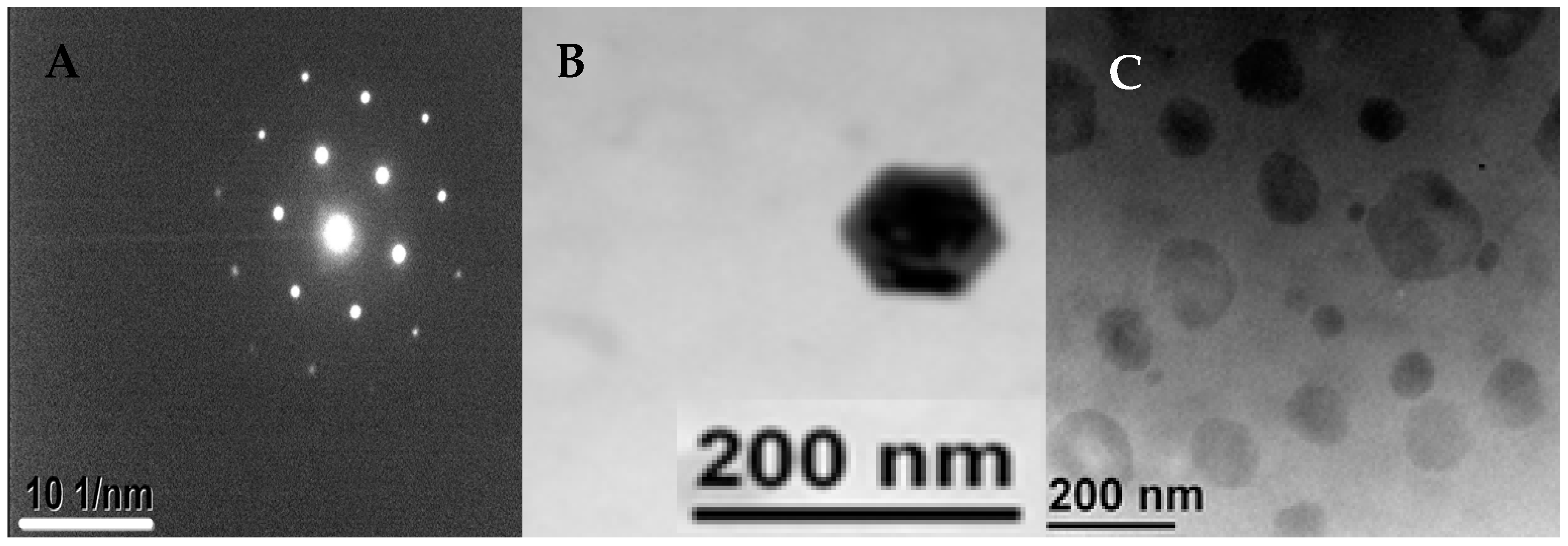
2.4.4. Transmission Electron Microscopy (TEM)
3. Results and Discussion
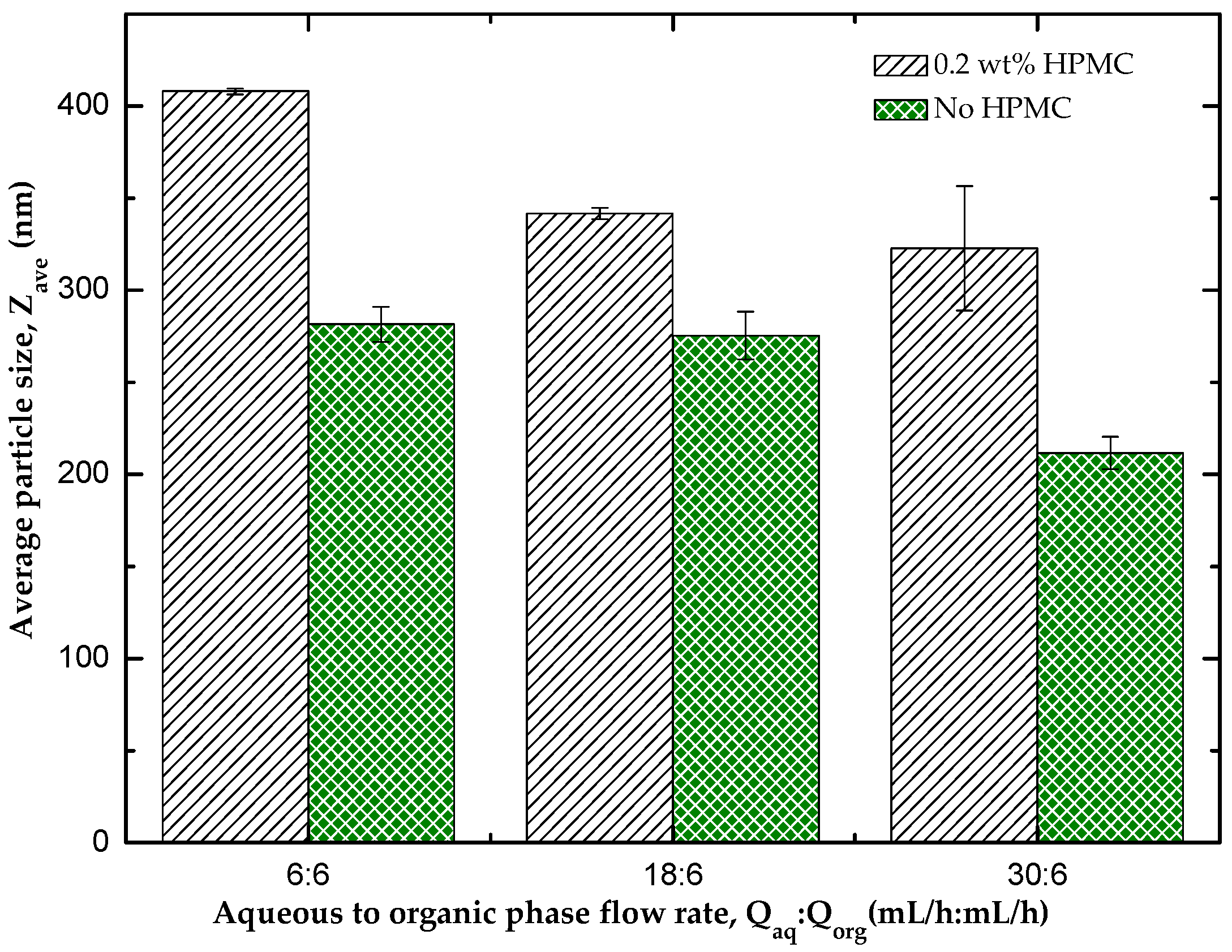
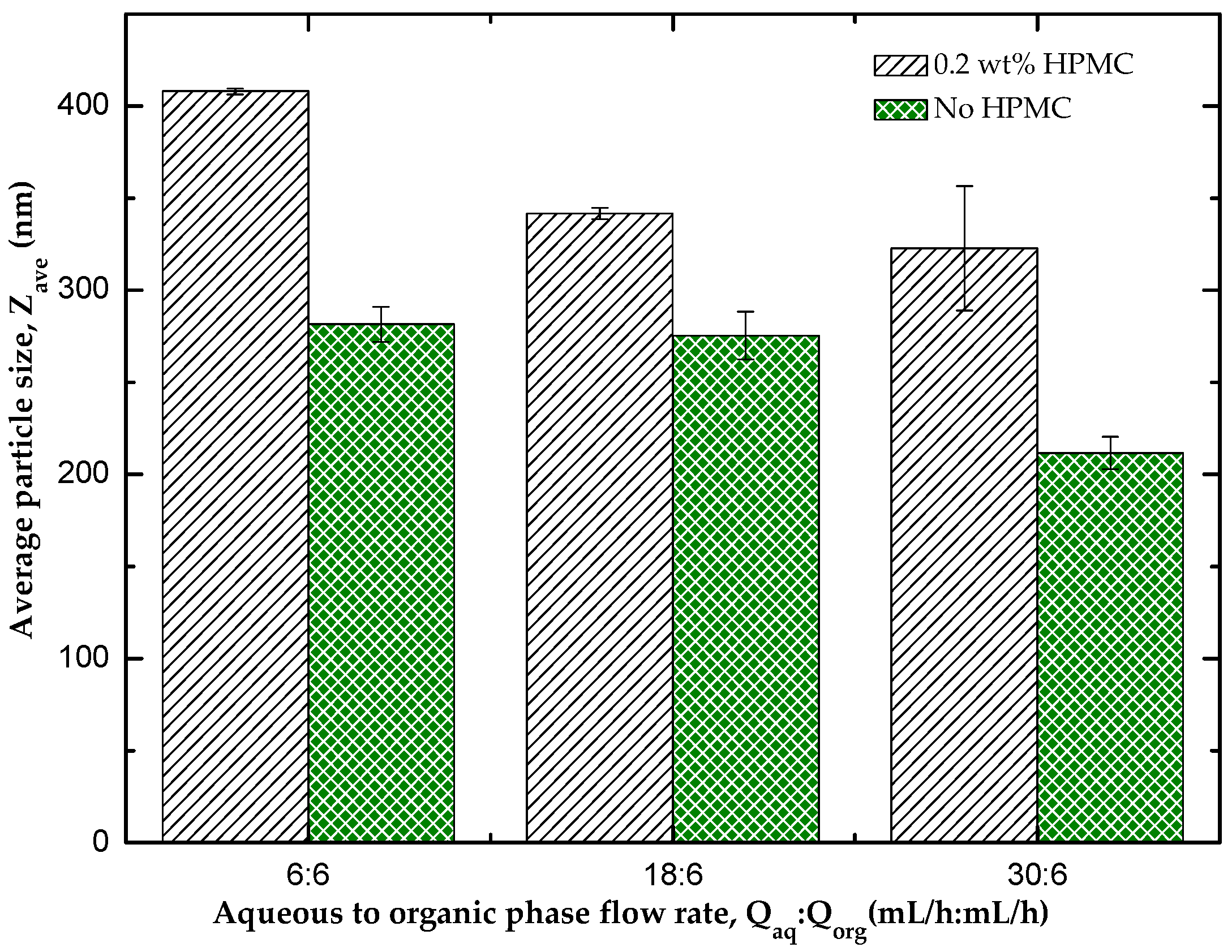
3.1. Effect of HPMC on the Particle Size of the Nanocrystals
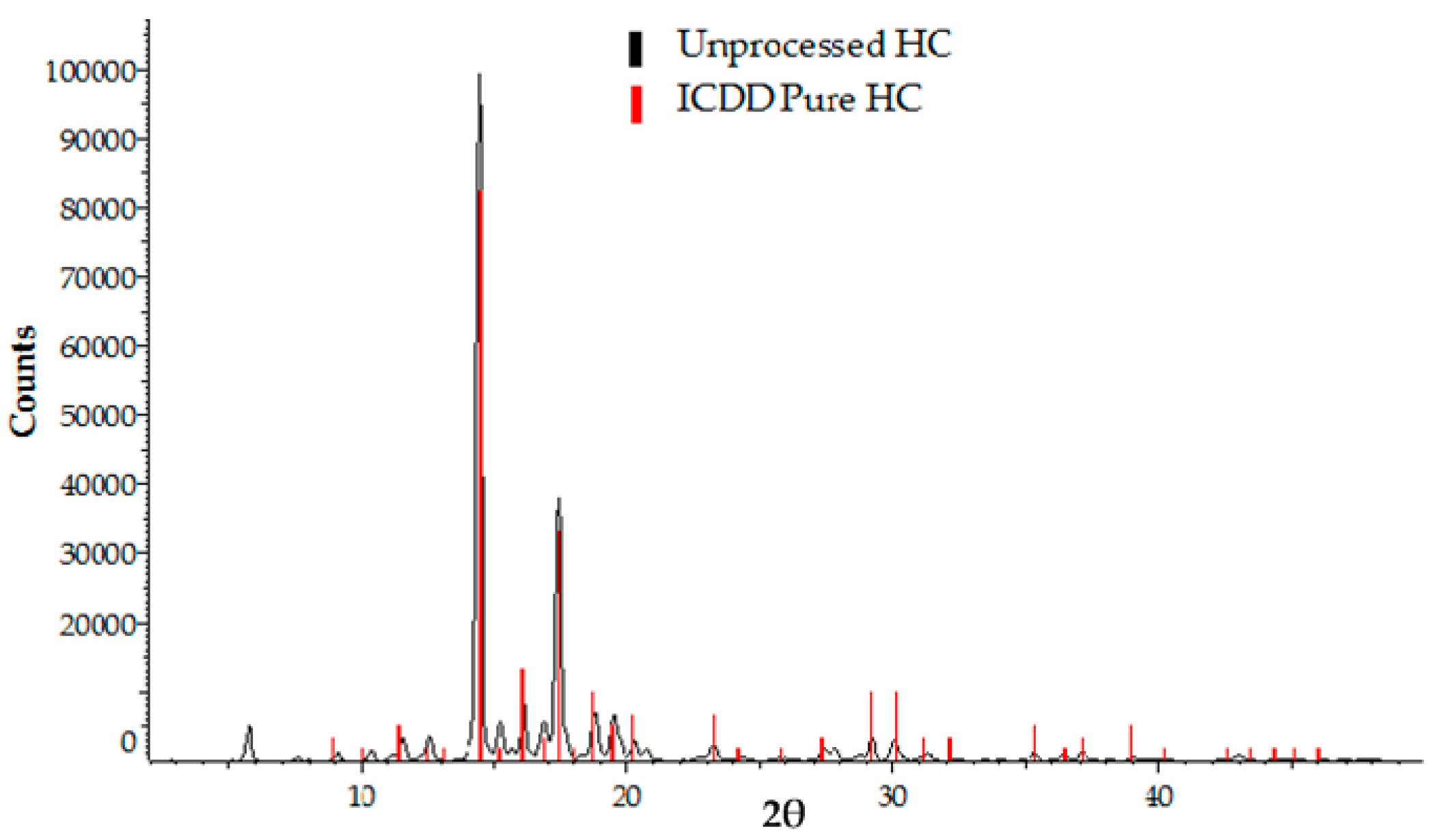
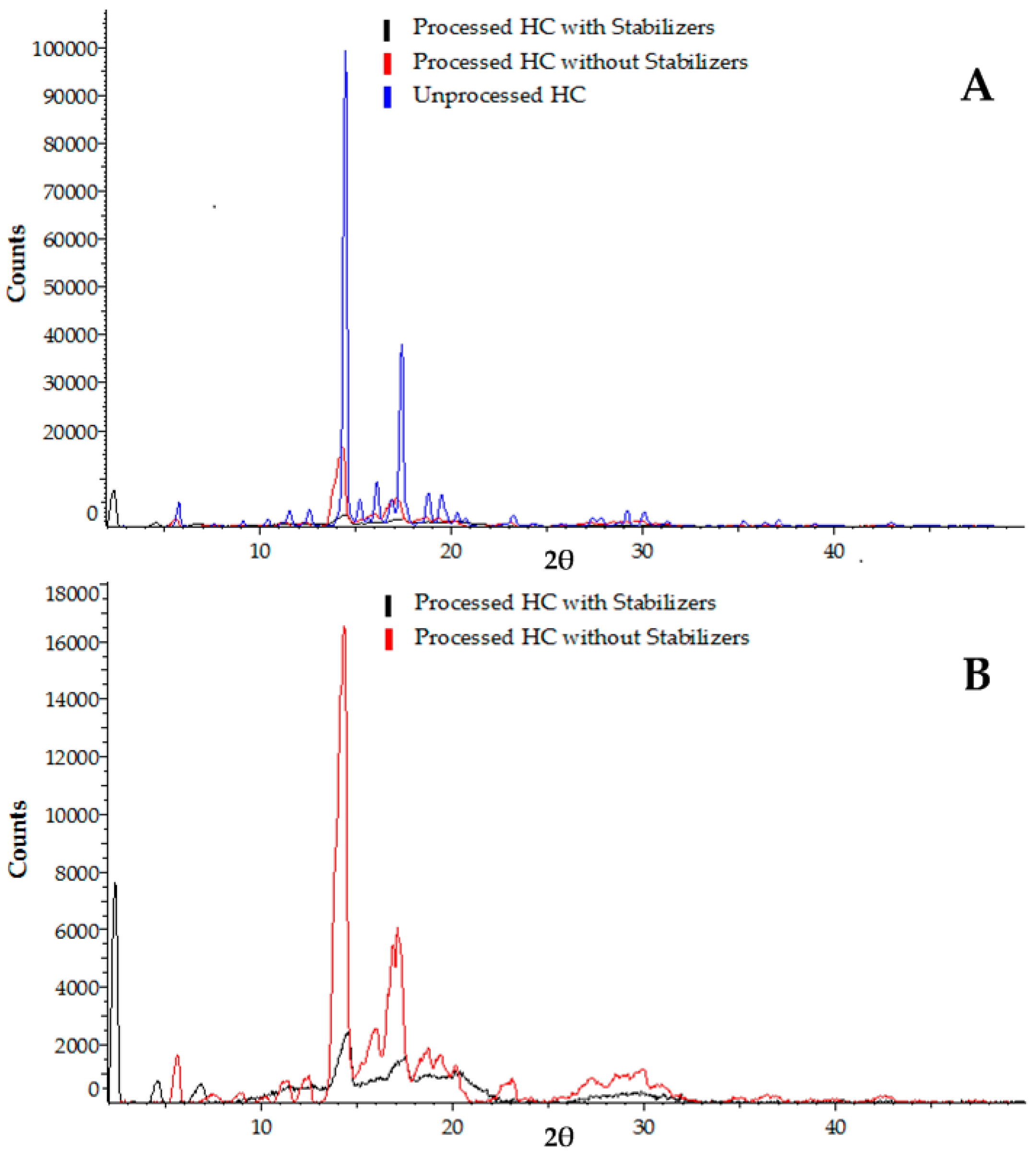
3.2. Analysis of Crystallinity of Hydrocortisone Nanocrystals
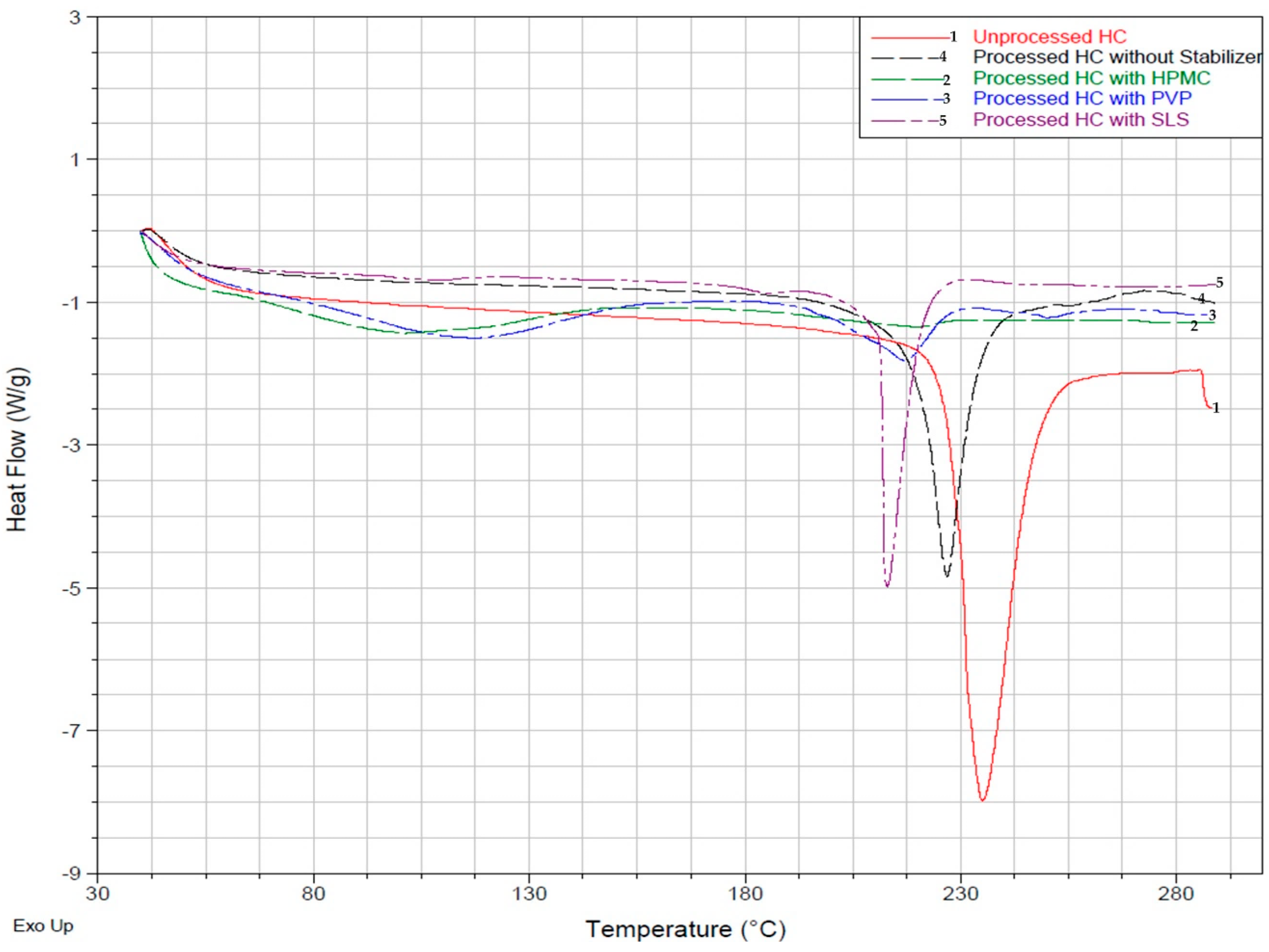
3.2.1. Differential Scanning Calorimetry (DSC) Analysis
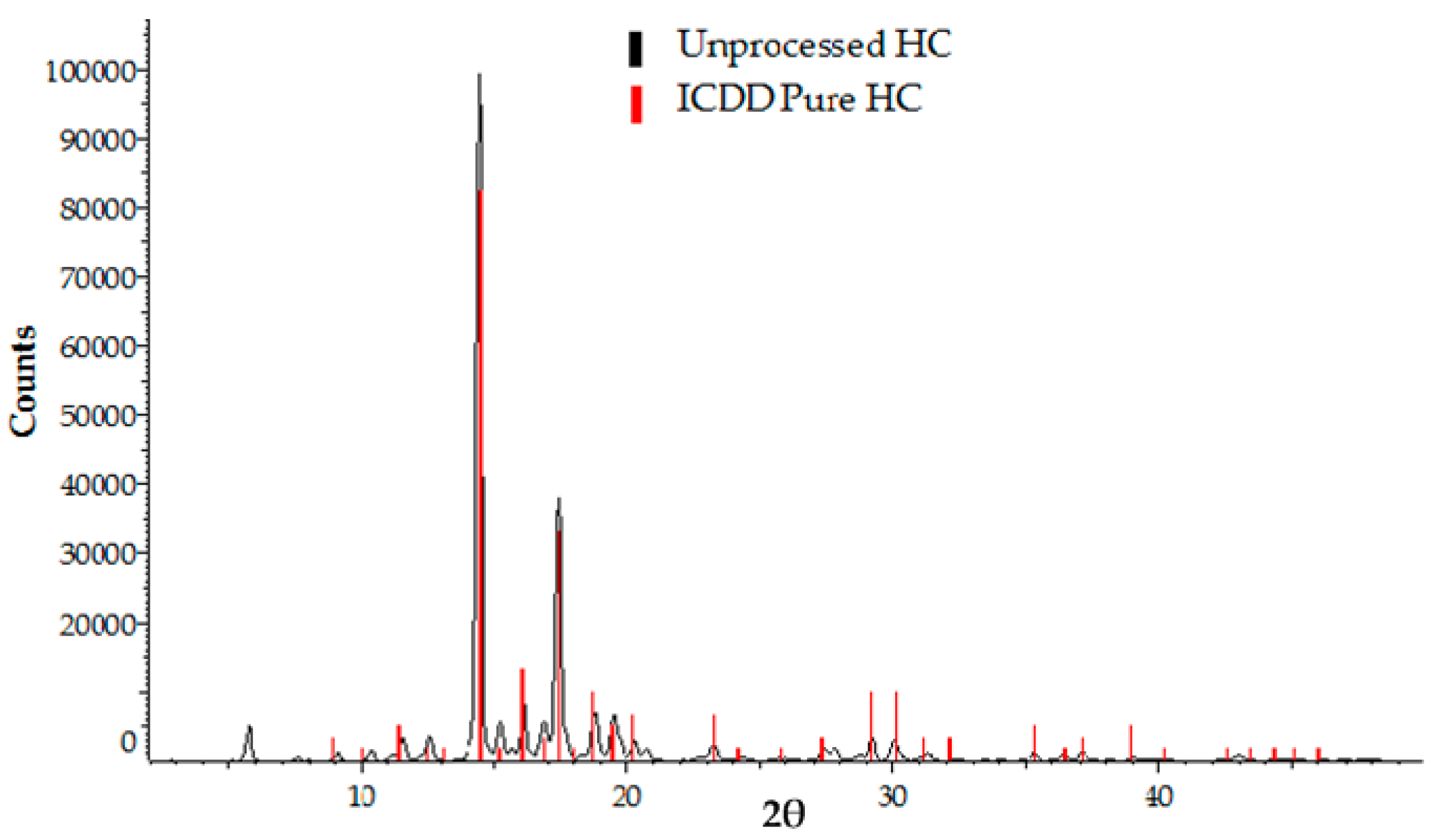
3.2.2. X-ray Powder Diffraction (XRPD) Analysis
3.3. Morphology of the Produced Nanocrystals
3.4. The Effect of Operating Parameters and Geometry on the Size of Nanocrystals
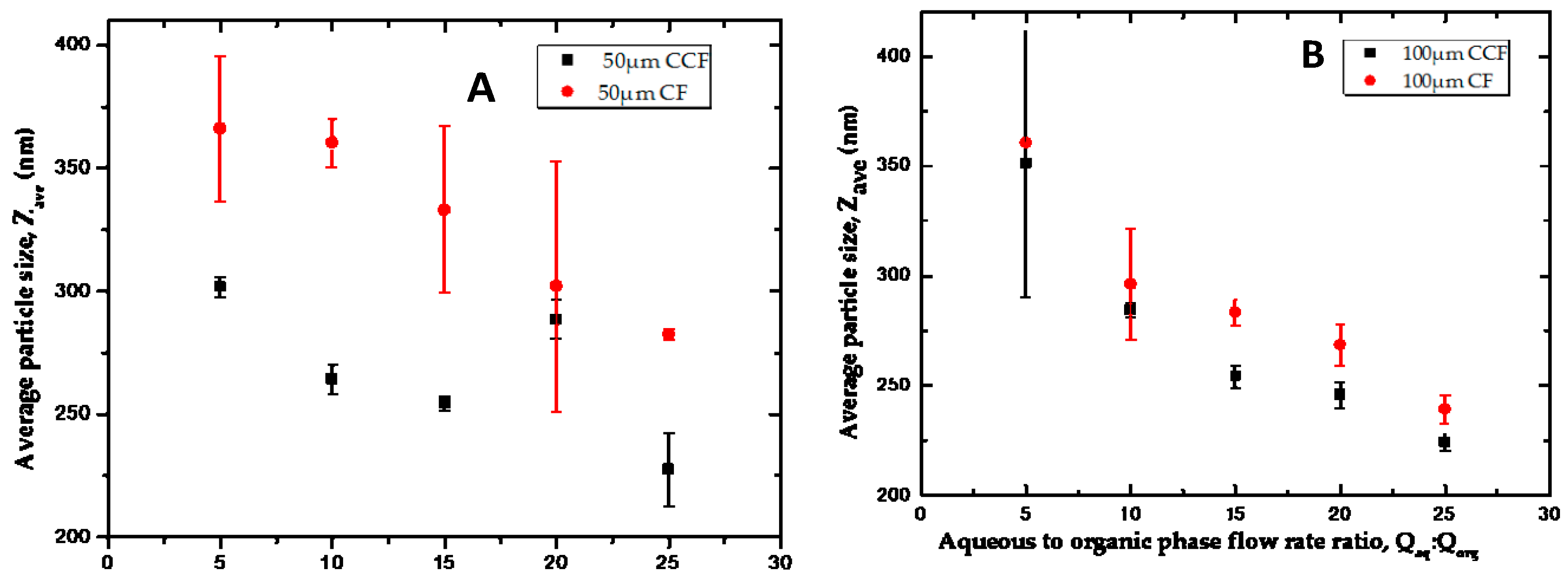
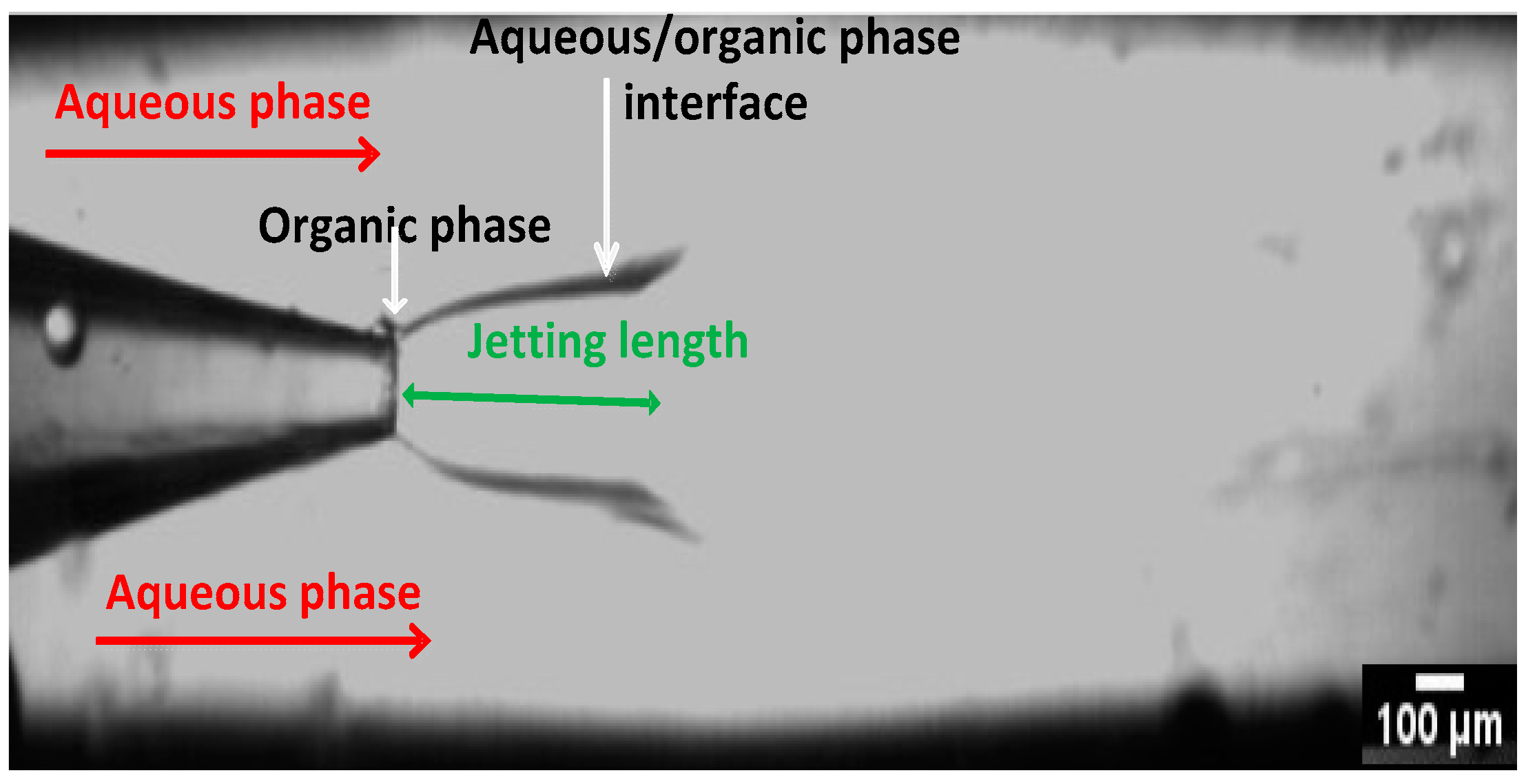
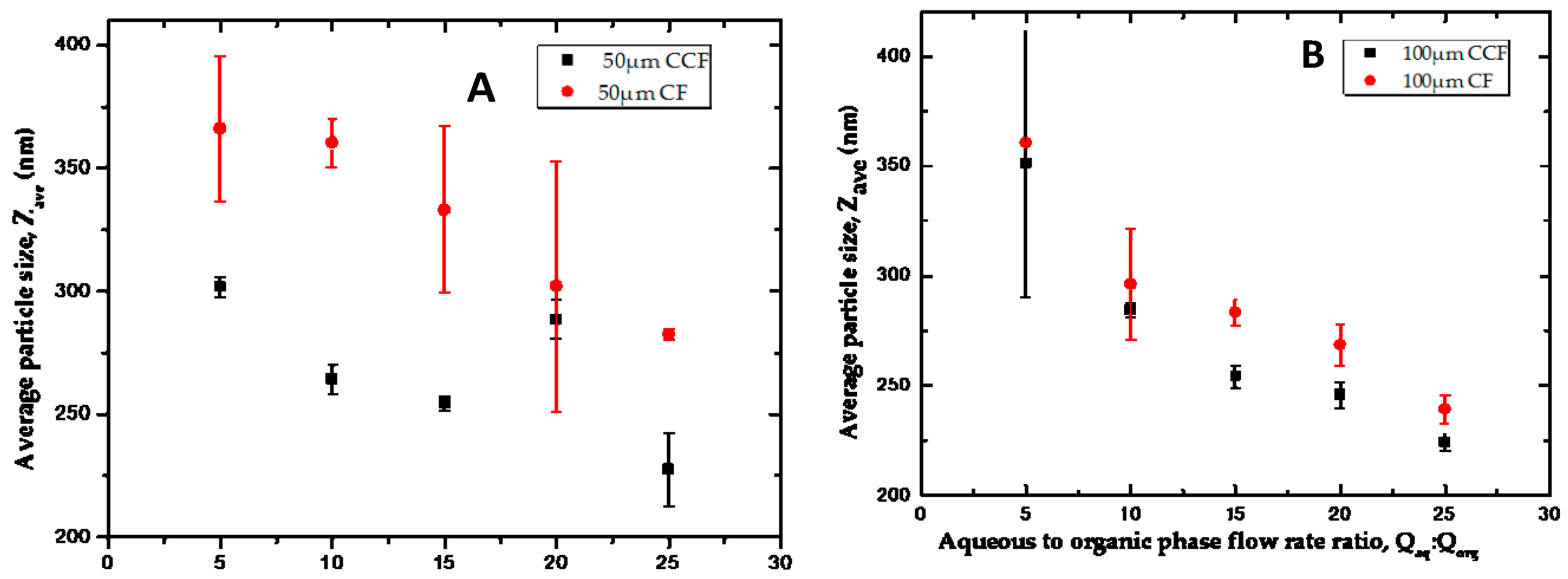
3.4.1. Counter Current vs. Co-current Flow
3.4.2. The Effect of Flow Rate Ratio
3.4.3. The Effect of Device Geometry
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Squires, T.; Quake, S. Microfluidics: Fluid physics at the nanoliter scale. Rev. Mod. Phys. 2005, 77, 977–1026. [Google Scholar] [CrossRef]
- Van der Woerd, M.; Ferree, D.; Pusey, M. The promise of macromolecular crystallization in microfluidic chips. J. Struct. Biol. 2003, 142, 180–187. [Google Scholar] [CrossRef]
- Zhao, C.X.; Middelberg, A.P.J. Two-Phase microfluidic flows. Chem. Eng. Sci. 2011, 66, 1394–1411. [Google Scholar] [CrossRef]
- Yang, S.; Guo, F.; Kiraly, B.; Mao, X.; Lu, M.; Leong, K.W.; Huang, T.J. Microfluidic synthesis of multifunctional Janus particles for biomedical applications. Lab Chip 2012, 12, 2097–2102. [Google Scholar] [CrossRef] [PubMed]
- Masand, S.N.; Mignone, L.; Zahn, J.D.; Shreiber, D.I. Nanoporous membrane-sealed microfluidic devices for improved cell viability. Biomed. Microdevices 2011, 13, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.S.M.; York, P.; Blagden, N. Preparation of hydrocortisone nanosuspension through a bottom-up nanoprecipitation technique using microfluidic reactors. Int. J. Pharm. 2009, 375, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.S.M.; York, P.; Ali, A.M.A.; Blagden, N. Hydrocortisone nanosuspensions for ophthalmic delivery: A comparative study between microfluidic nanoprecipitation and wet milling. J. Control. Release 2011, 149, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Vladisavljević, G.; Kobayashi, I.; Nakajima, M. Production of uniform droplets using membrane, microchannel and microfluidic emulsification devices. Microfluid. Nanofluid. 2012, 13, 151–178. [Google Scholar] [CrossRef] [Green Version]
- Vladisavljević, G.T.; Laouini, A.; Charcosset, C.; Fessi, H.; Bandulasena, H.C.; Holdich, R.G. Production of liposomes using microengineered membrane and co-flow microfluidic device. Colloids Surf. 2014, 458, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Anna, S.L.; Bontoux, N.; Stone, H.A. Formation of dispersions using “flow focusing” in microchannels. Appl. Phys. Lett. 2003, 82, 364–366. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Kikuchi, Y.; Nakajima, M. Regular-Sized cell creation in microchannel emulsification by visual microprocessing method. J. Am. Oil Chem. Soc. 1997, 74, 317–321. [Google Scholar] [CrossRef]
- Thorsen, T.; Roberts, R.W.; Arnold, F.H.; Quake, S.R. Dynamic pattern formation in a vesicle-generating microfluidic device. Phys. Rev. Lett. 2001, 86, 4163–4166. [Google Scholar] [CrossRef] [PubMed]
- Utada, A.; Lorenceau, E.; Link, D. Monodisperse double emulsions generated from a microcapillary device. Science 2005, 308, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Chen, A.; Colombo, P.; Martinez, C.; Meccanica, I.; Universita, S.M. Ceramic microparticles and capsules via microfluidic processing of a preceramic polymer. J. R. Soc. Interface 2010, 7, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Frenz, L.; El Harrak, A.; Pauly, M.; Bégin-Colin, S.; Griffiths, A.D.; Baret, J.-C. Droplet-based microreactors for the synthesis of magnetic iron oxide nanoparticles. Angew. Chem. Int. Ed. Engl. 2008, 47, 6817–6820. [Google Scholar] [CrossRef] [PubMed]
- Gómez-de Pedro, S.; Puyol, M.; Alonso-Chamarro, J. Continuous flow synthesis of nanoparticles using ceramic microfluidic devices. Nanotechnology 2010, 21, 415603. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Huang, K.S.; Lin, Y.S.; Lu, K.; Tzeng, C.C.; Wang, E.C.; Lin, C.-H.; Hsu, W.Y.; Chang, J.Y. Microfluidic assisted synthesis of multi-functional polycaprolactone microcapsules: Incorporation of CdTe quantum dots, Fe3O4 superparamagnetic nanoparticles and tamoxifen anticancer drugs. Lab Chip 2009, 9, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Fisher, J.; Lee, A.; Cristini, V.; Lee, A. Design of microfluidic channel geometries for the control of droplet volume, chemical concentration, and sorting. Lab Chip 2004, 4, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Tice, J.D.; Ismagilov, R.F. Formation of droplets of alternating composition in microfluidic channels and applications to indexing of concentrations in droplet-based assays. Anal. Chem. 2007, 76, 4977–4982. [Google Scholar] [CrossRef] [PubMed]
- Nisisako, T.; Torii, T.; Higuchi, T. Droplet formation in a microchannel network. Lab Chip 2002, 2, 24–26. [Google Scholar] [CrossRef] [PubMed]
- DeMello, A.J. Control and detection of chemical reactions in microfluidic systems. Nature 2006, 442, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Karnik, R.; Gu, F.; Basto, P.; Cannizzaro, C.; Dean, L.; Kyei-Manu, W.; Langer, R.; Farokhzad, O.C. Microfluidic platform for controlled synthesis of polymeric nanoparticles. Nano Lett. 2008, 8, 2906–2912. [Google Scholar] [CrossRef] [PubMed]
- Shestopalov, I.; Tice, J.D.; Ismagilov, R.F. Multi-Step synthesis of nanoparticles performed on millisecond time scale in a microfluidic droplet-based system. Lab Chip 2004, 4, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Krishnadasan, S.; Brown, R.J.C.; deMello, A.J.; deMello, J.C. Intelligent routes to the controlled synthesis of nanoparticles. Lab Chip 2007, 7, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Marre, S.; Jensen, K.F. Synthesis of micro and nanostructures in microfluidic systems. Chem. Soc. Rev. 2010, 39, 1183–1202. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Suresh, P.K. Nanosuspension: A new vehicle for the improvement of the delivery of drugs to the ocular surface. Application to amphotericin B. Nanomedicine 2011, 7, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Orive, G.; Hernández, R.M.; Gascón, A.R.; Domı́nguez-Gil, A.; Pedraz, J.L. Drug delivery in biotechnology: Present and future. Curr. Opin. Biotechnol. 2003, 14, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Kassem, M.A.; Abdel Rahman, A.A.; Ghorab, M.M.; Ahmed, M.B.; Khalil, R.M. Nanosuspension as an ophthalmic delivery system for certain glucocorticoid drugs. Int. J. Pharm. 2007, 340, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Labhasetwar, V. Nanotechnology for drug and gene therapy: The importance of understanding molecular mechanisms of delivery. Curr. Opin. Biotechnol. 2005, 16, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Langer, R. New methods of drug delivery. Science 1990, 249, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Tuomela, A.; Saarinen, J.; Strachan, C.J.; Hirvonen, J.; Peltonen, L. Production, applications and in vivo fate of drug nanocrystals. J. Drug Deliv. Sci. Technol. 2016, 34, 21–31. [Google Scholar] [CrossRef]
- Gao, L.; Liu, G.; Ma, J.; Wang, X.; Zhou, L.; Li, X. Drug nanocrystals: In vivo performances. J. Control. Release 2012, 160, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, Y.; Wu, W. Injected nanocrystals for targeted drug delivery. Acta Pharm. Sin. B. 2016, 2, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Junghanns, J.U.A.H.; Müller, R.H. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295–309. [Google Scholar]
- Shegokar, R.; Müller, R.H. Nanocrystals: Industrially feasible multifunctional formulation technology for poorly soluble actives. Int. J. Pharm. 2010, 399, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Kaialy, W.; Al Shafiee, M. Recent advances in the engineering of nanosized active pharmaceutical ingredients: Promises and challenges. Adv. Colloid Interface Sci. 2016, 228, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.D.; Mallet, F.P.; Ricard, F.; Heng, J.Y.Y. Pharmaceutical nanocrystals. Curr. Opin. Chem. Eng. 2012, 1, 102–107. [Google Scholar] [CrossRef]
- Müller, R.H.; Gohla, S.; Keck, C.M. State of the art of nanocrystals—Special features, production, nanotoxicology aspects and intracellular delivery. Eur. J. Pharm. Biopharm. 2011, 78, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.; Müller, R.H.; Möschwitzer, J.P. Bottom-up approaches for preparing drug nanocrystals: Formulations and factors affecting particle size. Int. J. Pharm. 2013, 453, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Möschwitzer, J.P. Drug nanocrystals in the commercial pharmaceutical development process. Int. J. Pharm. 2013, 453, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Vladisavljević, G.T.; Shum, H.C.; Weitz, D.A. Control over the shell thickness of core/shell drops in three-phase glass capillary devices. UK Colloids 2011 2012, 115–118. [Google Scholar]
- Deng, N.N.; Wang, W.; Ju, X.J.; Xie, R.; Weitz, D.A.; Chu, L.Y. Wetting-Induced formation of controllable monodisperse multiple emulsions in microfluidics. Lab Chip 2013, 13, 4047–4052. [Google Scholar] [CrossRef] [PubMed]
- Othman, R.; Vladisavljević, G.T.; Bandulasena, H.H.; Nagy, Z.K. Production of polymeric nanoparticles by micromixing in a co-flow microfluidic glass capillary device. Chem. Eng. J. (Loughb. Engl.) 2015, 280, 316–329. [Google Scholar] [CrossRef] [Green Version]
- Othman, R.; Vladisavljević, G.T.; Thomas, N.L.; Nagy, Z.K. Fabrication of composite poly (d, l-lactide)/montmorillonite nanoparticles for controlled delivery of acetaminophen by solvent-displacement method using glass capillary microfluidics. Colloids Surf. B 2016, 141, 187–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laouini, A.; Koutroumanis, K.P.; Charcosset, C.; Georgiadou, S.; Fessi, H.; Holdich, R.G.; Vladisavljević, G.T. pH-Sensitive micelles for targeted drug delivery prepared using a novel membrane contactor method. ACS Appl. Mater. Interfaces 2013, 5, 8939–8947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Chowdhury, D.; Vladisavljević, G.T.; Koutroumanis, K.; Georgiadou, S. Production of fluconazole-Loaded polymeric micelles using membrane and microfluidic dispersion devices. Membranes 2016, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidya, B.; Chakera, A.J.; Dick, C. Addison’s disease. BMJ 2009, 339, b2385. [Google Scholar] [CrossRef] [PubMed]
- Wass, J.A.H.; Arlt, W. How to avoid precipitating an acute adrenal crisis. BMJ 2012, 345, e6333. [Google Scholar] [CrossRef] [PubMed]
- Hyvönen, S.; Peltonen, L.; Karjalainen, M.; Hirvonen, J. Effect of nanoprecipitation on the physicochemical properties of low molecular weight poly(L-lactic acid) nanoparticles loaded with salbutamol sulphate and beclomethasone dipropionate. Int. J. Pharm. 2005, 295, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Vaisman, L.; Wagner, H.D.; Marom, G. The role of surfactants in dispersion of carbon nanotubes. Adv. Colloid Interface Sci. 2006, 128–130, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Bilati, U.; Allémann, E.; Doelker, E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur. J. Pharm. Sci. 2005, 24, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.S.M.; York, P.; Blagden, N.; Soltanpour, S.; Acree, W.E.; Jouyban, A. Solubility of Budesonide, Hydrocortisone, and Prednisolone in Ethanol + Water Mixtures at 298.2 K. J. Chem. Eng. Data 2010, 55, 578–582. [Google Scholar] [CrossRef]
- Capretto, L.; Cheng, W.; Carugo, D.; Katsamenis, O.L.; Hill, M.; Zhang, X. Mechanism of co-nanoprecipitation of organic actives and block copolymers in a microfluidic environment. Nanotechnology 2012, 23, 375602. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Thomas, N.L.; Huang, W. Water vapour permeability of poly (lactic acid) nanocomposites. J. Memb. Sci. 2013, 445, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Jaipal, A.; Pandey, M.M.; Charde, S.Y.; Raut, P.P.; Prasanth, K.V.; Prasad, R.G. Effect of HPMC and mannitol on drug release and bioadhesion behavior of buccal discs of buspirone hydrochloride: In-Vitro and in vivo pharmacokinetic studies. Saudi Pharm. J. 2015, 23, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Priya James, H.; John, R.; Alex, A.; Anoop, K.R. Smart polymers for the controlled delivery of drugs—A concise overview. Acta Pharm. Sin. B 2014, 4, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.; Garside, J. From Molecules to Crystallizers; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Teja, S.B.; Patil, S.P.; Shete, G.; Patel, S.; Bansal, A.K. Drug-Excipient behavior in polymeric amorphous solid dispersions. J. Excip. Food Chem. 2013, 4, 70–94. [Google Scholar]
- Chen, J.; Wang, J.; Zhang, Y.; Wu, H.; Cheng, W.; Guo, Z. Crystal growth, structure and morphology of hydrocortisone methanol solvate. J. Cryst. Growth 2004, 265, 266–273. [Google Scholar] [CrossRef]
- Aoki, N.; Mae, K. Effects of channel geometry on mixing performance of micromixers using collision of fluid segments. Chem. Eng. J. 2006, 118, 189–197. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, J.X.; Wang, Q.A.; Chen, J.F.; Yun, J. Controlled liquid antisolvent precipitation of hydrophobic pharmaceutical nanoparticles in a microchannel reactor. Ind. Eng. Chem. Res. 2007, 46, 8229–8235. [Google Scholar] [CrossRef]
- Othman, R.; Vladisavljević, G.T.; Nagy, Z.K. Preparation of biodegradable polymeric nanoparticles for pharmaceutical applications using glass capillary microfluidics. Chem. Eng. Sci. 2015, 137, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Jahn, A.; Stavis, S.M.; Hong, J.S.; Vreeland, W.N.; DeVoe, D.L.; Gaitan, M. Microfluidic mixing and the formation of nanoscale lipid vesicles. ACS Nano 2010, 4, 2077–2087. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flow Rates (Qaq:Qorg, mL/h:mL/h) | 50 μm Orifice Device | 100 μm Orifice Device |
|---|---|---|
| PDI | PDI | |
| 6:6 | 0.253 ± 0.005 | 0.236 ± 0.039 |
| 18:6 | 0.405 ± 0.038 | 0.203 ± 0.038 |
| 30:6 | 0.612 ± 0.086 | 0.327 ± 0.023 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Odetade, D.F.; Vladisavljevic, G.T. Microfluidic Fabrication of Hydrocortisone Nanocrystals Coated with Polymeric Stabilisers. Micromachines 2016, 7, 236. https://doi.org/10.3390/mi7120236
Odetade DF, Vladisavljevic GT. Microfluidic Fabrication of Hydrocortisone Nanocrystals Coated with Polymeric Stabilisers. Micromachines. 2016; 7(12):236. https://doi.org/10.3390/mi7120236
Chicago/Turabian StyleOdetade, David F., and Goran T. Vladisavljevic. 2016. "Microfluidic Fabrication of Hydrocortisone Nanocrystals Coated with Polymeric Stabilisers" Micromachines 7, no. 12: 236. https://doi.org/10.3390/mi7120236
APA StyleOdetade, D. F., & Vladisavljevic, G. T. (2016). Microfluidic Fabrication of Hydrocortisone Nanocrystals Coated with Polymeric Stabilisers. Micromachines, 7(12), 236. https://doi.org/10.3390/mi7120236