DNA Methylation Predicts the Response of Triple-Negative Breast Cancers to All-Trans Retinoic Acid


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
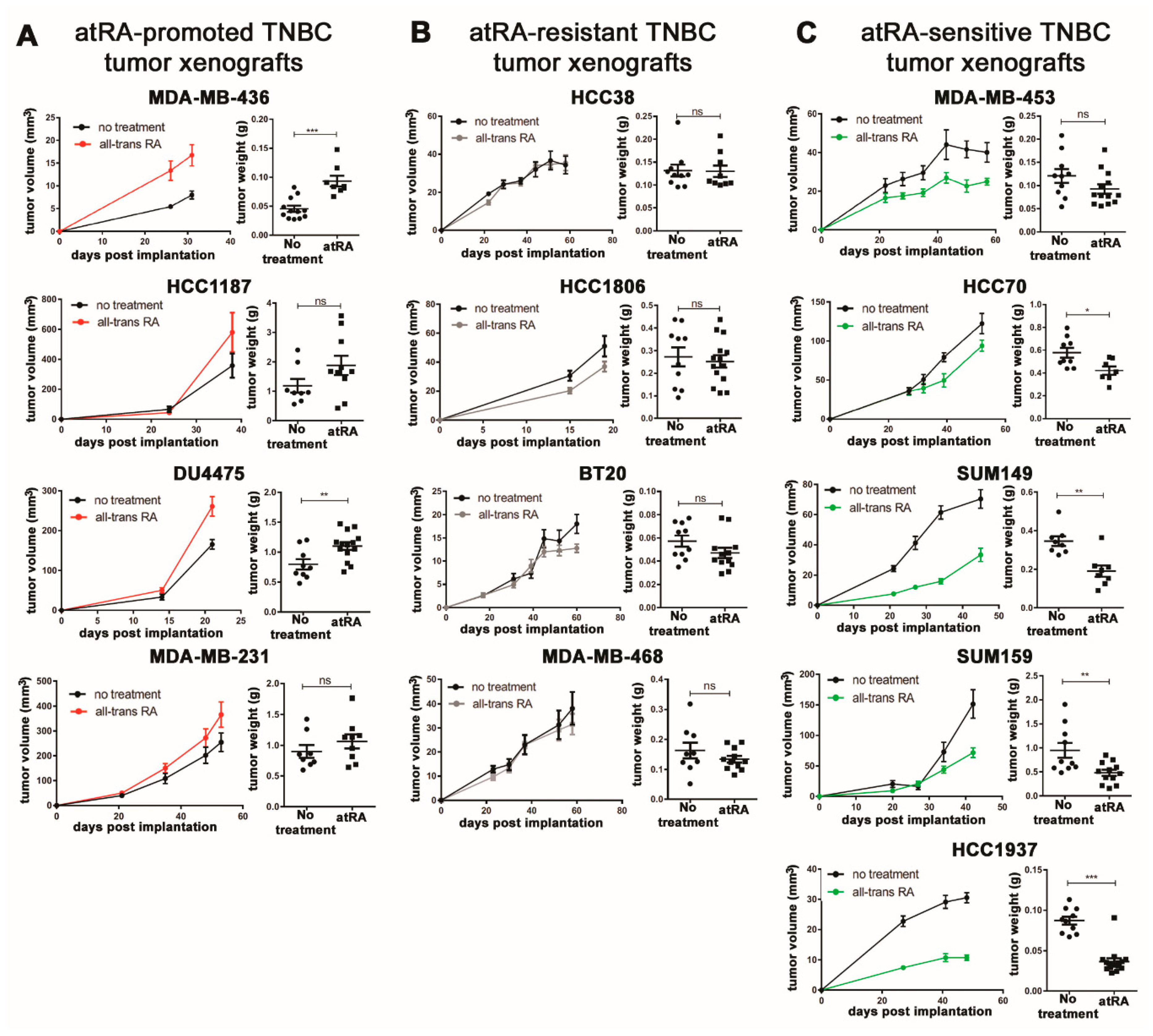
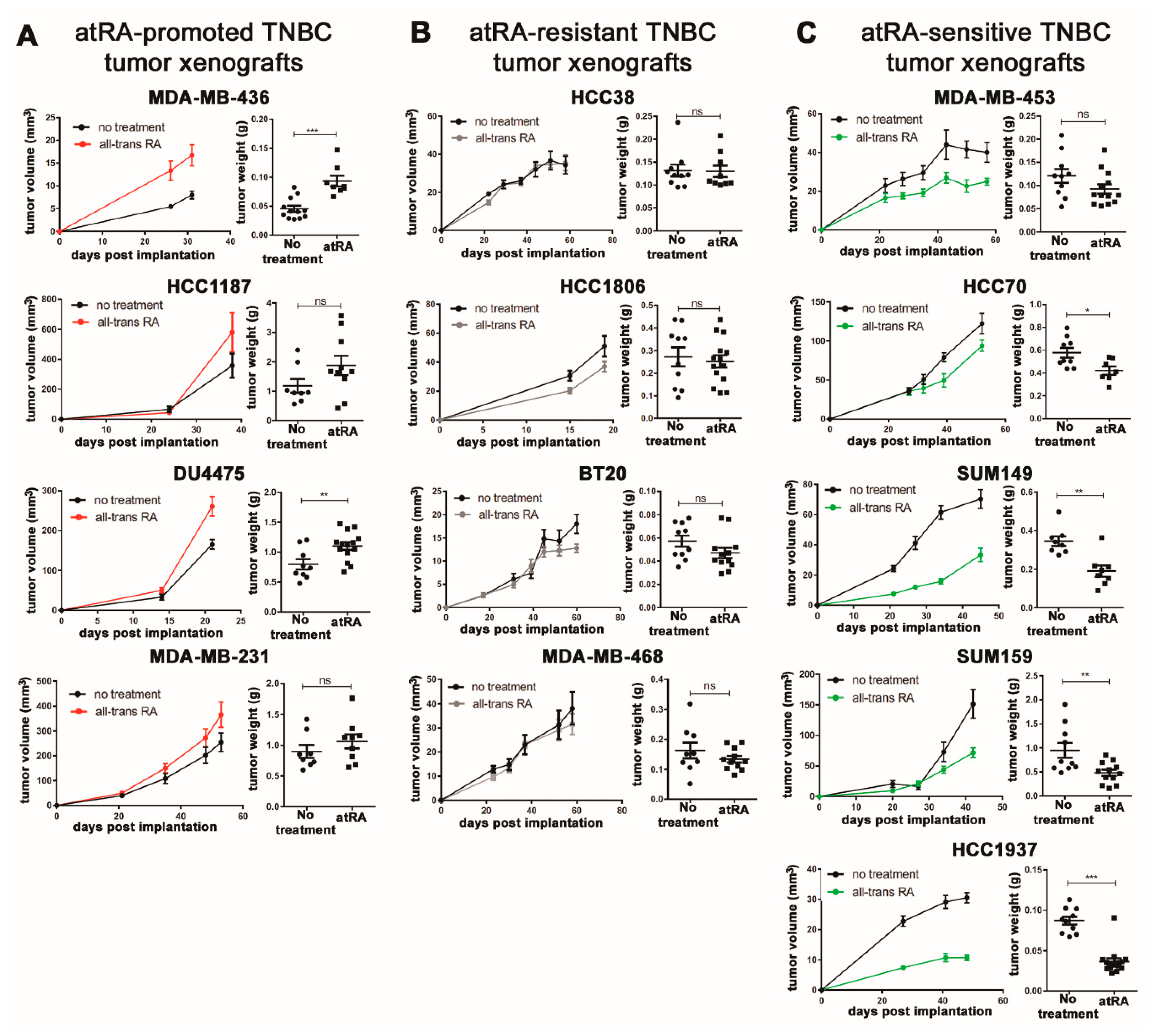
2.1. Breast Cancer Cell Lines Display A Wide Range of In Vivo Responses to atRA
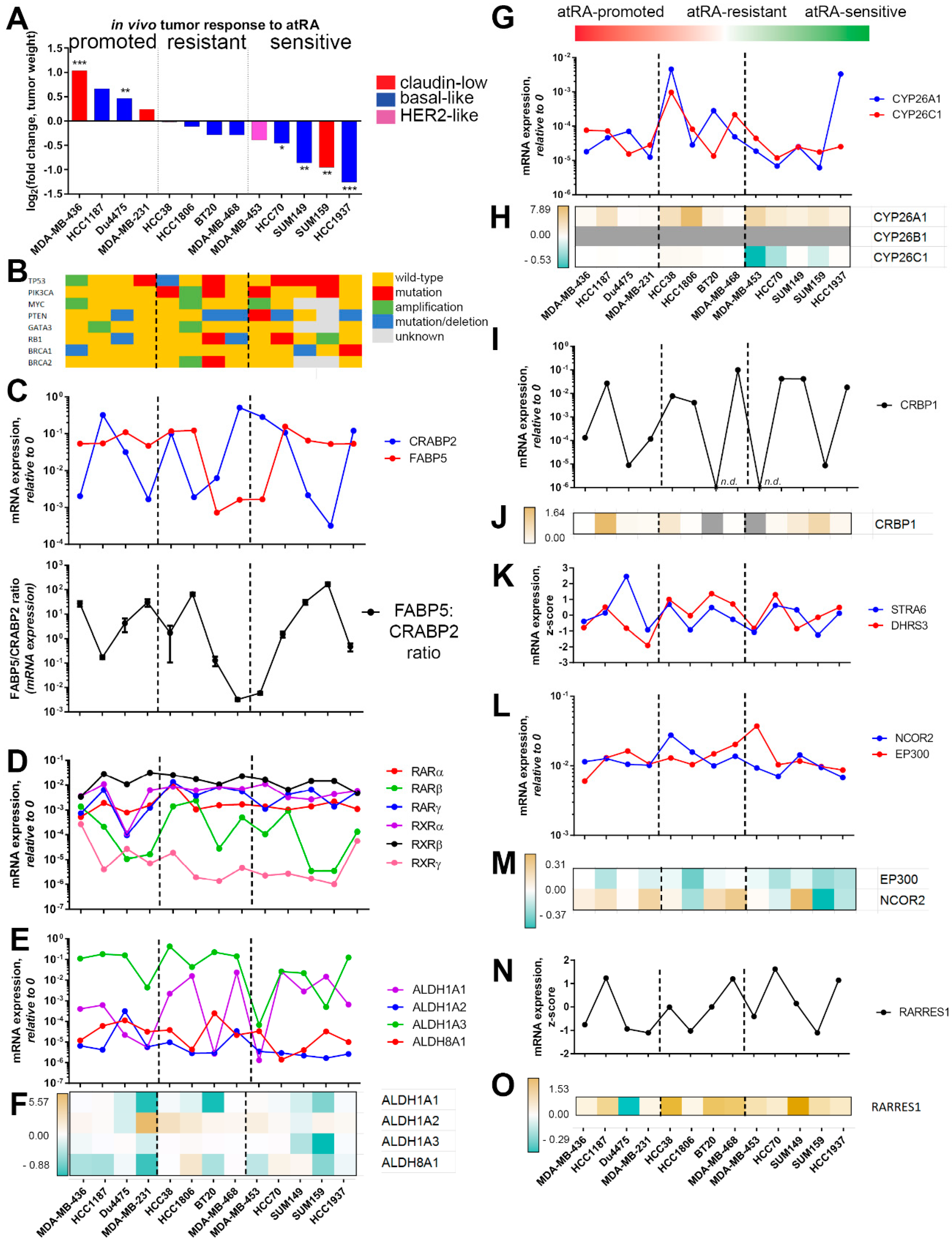
2.2. Expression of Retinoid Pathway Genes Does Not Correlate with atRA Sensitivity
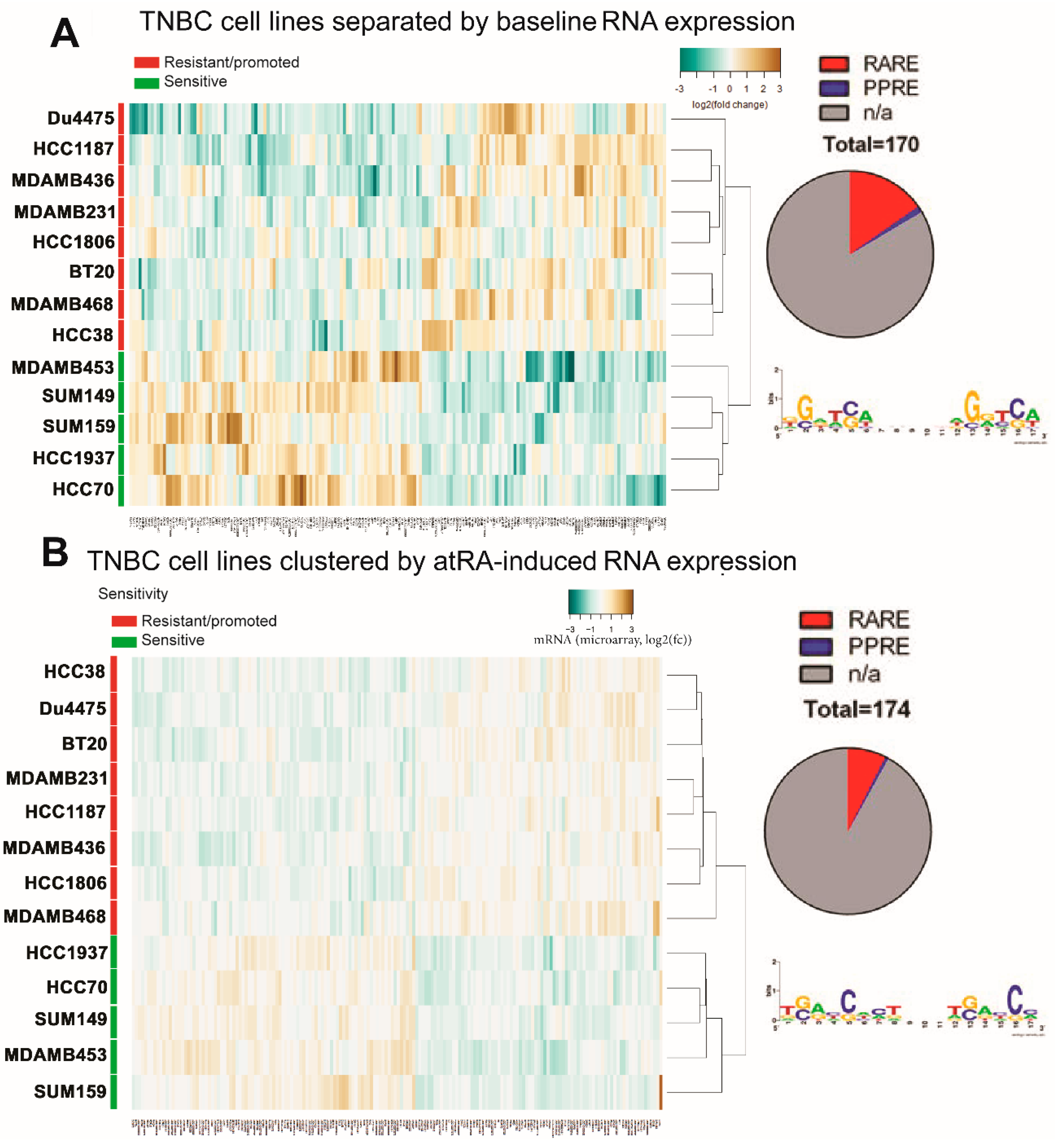
2.3. Differential Gene Expression Is Identified in atRA-Sensitive Cell Lines
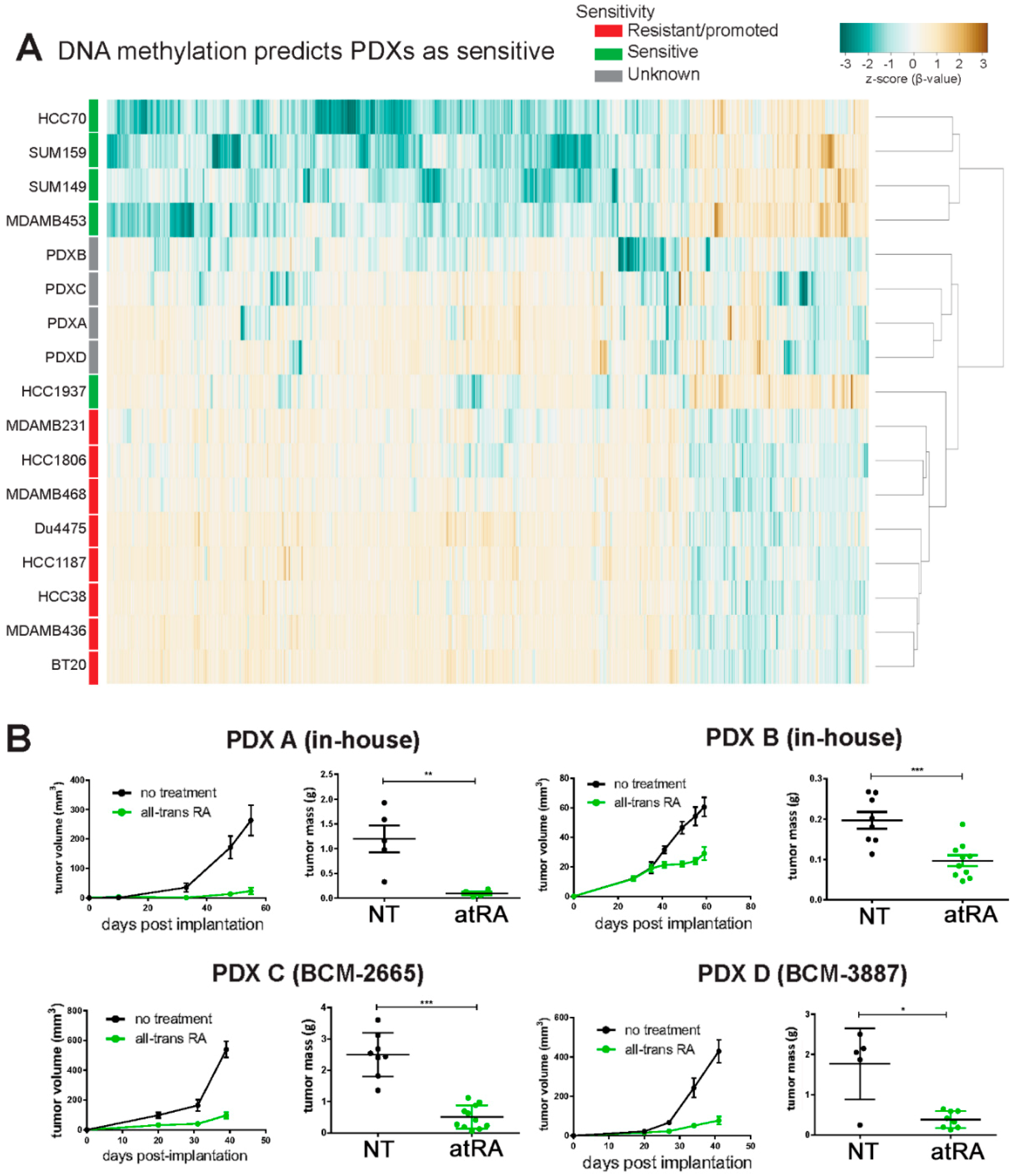
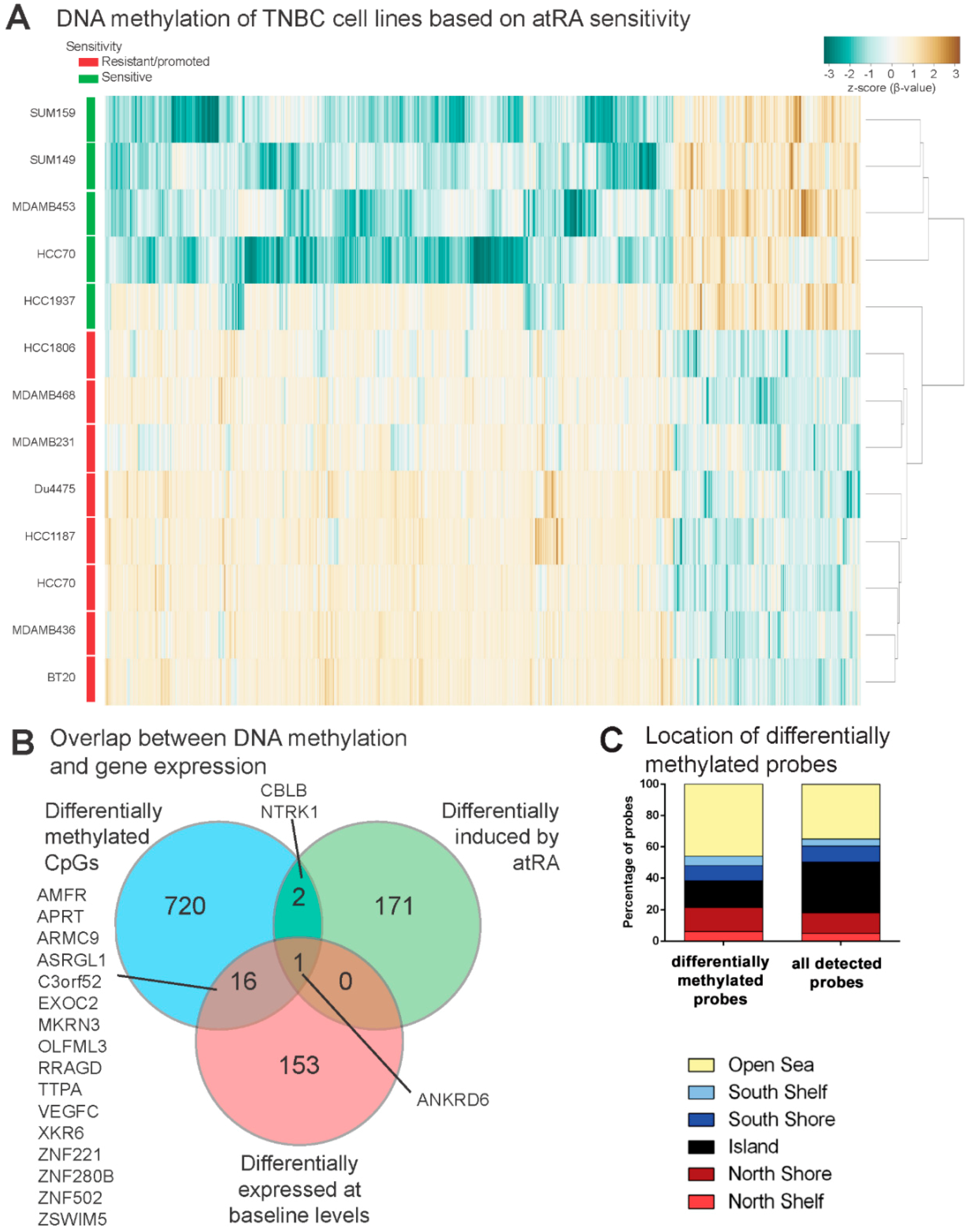
2.4. DNA Methylation Contributes to Differential Gene Expression between atRA-Responsive and -Resistant TNBC Cell Lines
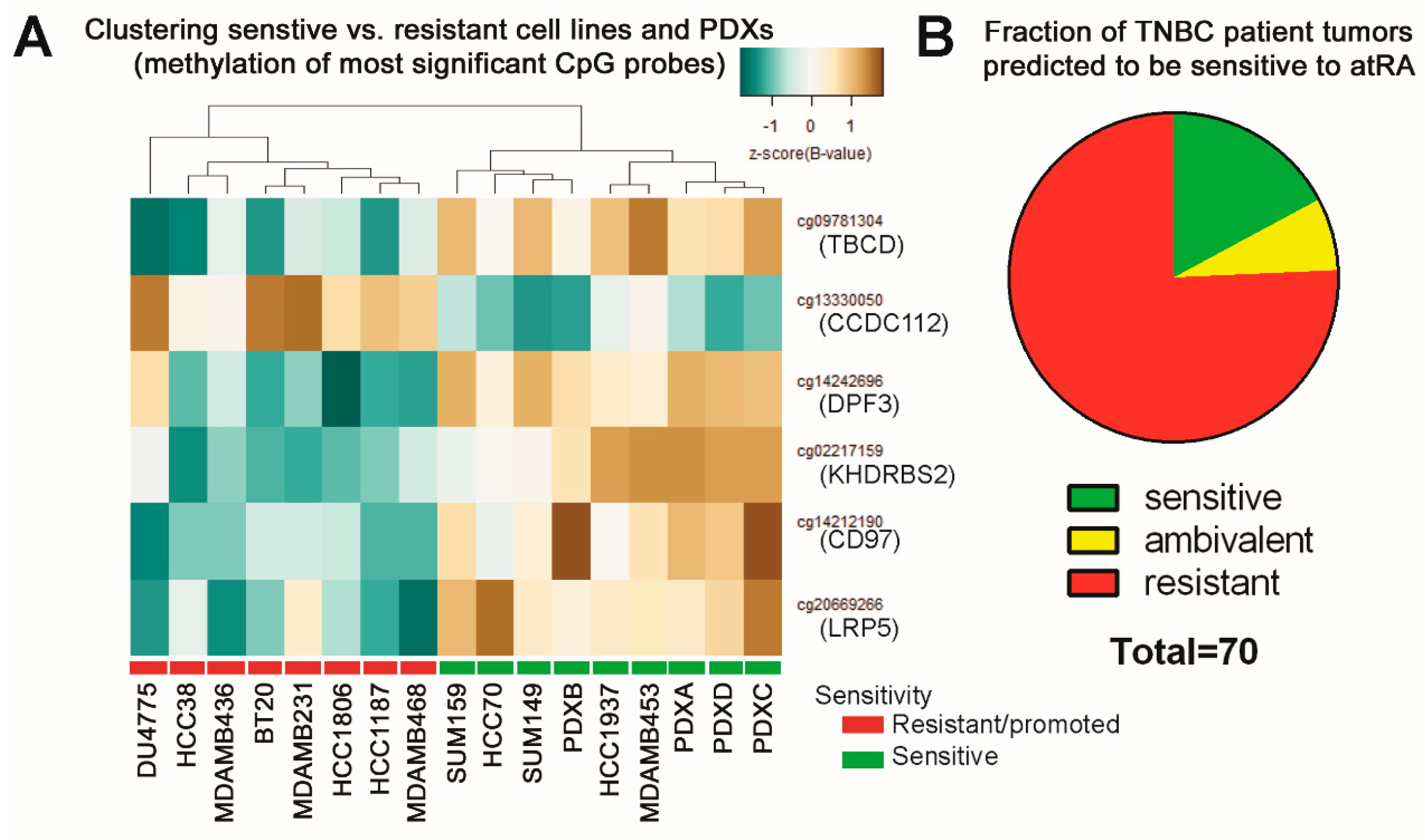
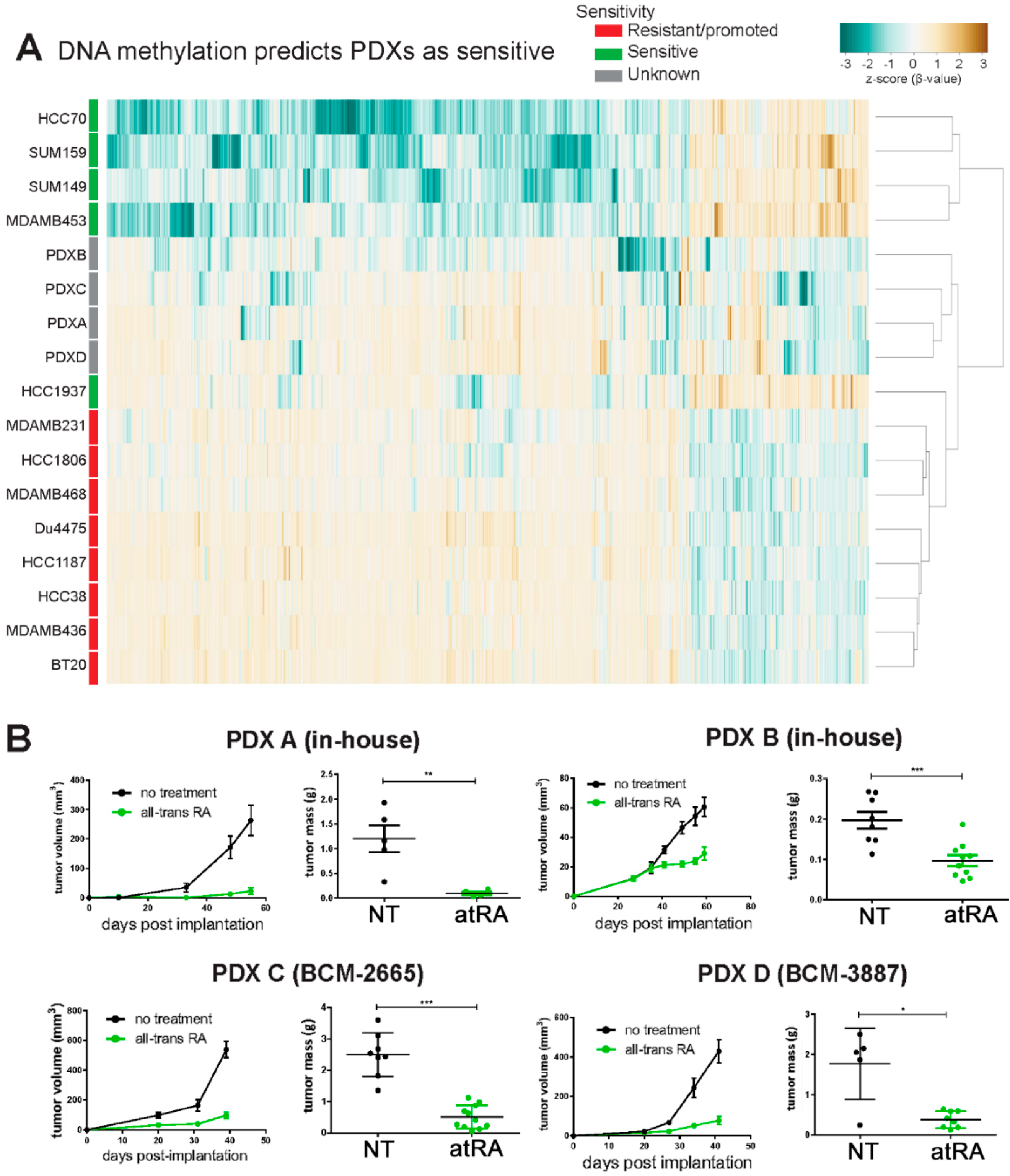
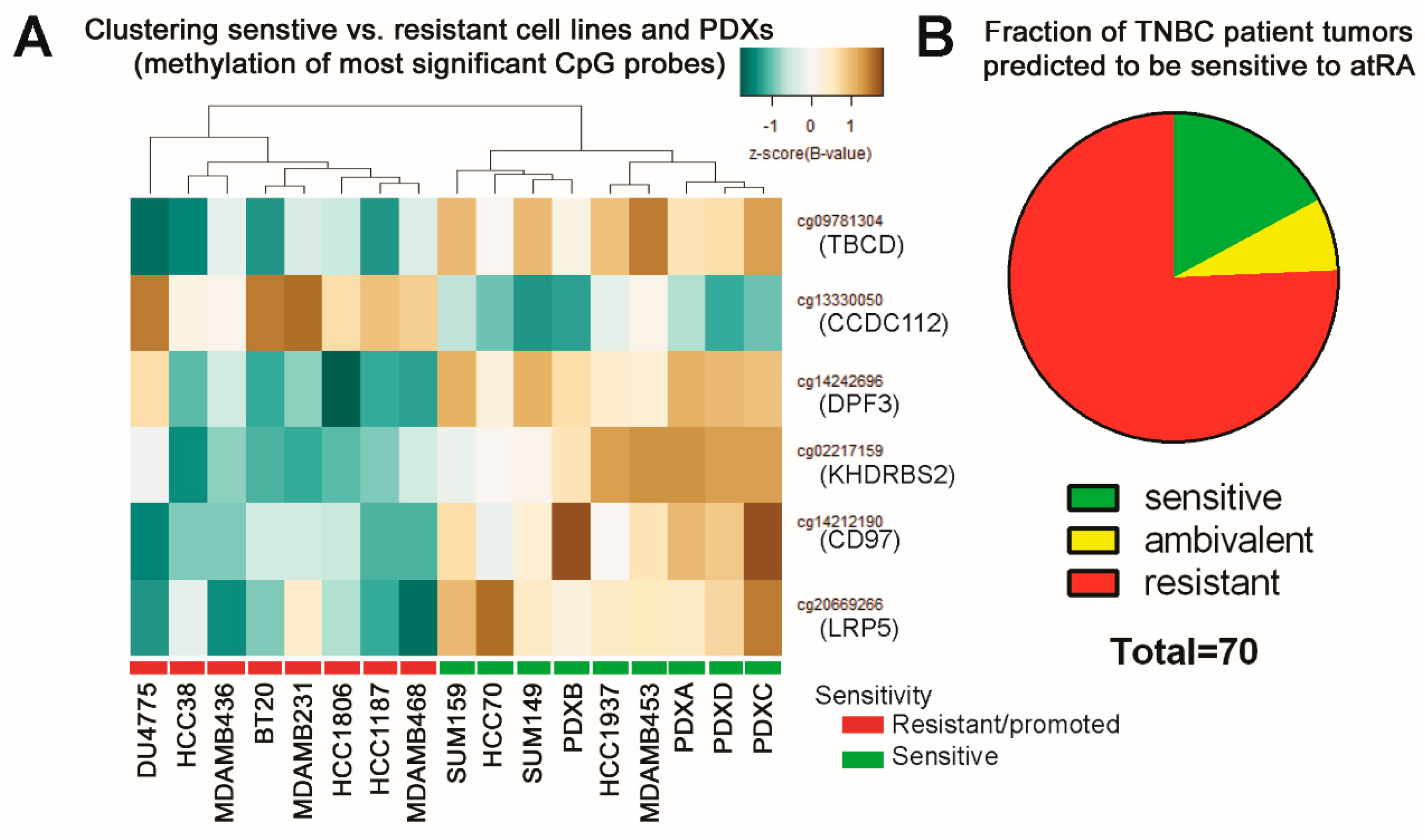
2.5. DNA Methylation Predicts Sensitivity of Four TNBC PDXs
2.6. Predicting Sensitive Patients from TCGA
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Cell-Line Xenografts
4.3. Patient-Derived Xenografts
4.4. Q Relative Real-Time PCR
4.5. Preparation of Cells from PDXs
4.6. Preparation of Cell Line Samples for Arrays and Analysis
4.7. Preparation of Patient-Derived Xenografts for Arrays and Analysis
4.8. Subtyping of Patient-Derived Xenografts
4.9. cBioPortal Analyses
4.10. Statistical Analyses
4.11. Study Approval
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of triple-negative breast cancer molecular subtypes: Implications for neoadjuvant chemotherapy selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Perou, C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Vidula, N.; Bardia, A. Targeted therapy for metastatic triple negative breast cancer: The next frontier in precision oncology. Oncotarget 2017, 8, 106167–106168. [Google Scholar] [CrossRef] [PubMed]
- Kalimutho, M.; Parsons, K.; Mittal, D.; López, J.A.; Srihari, S.; Khanna, K.K. Targeted Therapies for Triple-Negative Breast Cancer: Combating a Stubborn Disease. Trends Pharmacol. Sci. 2015, 36, 822–846. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.E.; Ye, Y.C.; Chen, S.R.; Chai, J.R.; Lu, J.X.; Zhoa, L.; Gu, L.J.; Wang, Z.Y. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [Google Scholar] [PubMed]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.P.; Matthay, K.K.; Villablanca, J.G.; Maurer, B.J. Retinoid therapy of high-risk neuroblastoma. Cancer Lett. 2003, 197, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Garattini, E.; Bolis, M.; Garattini, S.K.; Fratelli, M.; Centritto, F.; Paroni, G.; Gianni’, M.; Zanetti, A.; Pagani, A.; Fisher, J.N.; et al. Retinoids and breast cancer: From basic studies to the clinic and back again. Cancer Treat. Rev. 2014, 40, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Dean, C.A.; Liu, R.-Z.; Coyle, K.M.; Bydoun, M.; Wallace, M.; Clements, D.; Turner, C.; Mathenge, E.G.; Gujar, S.A.; et al. Aldehyde dehydrogenase 1A3 influences breast cancer progression via differential retinoic acid signaling. Mol. Oncol. 2015, 9, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Seewaldt, V.L.; Johnson, B.S.; Parker, M.B.; Collins, S.J.; Swisshelm, K. Expression of retinoic acid receptor beta mediates retinoic acid-induced growth arrest and apoptosis in breast cancer cells. Cell Growth Differ. 1995, 6, 1077–1088. [Google Scholar] [PubMed]
- Toma, S.; Raffo, P.; Isnardi, L. Effects of all-trans retinoic acid and 13-cis retinoic acid on breast cancer cell lines. In Vitamin A and Retinoids: An Update of Biological Aspects and Clinical Applications; Springer International Publishing: Birkhäuser, Basel, 2000; pp. 209–219. ISBN 978-3-0348-9574-3. [Google Scholar]
- Zhang, J.W.; Wang, J.Y.; Chen, S.J.; Chen, Z. Mechanisms of all-trans retinoic acid-induced differentiation of acute promyelocytic leukemia cells. J. Biosci. 2000, 25, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Song, Y.; Liu, Q.; Wu, Y.; He, R. Topical treatment of all- trans retinoic acid inhibits murine melanoma partly by promoting CD8+ T-cell immunity. Immunology 2017, 152, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Sutton, L.M.; Warmuth, M.A.; Petros, W.P.; Winer, E.P. Pharmacokinetics and clinical impact of all-trans retinoic acid in metastatic breast cancer: A phase II trial. Cancer Chemother. Pharmacol. 1997, 40, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Garattini, E.; Paroni, G.; Terao, M. Retinoids and breast cancer: New clues to increase their activity and selectivity. Breast Cancer Res. 2012, 14, 111. [Google Scholar] [CrossRef] [PubMed]
- Schenk, T.; Stengel, S.; Zelent, A. Unlocking the potential of retinoic acid in anticancer therapy. Br. J. Cancer 2014, 111, 2039–2045. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lee, M.O.; Wang, H.G.; Li, Y.; Hashimoto, Y.; Klaus, M.; Reed, J.C.; Zhang, X. Retinoic acid receptor beta mediates the growth-inhibitory effect of retinoic acid by promoting apoptosis in human breast cancer cells. Mol. Cell. Biol. 1996, 16, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Centritto, F.; Paroni, G.; Bolis, M.; Garattini, S.K.; Kurosaki, M.; Barzago, M.M.; Zanetti, A.; Fisher, J.N.; Scott, M.F.; Pattini, L.; et al. Cellular and molecular determinants of all-trans retinoic acid sensitivity in breast cancer: Luminal phenotype and RARα expression. EMBO Mol. Med. 2015, 7, 950–972. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kozono, S.; Kats, L.; Nechama, M.; Li, W.; Guarnerio, J.; Luo, M.; You, M.-H.; Yao, Y.; Kondo, A.; et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med. 2015, 21, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginestier, C.; Wicinski, J.; Cervera, N.; Monville, F.; Finetti, P.; Bertucci, F.; Wicha, M.S.; Birnbaum, D.; Charafe-Jauffret, E. Retinoid signaling regulates breast cancer stem cell differentiation. Cell Cycle 2009, 8, 3297–3302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, V.F.; Nguyen, N.; Jin, K.; Sadik, H.; Cho, S.; Korangath, P.; Han, L.; Foster, Y.M.N.; Zhou, X.C.; Zhang, Z.; et al. Combined treatment with epigenetic, differentiating, and chemotherapeutic agents cooperatively targets tumor-initiating cells in triple-negative breast cancer. Cancer Res. 2016, 76, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-J.; Kim, M.R.; Chen, Y.-S.; Yang, J.-Y.; Chang, C.-J. Retinoic acid directs breast cancer cell state changes through regulation of TET2-PKCζ pathway. Oncogene 2017, 36, 3193. [Google Scholar] [CrossRef] [PubMed]
- Takatsuka, J.; Takahashi, N.; de Luca, L.M. Retinoic acid metabolism and inhibition of cell proliferation: An unexpected liaison. Cancer Res. 1996, 56, 675–678. [Google Scholar] [PubMed]
- Tari, A.M.; Lim, S.-J.; Hung, M.-C.; Esteva, F.J.; Lopez-Berestein, G. Her2/neu induces all-trans retinoic acid (ATRA) resistance in breast cancer cells. Oncogene 2002, 21, 5224–5232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montesinos, P.; Sanz, M.A. The differentiation syndrome in patients with acute promyelocytic leukemia: Experience of the Pethema Group and review of the literature. Mediterr. J. Hematol. Infect. Dis. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Coyle, K.M.; Maxwell, S.; Thomas, M.L.; Marcato, P. Profiling of the transcriptional response to all-Trans retinoic acid in breast cancer cells reveals RARE-independent mechanisms of gene expression. Sci. Rep. 2017, 7, 16684. [Google Scholar] [CrossRef] [PubMed]
- Barnabas, N.; Cohen, D. Phenotypic and molecular characterization of MCF10DCIS and SUM breast cancer cell lines. Int. J. Breast Cancer 2013, 2013, 872743. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elstrodt, F.; Hollestelle, A.; Nagel, J.H.A.; Gorin, M.; Wasielewski, M.; van den Ouweland, A.; Merajver, S.D.; Ethier, S.P.; Schutte, M. BRCA1 mutation analysis of 41 human breast cancer cell lines reveals three new deleterious mutants. Cancer Res. 2006, 66, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Berry, D.C.; Shaw, N.S.; Travis, S.N.; Noy, N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 2007, 129, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Berry, D.C.; Toshkov, I.A.; Cheng, L.; Nikitin, A.Y.; Noy, N. Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPARbeta/delta to RAR. Proc. Natl. Acad. Sci. USA 2008, 105, 7546–7551. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Goodison, S.; Urquidi, V.; Tarin, D.; Lotan, R.; Tahara, E. Differential effects of retinoic acid on the growth of isogenic metastatic and non-metastatic breast cancer cell lines and their association with distinct expression of retinoic acid receptor β isoforms 2 and 4. Int. J. Oncol. 2003, 22, 623–629. [Google Scholar] [PubMed]
- Schneider, S.M.; Offterdinger, M.; Huber, H.; Grunt, T.W. Activation of retinoic acid receptor alpha is sufficient for full induction of retinoid responses in SK-BR-3 and T47D human breast cancer cells. Cancer Res. 2000, 60, 5479–5487. [Google Scholar] [PubMed]
- Coyle, K.M.; Murphy, J.P.; Vidovic, D.; Vaghar-Kashani, A.; Dean, C.A.; Sultan, M.; Clements, D.; Wallace, M.; Thomas, M.L.; Hundert, A.; et al. Breast cancer subtype dictates DNA methylation and ALDH1A3-mediated expression of tumor suppressor RARRES1. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Dean, C.A.; Da, P.; Araslanova, R.; Gillis, M.; Joshi, M.; Helyer, L.; Pan, L.; Leidal, A.; Gujar, S.; et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells 2011, 29, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croker, A.K.; Allan, A.L. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhi CD44+ human breast cancer cells. Breast Cancer Res. Treat. 2012, 133, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aran, D.; Sabato, S.; Hellman, A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013, 14, R21. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Hellman, A. DNA methylation of transcriptional enhancers and cancer predisposition. Cell 2013, 154, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Claerhout, S.; Prat, A.; Dobrolecki, L.E.; Petrovic, I.; Lai, Q.; Landis, M.D.; Wiechmann, L.; Schiff, R.; Giuliano, M.; et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013, 73, 4885–4897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Shah, S.R.; Illum, H.; Dowell, J. Vemurafenib: Targeted inhibition of mutated BRAF for treatment of advanced melanoma and its potential in other malignancies. Drugs 2012, 72, 2207–2222. [Google Scholar] [CrossRef] [PubMed]
- Taron, M.; Rosell, R.; Felip, E.; Mendez, P.; Souglakos, J.; Ronco, M.S.; Queralt, C.; Majo, J.; Sanchez, J.M.; Sanchez, J.J.; et al. BRCA1 mRNA expression levels as an indicator of chemoresistance in lung cancer. Hum. Mol. Genet. 2004, 13, 2443–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-Repair Gene MGMT and the Clinical Response of Gliomas to Alkylating Agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Craig, J.M. DNA methylation biomarkers: Cancer and beyond. Genes (Basel) 2014, 5, 821–864. [Google Scholar] [CrossRef] [PubMed]
- Shiwa, Y.; Hachiya, T.; Furukawa, R.; Ohmomo, H.; Ono, K.; Kudo, H.; Hata, J.; Hozawa, A.; Iwasaki, M.; Matsuda, K.; et al. Adjustment of cell-type composition minimizes systematic bias in blood DNA methylation profiles derived by DNA collection protocols. PLoS ONE 2016, 11, e0147519. [Google Scholar] [CrossRef] [PubMed]
- Cope, L.M.; Fackler, M.J.; Lopez-Bujanda, Z.; Wolff, A.C.; Visvanathan, K.; Gray, J.W.; Sukumar, S.; Umbricht, C.B. Do breast cancer cell lines provide a relevant model of the patient tumor methylome? PLoS ONE 2014, 9, e105545. [Google Scholar] [CrossRef] [PubMed]
- Ehrich, M.; Turner, J.; Gibbs, P.; Lipton, L.; Giovanneti, M.; Cantor, C.; Boom, D. van den Cytosine methylation profiling of cancer cell lines. Proc. Natl. Acad. Sci. USA 2008, 105, 4844–4849. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Park, S.-J.; Nakai, K. Differential landscape of non-CpG methylation in embryonic stem cells and neurons caused by DNMT3s. Sci. Rep. 2017, 7, 11295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.S.; Shin, W.J.; Lee, J.E.; Do, J.T. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes (Basel) 2017, 8, 148. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.; Ward, R.L.; Hesson, L.B. The evidence for functional non-CpG methylation in mammalian cells. Epigenetics 2014, 9, 823–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Cortez, C.C.; Yang, X.; Nichols, P.W.; Jones, P.A.; Liang, G. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum. Mol. Genet. 2011, 20, 4299–4310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, N.; Bosch, A.; Leibovitch, B.; Pereira, L.; Cubedo, E.; Yu, J.; Pierzchalski, K.; Jones, J.W.; Fishel, M.; Kane, M.; et al. Blocking the PAH2 domain of Sin3A inhibits tumorigenesis and confers retinoid sensitivity in triple negative breast cancer. Oncotarget 2016, 7, 43689–43702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlenk, R.F.; Döhner, K.; Kneba, M.; Götze, K.; Hartmann, F.; del Valle, F.; Kirchen, H.; Koller, E.; Fischer, J.T.; Bullinger, L.; et al. Gene mutations and response to treatment with all-trans retinoic acid in elderly patients with acute myeloid leukemia. Results from the AMLSG Trial AML HD98B. Haematologica 2009, 94, 54–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.-P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.; Hansen, K.D. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gendoo, D.M.A.; Ratanasirigulchai, N.; Schröder, M.S.; Paré, L.; Parker, J.S.; Prat, A.; Haibe-Kains, B. Genefu: An R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics 2016, 32, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coyle, K.M.; Dean, C.A.; Thomas, M.L.; Vidovic, D.; Giacomantonio, C.A.; Helyer, L.; Marcato, P. DNA Methylation Predicts the Response of Triple-Negative Breast Cancers to All-Trans Retinoic Acid. Cancers 2018, 10, 397. https://doi.org/10.3390/cancers10110397
Coyle KM, Dean CA, Thomas ML, Vidovic D, Giacomantonio CA, Helyer L, Marcato P. DNA Methylation Predicts the Response of Triple-Negative Breast Cancers to All-Trans Retinoic Acid. Cancers. 2018; 10(11):397. https://doi.org/10.3390/cancers10110397
Chicago/Turabian StyleCoyle, Krysta Mila, Cheryl A. Dean, Margaret Lois Thomas, Dejan Vidovic, Carman A. Giacomantonio, Lucy Helyer, and Paola Marcato. 2018. "DNA Methylation Predicts the Response of Triple-Negative Breast Cancers to All-Trans Retinoic Acid" Cancers 10, no. 11: 397. https://doi.org/10.3390/cancers10110397