
Photochemical Synthesis and Versatile Functionalization Method of a Robust Porous Poly(ethylene glycol methacrylate-co-allyl methacrylate) Monolith Dedicated to Radiochemical Separation in a Centrifugal Microfluidic Platform

Abstract
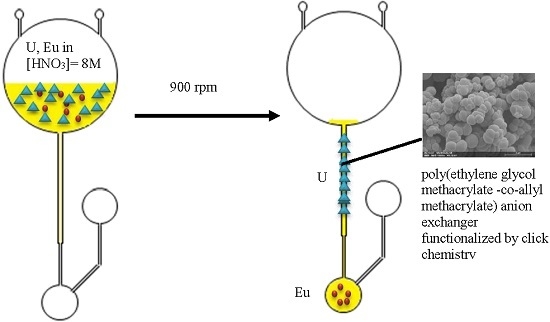
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of the Poly(ethylene glycol methacrylate methacrylate-co-allyl methacrylate) Monolith (also Referred to as Poly(EDMA-co-AMA) Monolith)
2.3. Functionalization of the Monolith
3. Results and Discussion
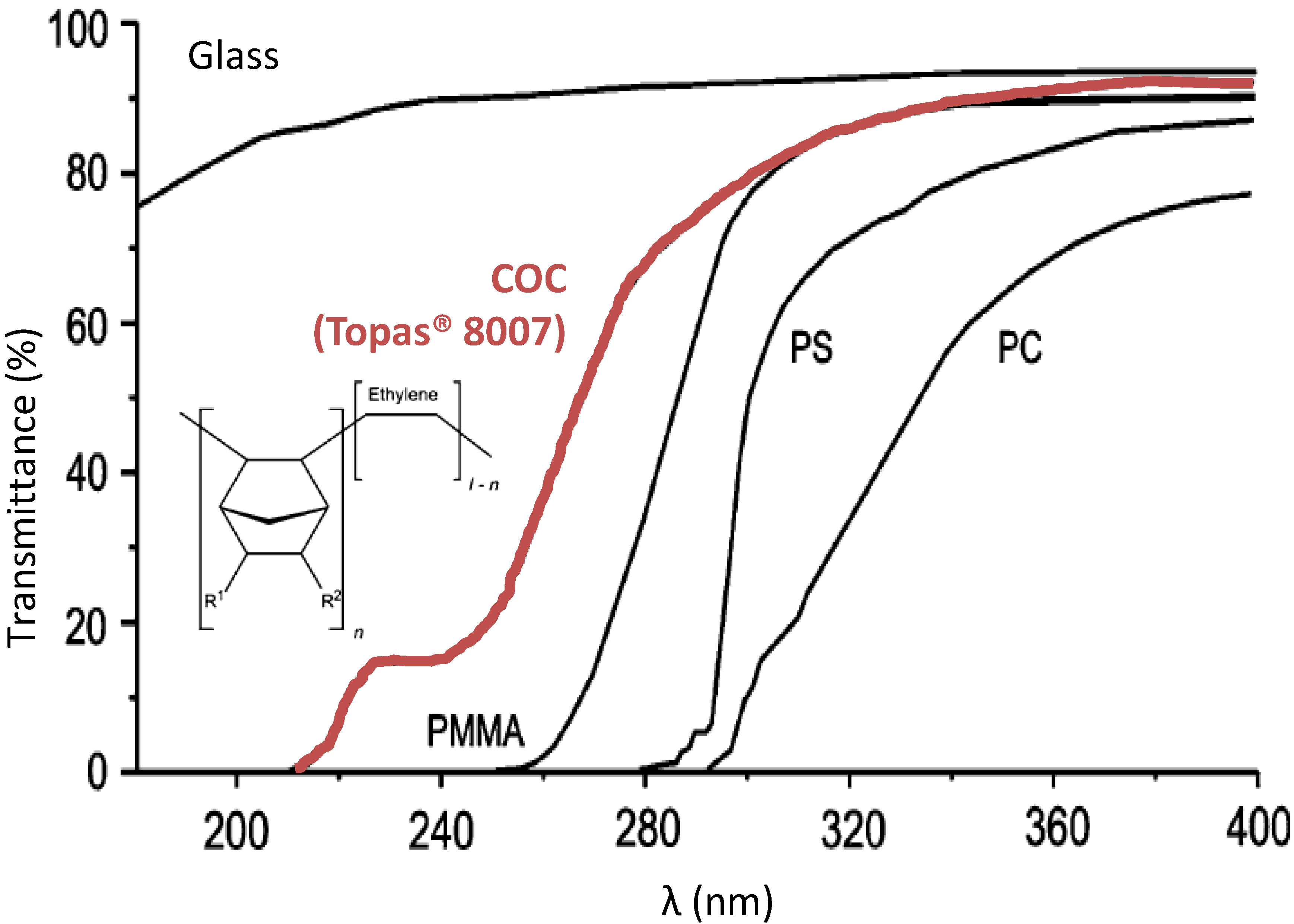
3.1. Monomers Choice and Optimization of the Monolith Synthesis
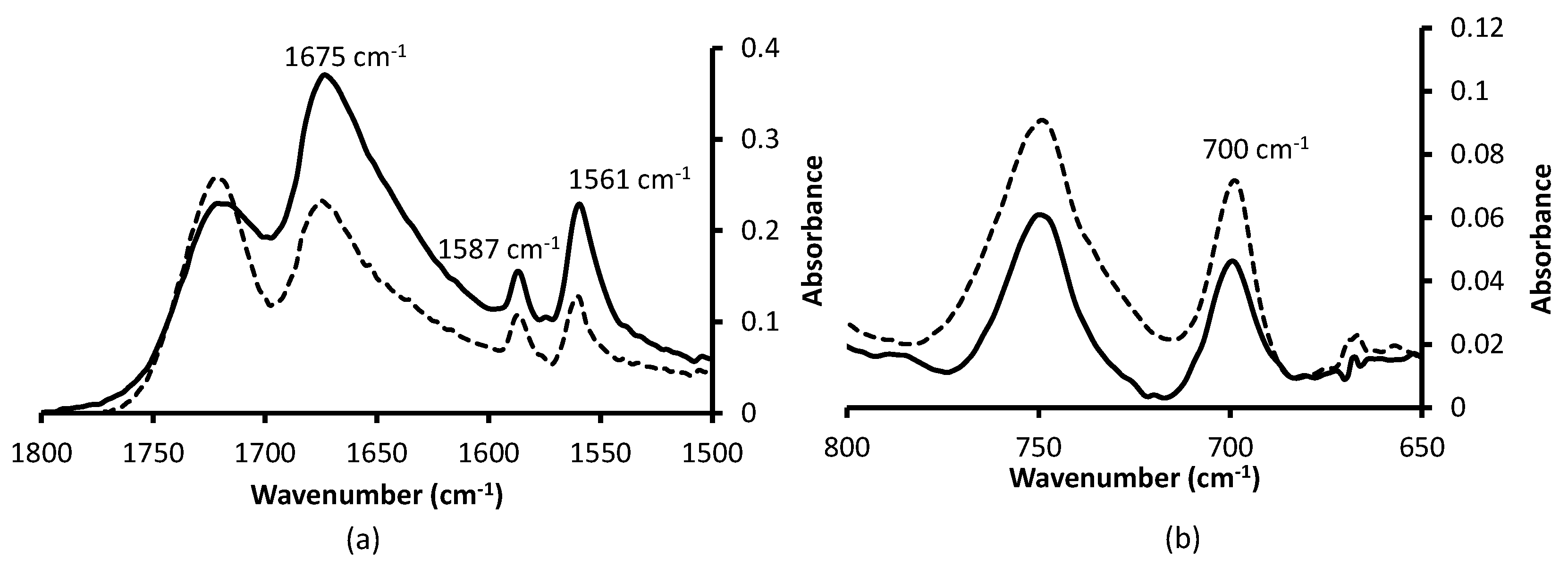
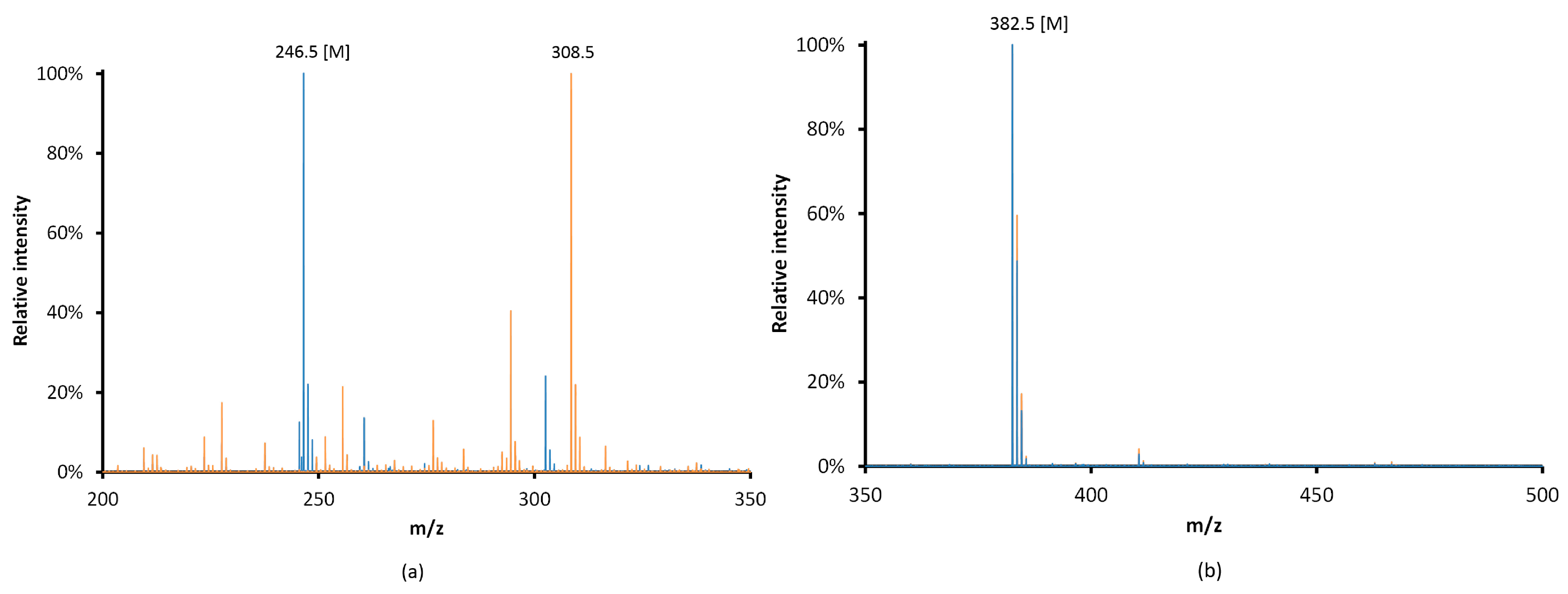
3.2. Choice and Optimization of a Robust and Versatile Functionalization Method of the Monolith
3.3. Robustness in Nitric Acid Medium
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Horwitz, E.P.; Chiarizia, R.; Dietz, M.L.; Diamond, H.; Nelson, D.M. Separation and preconcentration of actinides from acidic media by extraction chromatography. Anal. Chim. Acta 1993, 281, 361–372. [Google Scholar] [CrossRef]
- Janssens-Maenhout, G. The benefits of applying microsystems in radiochemistry. Nanotechnol. Percept. 2007, 3, 183–192. [Google Scholar] [CrossRef]
- Janssens-Maenhout, G.; Buyst, J.; Peerani, P. Reducing the radioactive doses of liquid samples taken from reprocessing plant vessels by volume reduction. Nucl. Eng. Des. 2007, 237, 880–886. [Google Scholar] [CrossRef]
- Toulhoat, P. Défis actuels et à venir en matière d’analyse de traces et d’ultra-traces. Oil Gas Sci. Technol. Rev. IFP 2005, 60, 967–977. [Google Scholar] [CrossRef]
- Gorkin, R.; Park, J.; Siegrist, J.; Amasia, M.; Lee, B.S.; Park, J.-M.; Kim, J.; Kim, H.; Madou, M.; Cho, Y.-K. Centrifugal microfluidics for biomedical applications. Lab Chip 2010, 10, 1758–1773. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.N.; Li, B.S. Lab-on-CD microfluidic platform for rapid separation and mixing of plasma from whole blood. Biomed. Microdevices 2014, 16, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.X.; Perebikovsky, A.; Moebius, J.; Kulinsky, L.; Madou, M. Lab-on-a-CD: A fully integrated molecular diagnostic system. J. Lab Autom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Madou, M.; Zoval, J.; Jia, G.; Kido, H.; Kim, J.; Kim, N. Lab on a CD. Annu. Rev. Biomed. Eng. 2006, 8, 601–628. [Google Scholar] [CrossRef] [PubMed]
- Amasia, M.; Madou, M. Large-volume centrifugal microfluidic device for blood plasma separation. Bioanalysis 2010, 2, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Strohmeier, O.; Keller, M.; Schwemmer, F.; Zehnle, S.; Mark, D.; von Stetten, F.; Zengerle, R.; Paust, N. Centrifugal microfluidic platforms: Advanced unit operations and application. Chem. Soc. Rev. 2015, 44, 6187–6229. [Google Scholar] [CrossRef] [PubMed]
- Ducrée, J.; Brenner, T.; Glatzel, T.; Zengerle, R. Ultrafast micromixing by coriolis-induce multi-lamination of centrifugal flow. In Proceedings of the 9th International Conference on New Actuators, Bremen, Germany, 14–16 June 2004; pp. 533–536.
- Kim, B.Y.; Yang, J.; Gong, M.; Flachsbart, B.R.; Shannon, M.R.; Bohn, V.R.; Sweedler, J.V. Multidimensional Separation of Chiral Amino Acid Mixtures in a Multilayered Three-Dimensional Hybrid Microfluidic/Nanofluidic Device. Anal. Chem. 2009, 81, 2715–2722. [Google Scholar] [CrossRef] [PubMed]
- Siegrist, J.; Gorkin, R.; Bastien, M.; Stewart, G.; Peytavi, R.; Kido, H.; Bergeron, M.; Madou, M. Validation of a centrifugal microfluidic sample lysis and homogenization platform for nucleic acid extraction with clinical samples. Lab Chip 2010, 10, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Lafleur, J.P.; Salin, E.D. Pre-concentration of trace metals on centrifugal microfluidic discs with direct determination by laser ablation inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 2009, 24, 1511–1516. [Google Scholar] [CrossRef]
- Bruchet, A.; Taniga, V.; Descroix, S.; Malaquin, L.; Goutelard, F.; Mariet, C. Centrifugal microfluidic platform for radiochemistry: Potentialities for the chemical analysis of nuclear spent fuels. Talanta 2013, 116, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Penrose, A.; Myers, P.; Bartle, K.; McCrossen, S. Development and assessment of a miniaturised centrifugal chromatograph for reversed-phase separations in micro-channels. Analyst 2004, 129, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Svec, F.; Lv, Y. Advances and recent trends in the field of monolithic columns for chromatography. Anal. Chem. 2015, 87, 250–273. [Google Scholar] [CrossRef] [PubMed]
- Nunes, P.S.; Ohlsson, P.D.; Ordeig, O.; Kutter, J.P. Cyclic olefin polymers: emerging materials for lab-on-a-chip applications. Microfluid. Nanofluid. 2010, 9, 145–161. [Google Scholar] [CrossRef]
- Svec, F.; Frechet, J.M.J. Continuous rods of macroporous polymer as high-performance liquid chromatography separation media. Anal. Chem. 1992, 64, 820–822. [Google Scholar] [CrossRef]
- Peters, E.C.; Petro, M.; Svec, F.; Fréchet, J.M. Molded rigid polymer monoliths as separation media for capillary electrochromatography. Anal. Chem. 1997, 69, 3646–3649. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, C.F.; Tsao, C.W.; Chang, C.C.; Chu, C.C.; DeVoe, D.L. Polymer microchips integrating solid-phase extraction and high-performance liquid chromatography using reversed-phase polymethacrylate monoliths. Anal. Chem. 2009, 81, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Svec, F.; Fréchet, J.M.J. Towards stationary phases for chromatography on a microchip: Molded porous polymer monoliths prepared in capillaries by photoinitiated in situ polymerization as separation media for electrochromatography. Electrophoresis 2000, 21, 120–127. [Google Scholar] [CrossRef]
- Yu, C.; Davey, M.H.; Svec, F.; Fréchet, J.M. Monolithic porous polymer for on-chip solid-phase extraction and preconcentration prepared by photoinitiated in situ polymerization within a microfluidic device. Anal. Chem. 2001, 73, 5088–5096. [Google Scholar] [CrossRef] [PubMed]
- Puangpila, C.; Nhujak, T.; El Rassi, Z. Investigation of neutral monolithic capillary columns with varying n-alkyl chain lengths in capillary electrochromatography. Electrophoresis 2012, 33, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Danquah, M.K.; Forde, M.F. Enhancing methacrylate-monolith-based downstream processes to champion plasmid DNA production. Biotechnol. Appl. Biochem. 2007, 48, 85–91. [Google Scholar] [PubMed]
- Stachowiak, T.B.; Rohr, T.; Hilder, E.F.; Peterson, D.S.; Yi, M.; Svec, F.; Fréchet, J.M. Fabrication of porous polymer monoliths covalently attached to the walls of channels in plastic microdevices. Electrophoresis 2003, 24, 3689–3693. [Google Scholar] [CrossRef] [PubMed]
- Connolly, D.; O’Shea, V.; Clark, P.; O’Connor, B.; Paull, B. Evaluation of photografted charged sites within polymer monoliths in capillary columns using contactless conductivity detection. J. Sep. Sci. 2007, 30, 3060–3068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tehfe, M.A. Study of a New Initiating Systems for Radical or Cationic Polymerization; Université de Haute Alsace: Mulhouse, France, 2011. [Google Scholar]
- Le Gac, S.; Carlier, J.; Camart, J.C.; Cren-Olivé, C.; Rolando, C. Monoliths for microfluidic devices in proteomics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 808, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Svec, F. Quest for organic polymer-based monolithic columns affording enhanced efficiency in high performance liquid chromatography separations of small molecules in isocratic mode. J. Chromatogr. A 2012, 1228, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, R.A.; Valdéz, A.E.G.; Aguilar, M.G.M.; Duarte, M.L.B. An effective method to prepare sucrose polymers by Thiol-Ene photopolymerization. Carbohydr. Polym. 2009, 78, 282–286. [Google Scholar] [CrossRef]
- Xie, S.; Svec, F.; Fréchet, J.M.J. Preparation of porous hydrophilic monoliths: Effect of the polymerization conditions on the porous properties of poly (acrylamide-co-N,N′-methylenebisacrylamide) monolithic rods. J. Polym. Sci. A Poly. Chem. 1997, 35, 1013–1021. [Google Scholar] [CrossRef]
- Yu, C.; Xu, M.; Svec, F.; Fréchet, J.M.J. Preparation of monolithic polymers with controlled porous properties for microfluidic chip applications using photoinitiated free-radical polymerization. J. Polym. Sci. A Poly. Chem. 2002, 40, 755–769. [Google Scholar] [CrossRef]
- Bruchet, A.; Dugas, V.; Mariet, C.; Goutelard, F.; Randon, J. Improved chromatographic performances of glycidyl methacrylate anion-exchange monolith for fast nano-ion exchange chromatography. J. Sep. Sci. 2011, 34, 2079–2087. [Google Scholar] [CrossRef] [PubMed]
- Rahmi, D.; Takasaki, Y.; Zhu, Y.; Kobayashi, H.; Konagaya, S.; Haraguchi, H.; Umemura, T. Preparation of monolithic chelating adsorbent inside a syringe filter tip for solid phase microextraction of trace elements in natural water prior to their determination by ICP-MS. Talanta 2010, 81, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Moyna, A.; Connolly, D.; Nesterenko, E.; Nesterenko, P.N.; Paull, B. Separation of selected transition metals by capillary chelation ion chromatography using acetyl-iminodiacetic acid modified capillary polymer monoliths. J. Chromatogr. A 2012, 1249, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Ueki, Y.; Umemura, T.; Iwashita, Y.; Odake, T.; Haraguchi, H.; Tsunoda, K. Preparation of low flow-resistant methacrylate-based monolithic stationary phases of different hydrophobicity and the application to rapid reversed-phase liquid chromatographic separation of alkylbenzenes at high flow rate and elevated temperature. J. Chromatogr. A 2006, 1106, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Rasband, W. ImageJ. Available online: http://imagej.nih.gov/ij/ (accessed on 8 March 2016).
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. Engl. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Marechal, A.; El-Debs, R.; Dugas, V.; Demesmay, C. Is click chemistry attractive for separation sciences? J. Sep. Sci. 2013, 36, 2049–2062. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Lin, Z.; Svec, F. “Thiol-ene” click chemistry: A facile and versatile route for the functionalization of porous polymer monoliths. Analyst 2012, 137, 4114–4118. [Google Scholar] [CrossRef] [PubMed]
- Khire, V.S.; Harant, A.W.; Watkins, A.W.; Anseth, K.S.; Bowman, C.N. Ultrathin patterned polymer films on surfaces using thiol-ene polymerizations. Macromolecules 2006, 39, 5081–5086. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Bowman, C.N. Thiol-ene click chemistry. Angew. Chem. Int. Ed. Engl. 2010, 49, 1540–1573. [Google Scholar] [CrossRef] [PubMed]
- Faris, J.P.; Buchanan, R.F. Anion exchange characteristics of the elements in nitric acid medium. Anal. Chem. 1964, 36, 1157–1158. [Google Scholar] [CrossRef]
- Kressin, I.K.; Waterbury, G.R. The quantitative separation of Plutonium from various ions by anion exchange. Anal. Chem. 1962, 34, 1598–1601. [Google Scholar] [CrossRef]
- Ryan, J.L. Species involved in the anion-exchange absorption of quadrivalent actinide nitrates. J. Phys. Chem. 1960, 64, 1375–1385. [Google Scholar]
- Chiarizia, R.; Gatrone, R.C.; Horwitz, E.P. Am(III) and Eu(III) extraction by Aliquat-336 and benzyl substituted quaternary ammonium salts from nitrate and thiocyanate solutions. Solvent Extr. Ion Exch. 1995, 13, 615–645. [Google Scholar]
- Skoog, D.A.; West, D.M.; Holler, F.J. Chimie Analytique; Boeck and Larcier: Bruxelles, Belgium, 1997. [Google Scholar]
- Silverstein, R.M.; Basler, G.C.; Morill, T.C. Identification Spectrométrique de Composés Organiques; Boeck and Larcier: Bruxelles, Belgium, 1998. [Google Scholar]
- Adriens, A.G.; Fassett, J.D.; Kelly, W.R.; Simons, D.S.; Adams, F.C. Determination of uranium and thorium concentrations in soils. Comparison of isotope dilution-secondary ion mass spectrometry and isotope dilution-thermal ionization mass spectrometry. Anal. Chem. 1992, 64, 2945–2950. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Losno, M.; Ferrante, I.; Brennetot, R.; Varlet, J.; Blanc, C.; Grenut, B.; Amblard, E.; Descroix, S.; Mariet, C. Photochemical Synthesis and Versatile Functionalization Method of a Robust Porous Poly(ethylene glycol methacrylate-co-allyl methacrylate) Monolith Dedicated to Radiochemical Separation in a Centrifugal Microfluidic Platform. Micromachines 2016, 7, 45. https://doi.org/10.3390/mi7030045
Losno M, Ferrante I, Brennetot R, Varlet J, Blanc C, Grenut B, Amblard E, Descroix S, Mariet C. Photochemical Synthesis and Versatile Functionalization Method of a Robust Porous Poly(ethylene glycol methacrylate-co-allyl methacrylate) Monolith Dedicated to Radiochemical Separation in a Centrifugal Microfluidic Platform. Micromachines. 2016; 7(3):45. https://doi.org/10.3390/mi7030045
Chicago/Turabian StyleLosno, Marion, Ivan Ferrante, René Brennetot, Jérôme Varlet, Cécile Blanc, Bernard Grenut, Etienne Amblard, Stéphanie Descroix, and Clarisse Mariet. 2016. "Photochemical Synthesis and Versatile Functionalization Method of a Robust Porous Poly(ethylene glycol methacrylate-co-allyl methacrylate) Monolith Dedicated to Radiochemical Separation in a Centrifugal Microfluidic Platform" Micromachines 7, no. 3: 45. https://doi.org/10.3390/mi7030045