The Role of Arrestin Domain-Containing 3 in Regulating Endocytic Recycling and Extracellular Vesicle Sorting of Integrin β4 in Breast Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
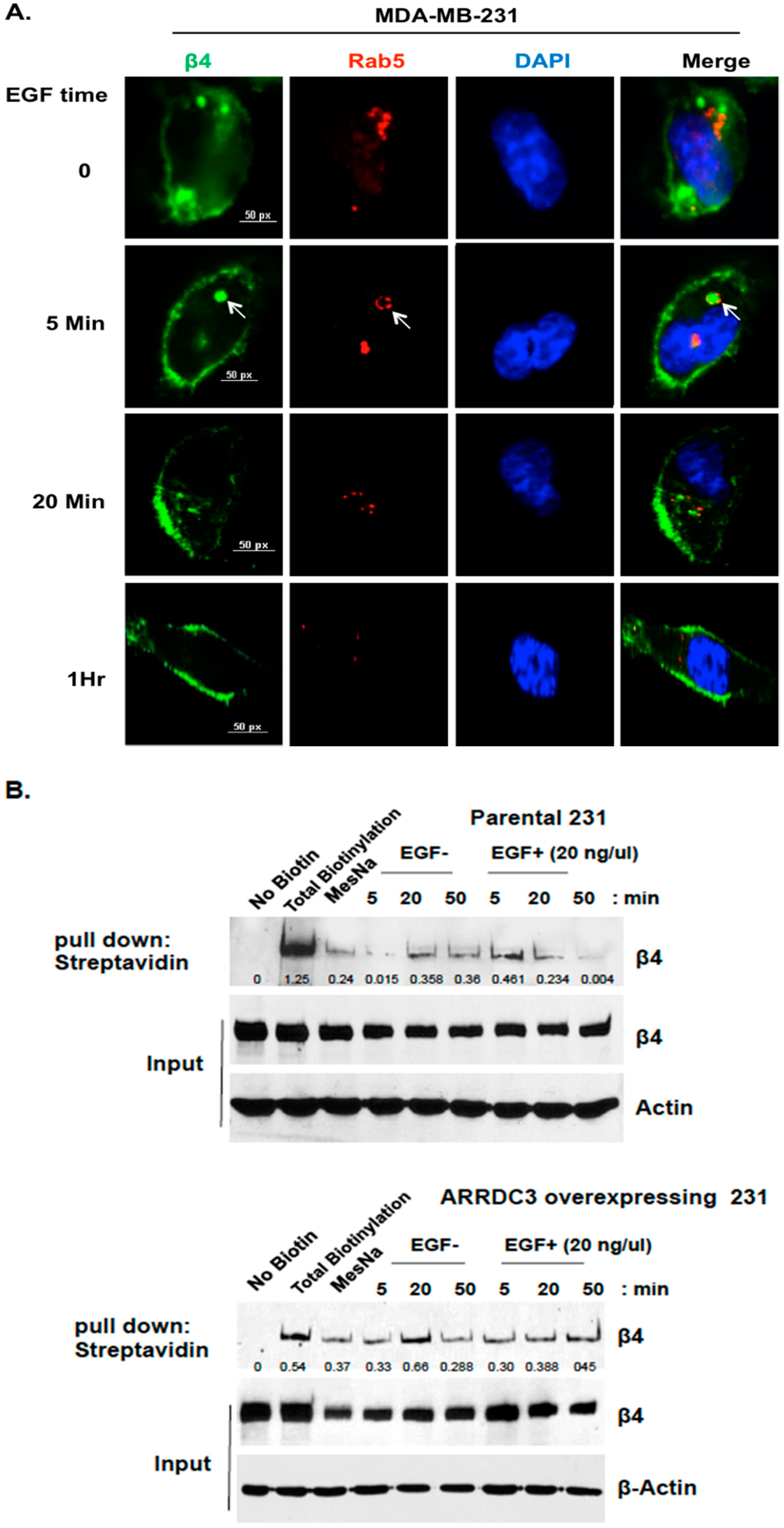
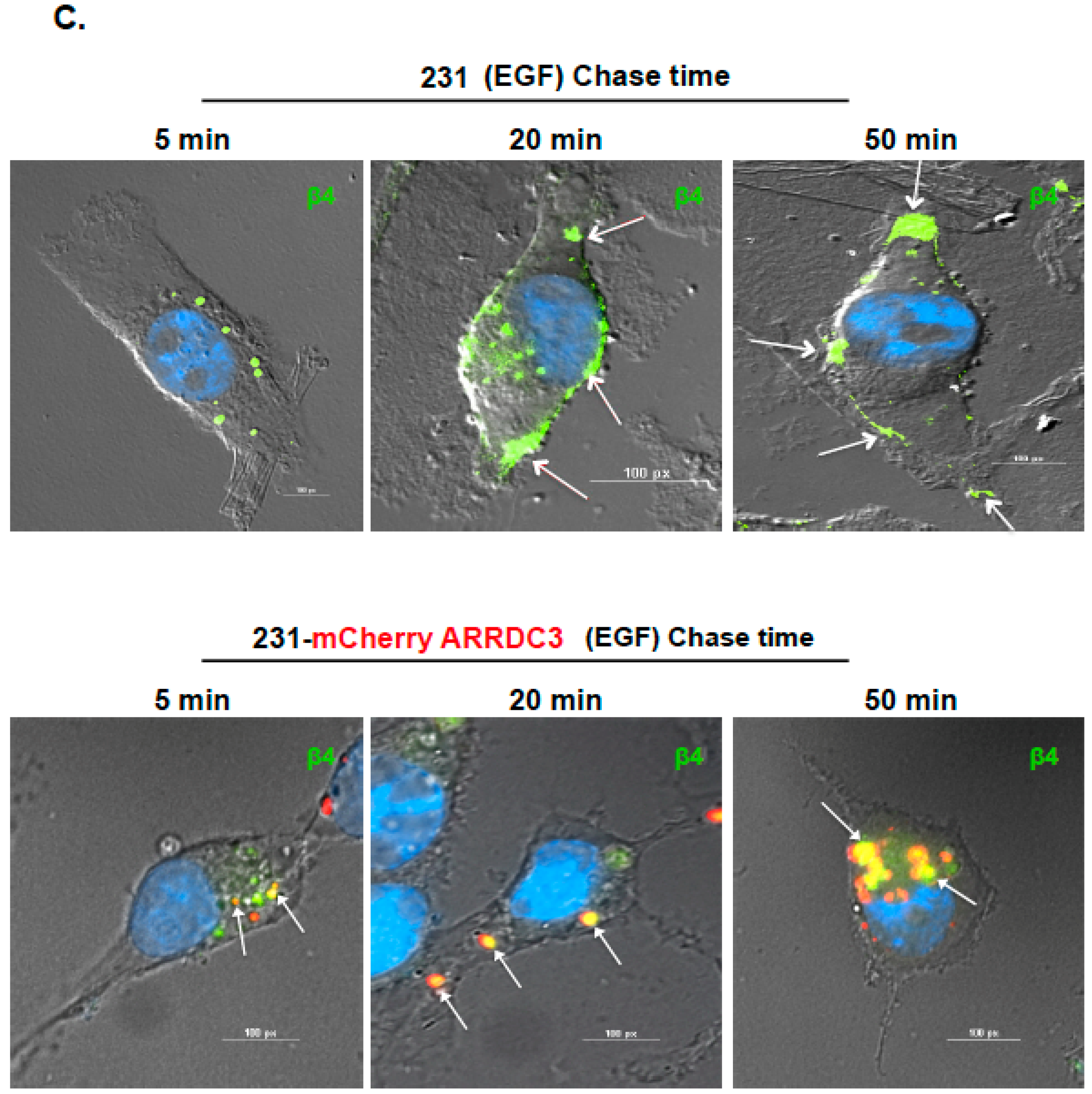
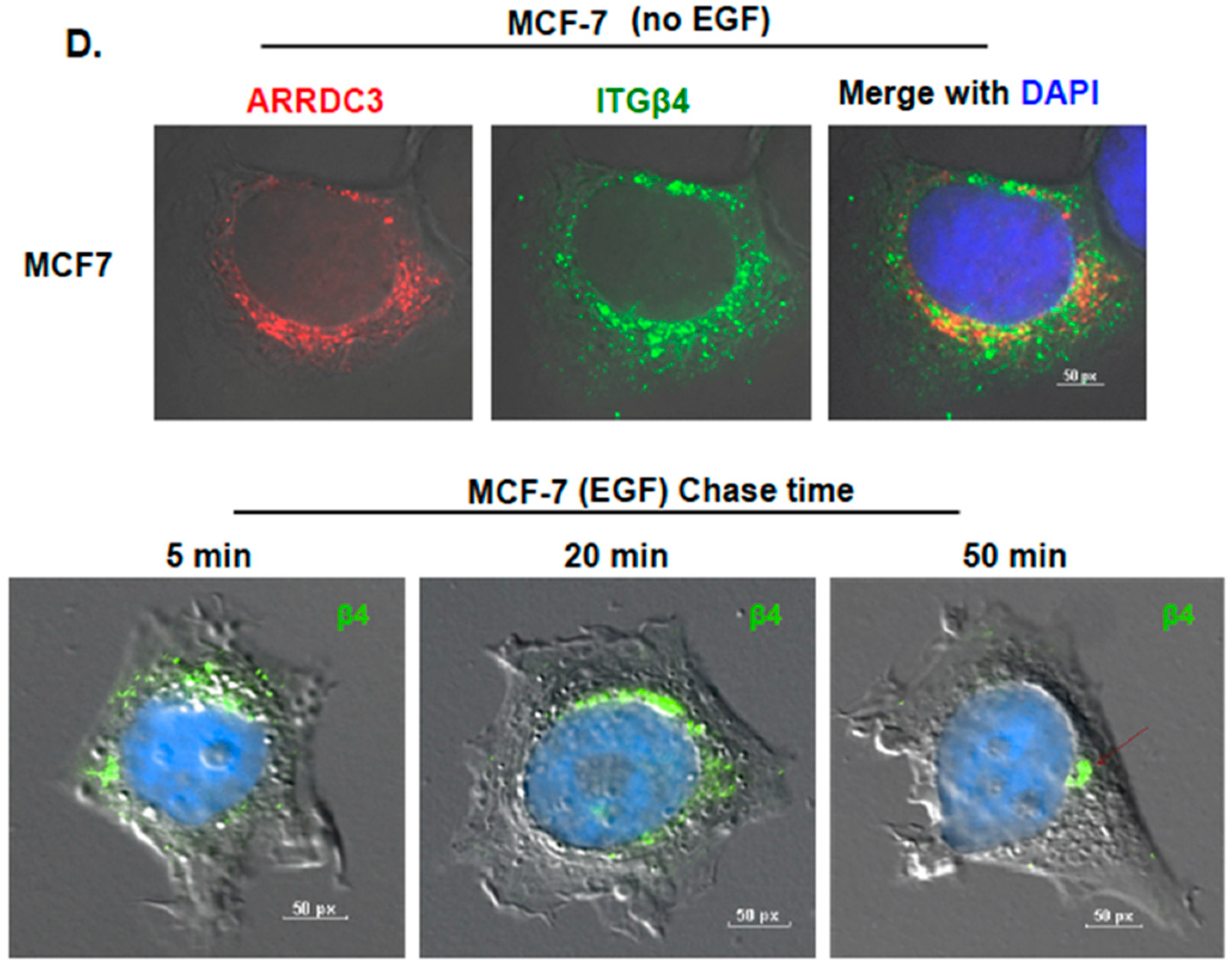
2.1. ARRDC3 Inhibits EGF-Driven Endocytic Recycling of ITG β4 in Triple Negative Breast Cancer (TNBC) Cells
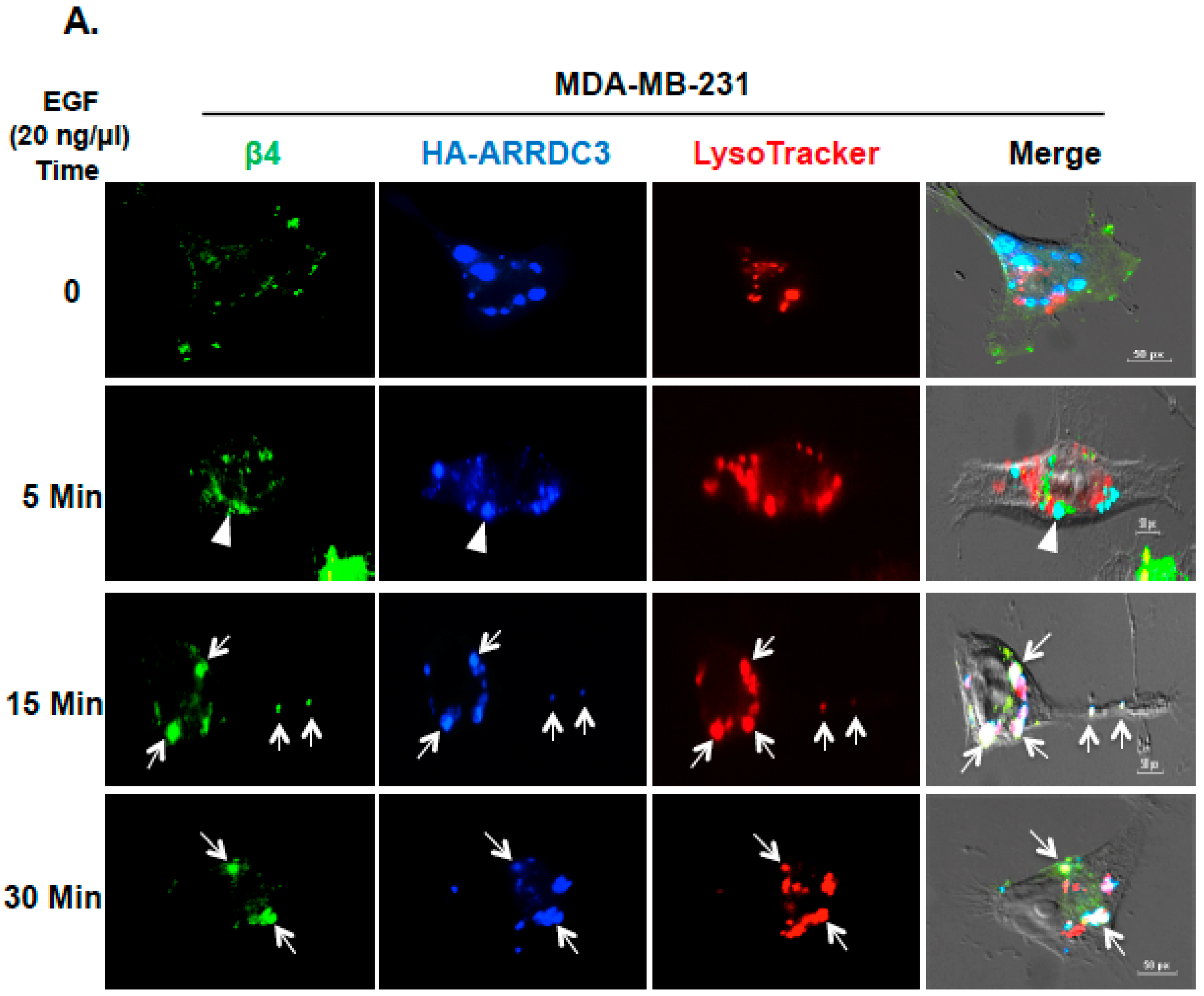
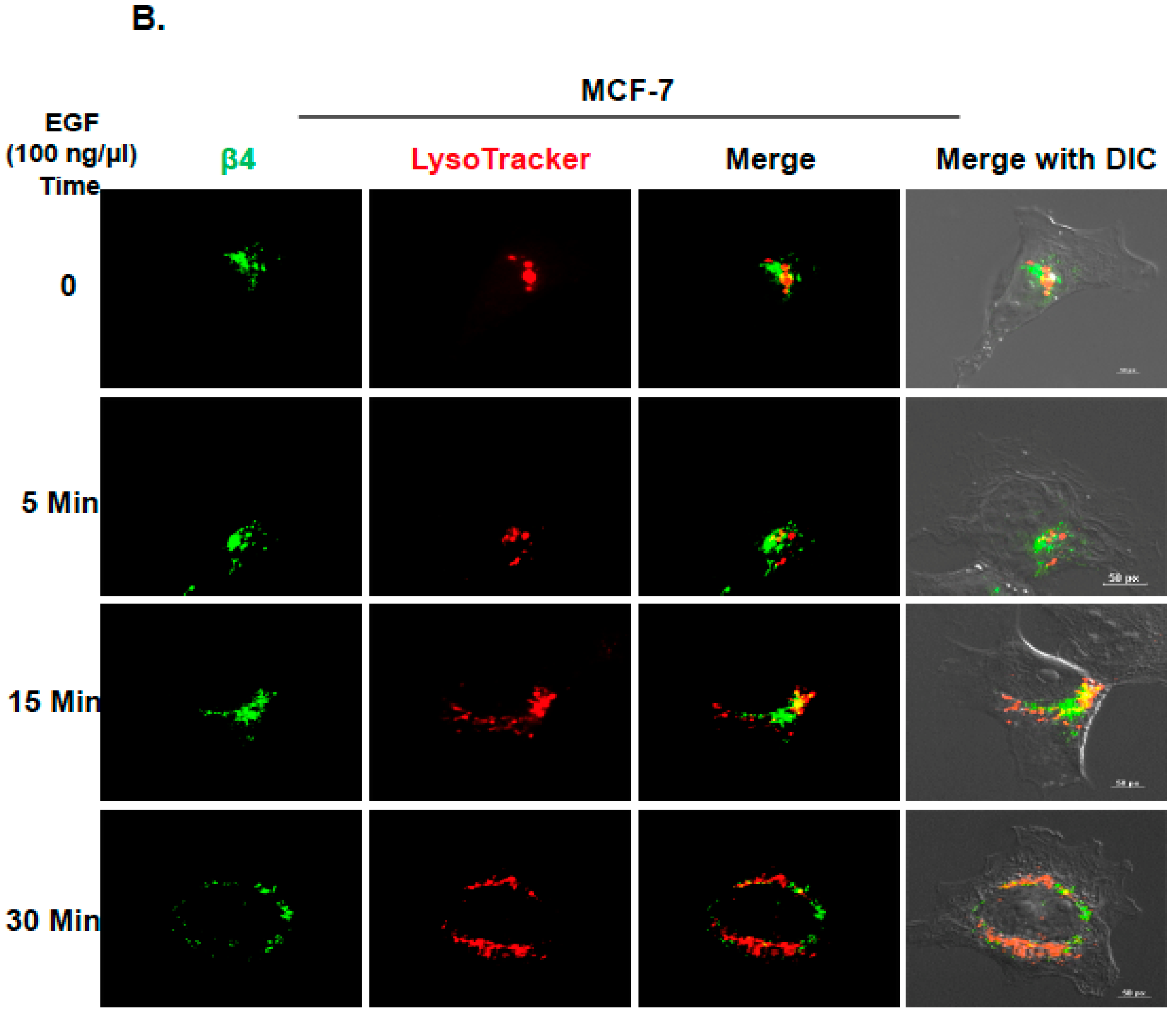
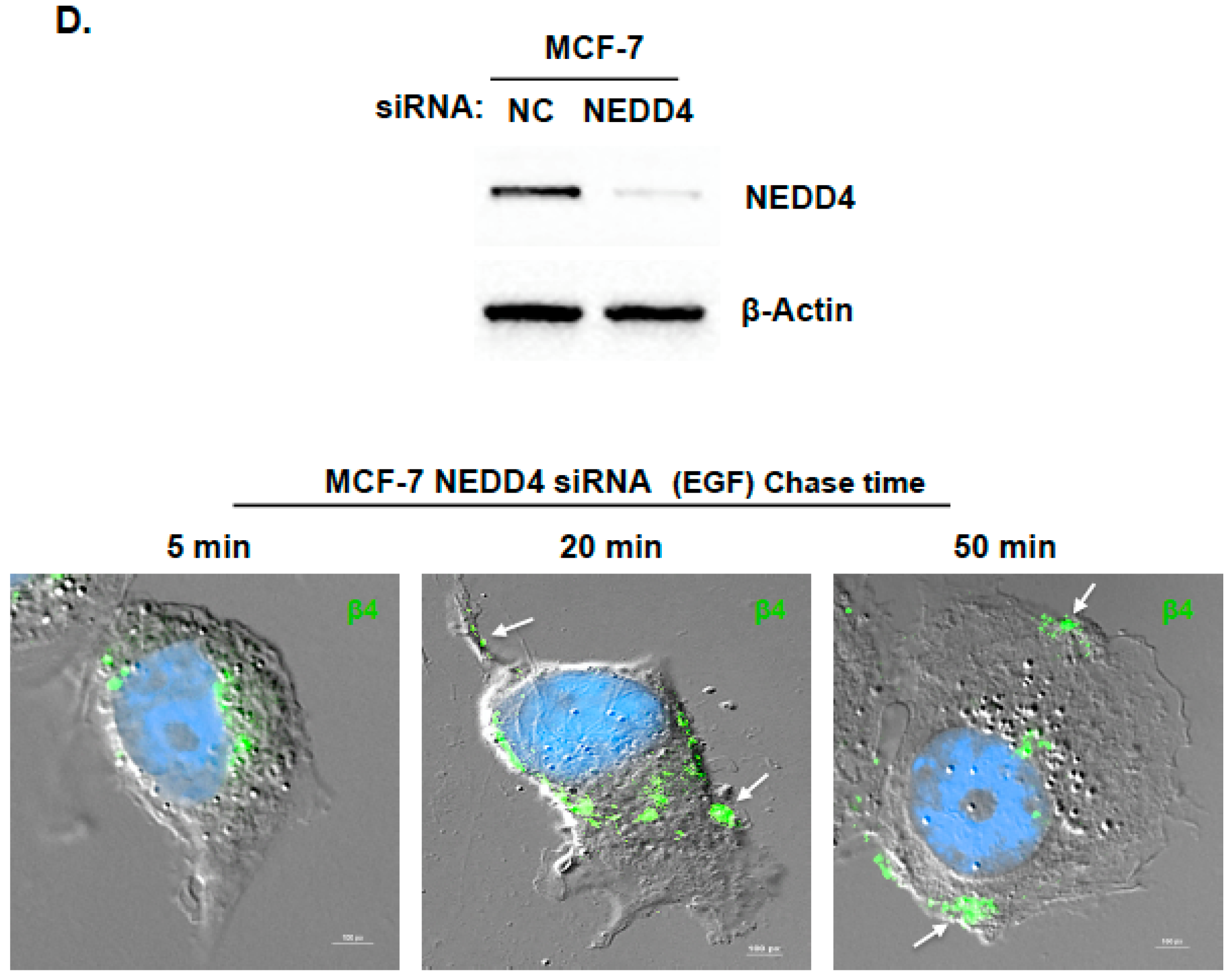
2.2. Inhibition of ITG β4 Recycling Is Linked to ARRDC3-Dependent Ubiquitination and Lysosomal Targeting of ITG β4
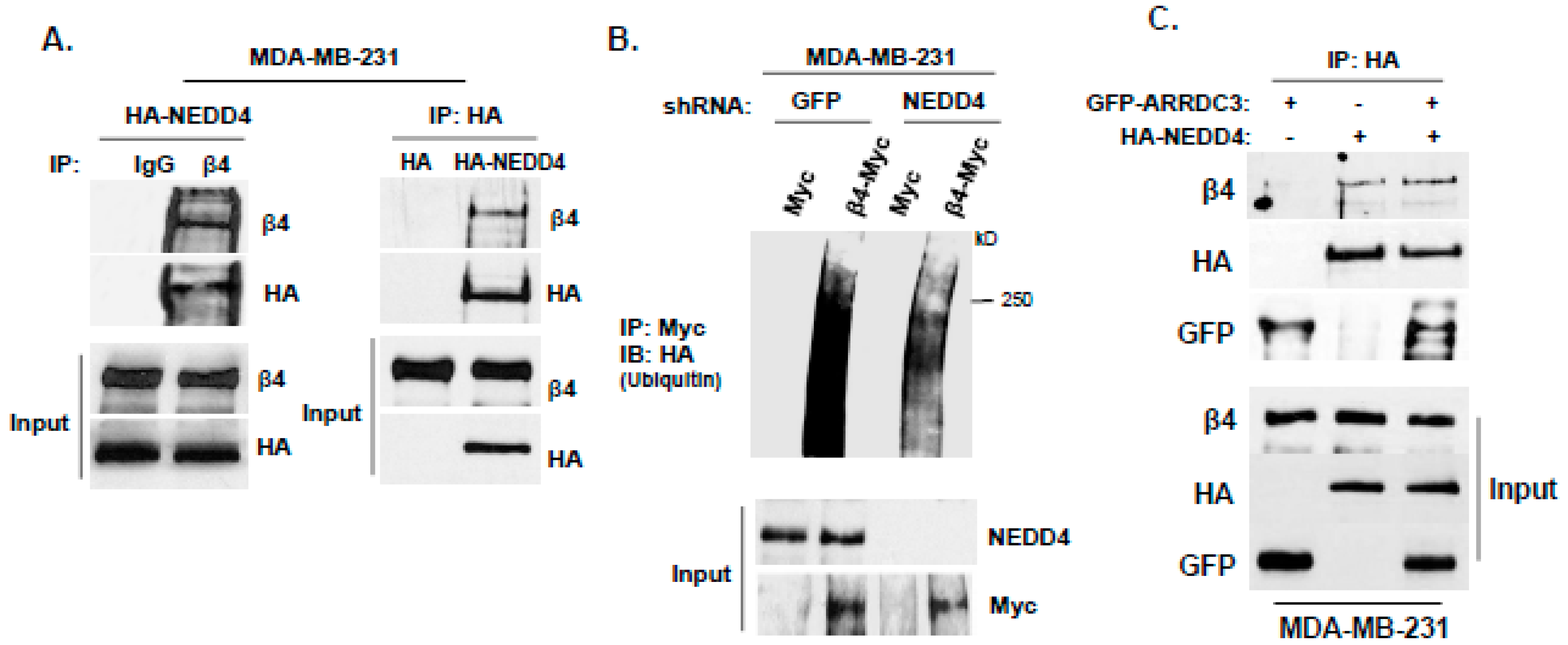
2.3. ARRDC3 Serves as an Adaptor Molecule for E3 Ligase NEDD4 and ITG β4 to Mediate ITG β4 Ubiquitination
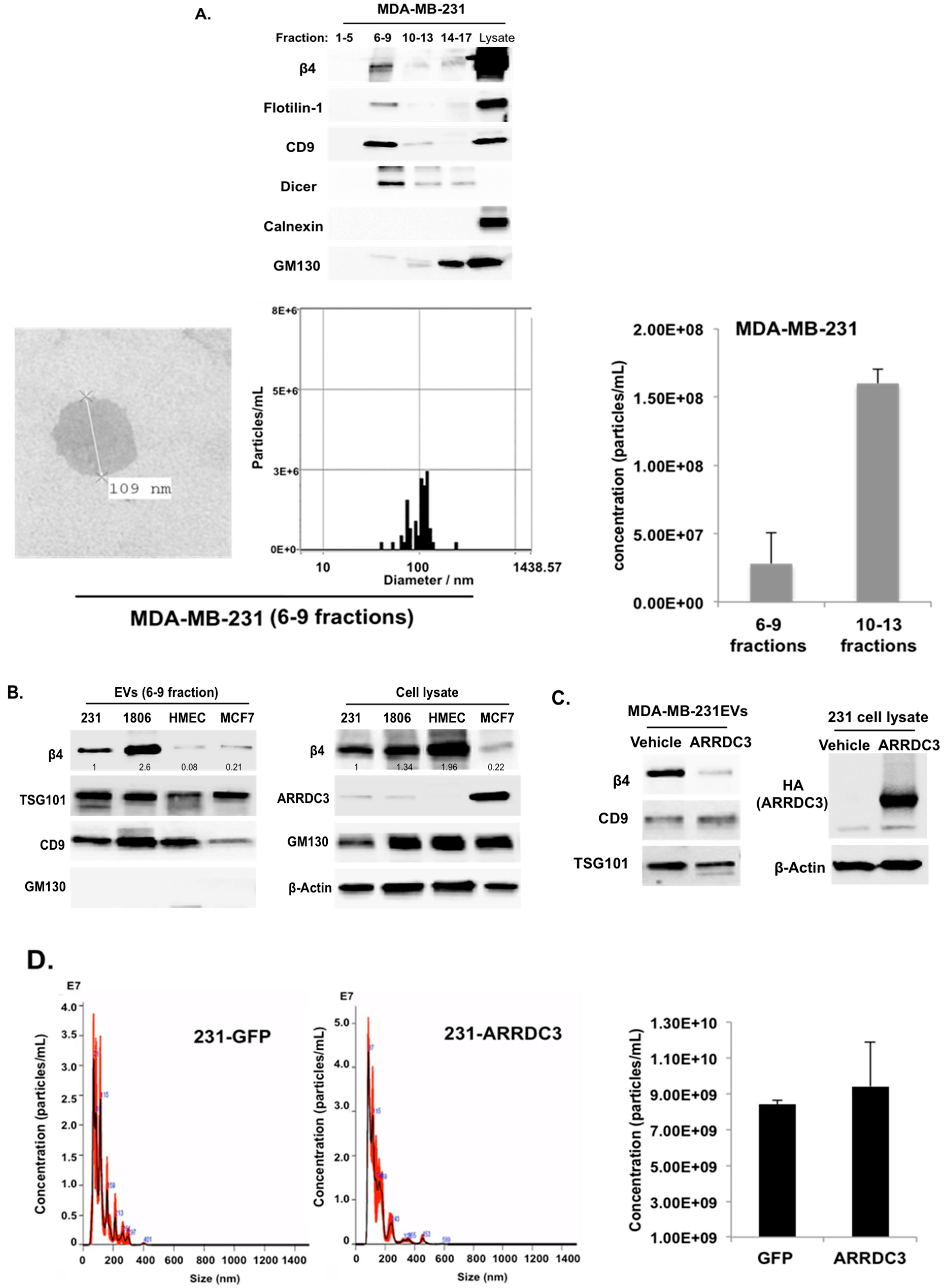
2.4. ARRDC3 Prevents the Sorting of ITG β4 into Extracellular Vesicles (EVs) without Affecting Overall EV Secretion from TNBC Cells
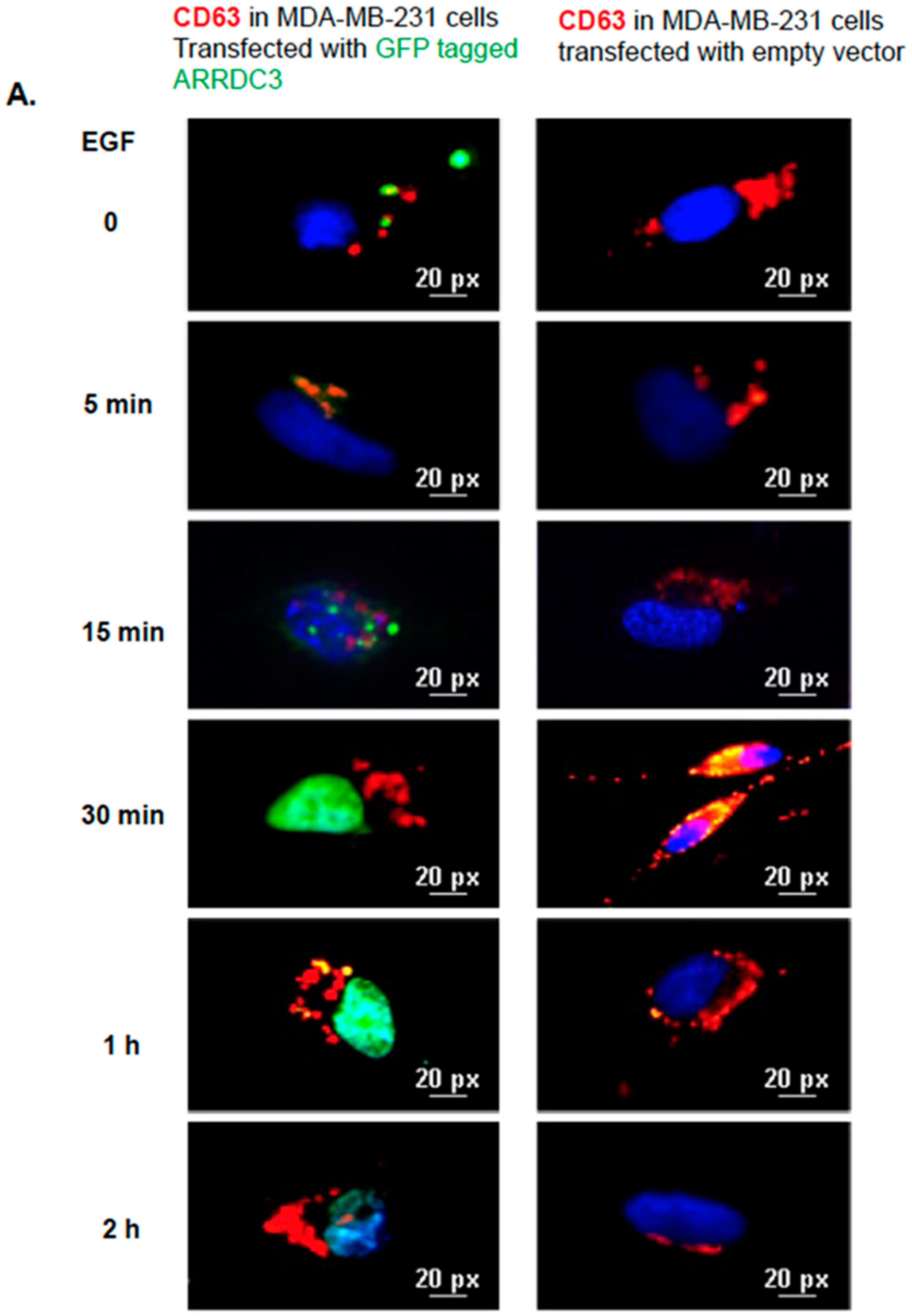
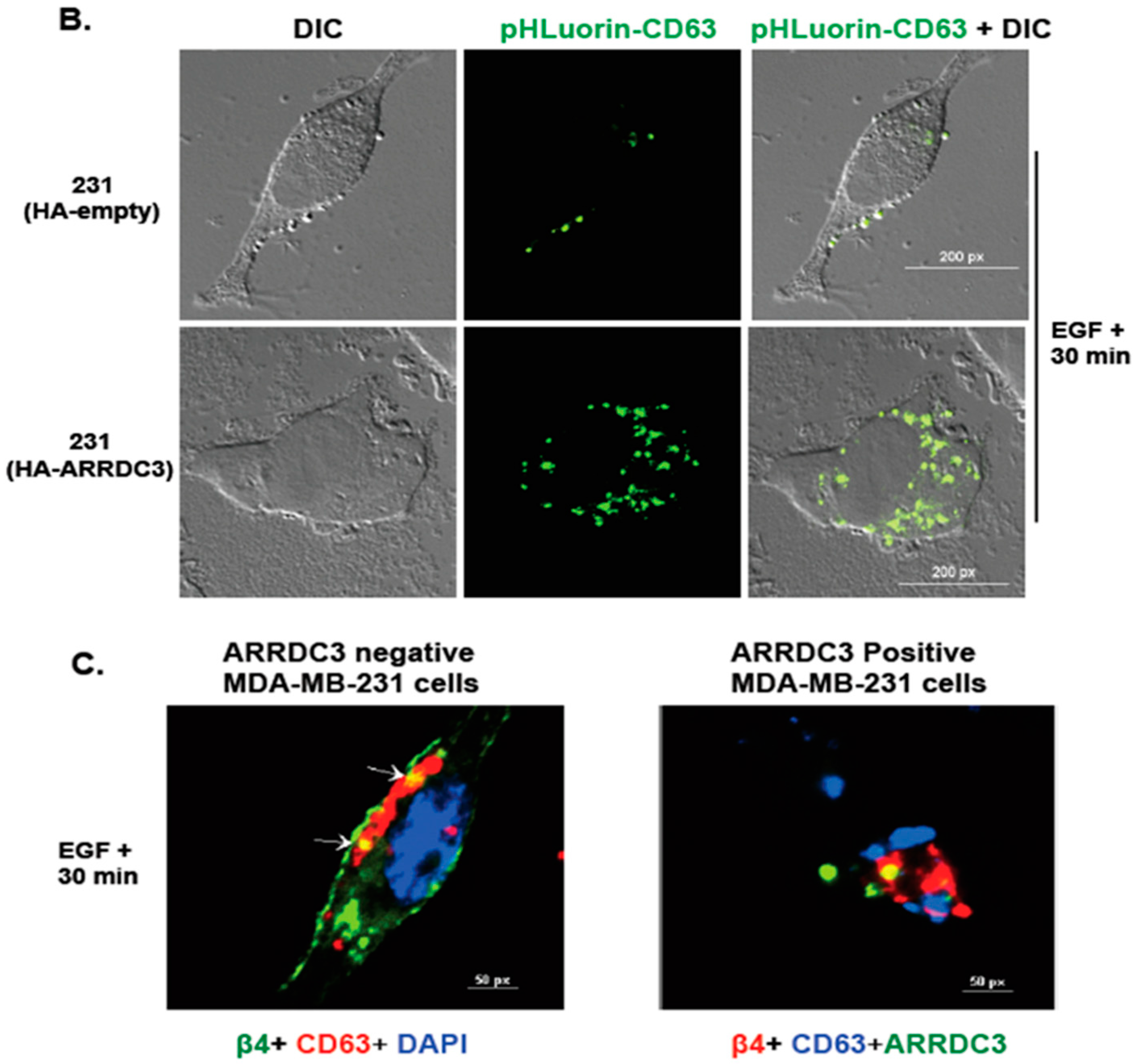
2.5. ARRDC3 Prevents EGF-Driven Fusion of CD63 Positive EVs at Plasma Membrane
2.6. ARRDC3 Reduces the Tumorigenic and Metastatic Potentials of TNBC Cell-Derived EVs
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. Western Blot Analysis
4.3. Ubiquitination and Co-Immunoprecipitation Assay
4.4. Assays That Monitor Intracellular Trafficking of ITG β4
4.4.1. Biotin-IP Based Integrin Recycling Assay
4.4.2. Immunofluorescence-Based Integrin Recycling Assay
4.5. EVs Purification
4.6. Transmission Electron Microscopy (TEM)
4.7. Proliferation and Transwell Migration Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ARRDC3 | Arrestin domain-containing 3 |
| ITG4 | Integrin 4 |
| TNBC | Triple negative breast cancer |
| Evs | Extracellular vesicles |
| NEDD4 | Neural precursor cell expressed, developmentally down-regulated 4 |
| E3 | Ubiquitin protein ligase 3 |
| IP | Immunoprecipitation |
References
- Lipscomb, E.A.; Mercurio, A.M. Mobilization and activation of a signaling competent alpha6beta4integrin underlies its contribution to carcinoma progression. Cancer Metast. Rev. 2005, 24, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, A.M.; Rabinovitz, I.; Shaw, L.M. The α6β4 integrin and epithelial cell migration. Curr. Opin. Cell Biol. 2001, 13, 541–545. [Google Scholar] [CrossRef]
- Rabinovitz, I.; Toker, A.; Mercurio, A.M. Protein kinase C-dependent mobilization of the alpha6beta4 integrin from hemidesmosomes and its association with actin-rich cell protrusions drive the chemotactic migration of carcinoma cells. J. Cell Biol. 1999, 146, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Germain, E.C.; Santos, T.M.; Rabinovitz, I. Phosphorylation of a novel site on the β4 integrin at the trailing edge of migrating cells promotes hemidesmosome disassembly. Mol. Biol. Cell 2009, 20, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsen, K.; Litjens, S.H.M.; Kuikman, I.; Margadant, C.; van Rheenen, J.; Sonnenberg, A. Serine phosphorylation of the integrin β4 subunit is necessary for epidermal growth factor receptor induced hemidesmosome disruption. Mol. Biol. Cell 2007, 18, 3512–3522. [Google Scholar] [CrossRef] [PubMed]
- Gagnoux-Palacios, L.; Dans, M.; van’t Hof, W.; Mariotti, A.; Pepe, A.; Meneguzzi, G.; Resh, M.D.; Giancotti, F.G. Compartmentalization of integrin α6β4 signaling in lipid rafts. J. Cell Biol. 2003, 162, 1189–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caswell, P.T.; Vadrevu, S.; Norman, J.C. Integrins: Masters and slaves of endocytic transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Paul, N.R.; Jacquemet, G.; Caswell, P.T. Endocytic Trafficking of Integrins in Cell Migration. Curr. Biol. 2015, 25, R1092–R1105. [Google Scholar] [CrossRef] [PubMed]
- Pellinen, T.; Ivaska, J. Integrin traffic. J. Cell Sci. 2006, 119, 3723–3731. [Google Scholar] [CrossRef] [Green Version]
- Caswell, P.T.; Chan, M.; Lindsay, A.J.; McCaffrey, M.W.; Boettiger, D.; Norman, J.C. Rab-coupling protein coordinates recycling of α5β1 integrin and EGFR1 to promote cell migration in 3D microenvironments. J. Cell Biol. 2008, 183, 143–155. [Google Scholar] [CrossRef]
- De Franceschi, N.; Hamidi, H.; Alanko, J.; Sahgal, P.; Ivaska, J. Integrin traffic—The update. J. Cell Sci. 2015, 128, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Dozynkiewicz, M.A.; Jamieson, N.B.; Macpherson, I.; Grindlay, J.; van den Berghe, P.V.E.; von Thun, A.; Morton, J.P.; Gourley, C.; Timpson, P.; Nixon, C.; et al. Rab25 and CLIC3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev. Cell 2012, 22, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruivo, C.F.; Adem, B.; Silva, M.; Melo, S.A. The Biology of Cancer Exosomes: Insights and New Perspectives. Cancer Res. 2017, 77, 6480–6488. [Google Scholar] [CrossRef] [PubMed]
- Bebelman, M.P.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer—Implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Brumbaugh, J.; Bonavida, B. Exosomes derived from cancerous and non-cancerous cells regulate the anti-tumor response in the tumor microenvironment. Genes Cancer 2018, 9, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Antonyak, M.A.; Li, B.; Boroughs, L.K.; Johnson, J.L.; Druso, J.E.; Bryant, K.L.; Holowka, D.A.; Cerione, R.A. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4852–4857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- Soung, Y.H.; Ford, S.; Zhang, V.; Chung, J. Exosomes in Cancer Diagnostics. Cancers 2017, 9, 8. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabhan, J.F.; Pan, H.; Lu, Q. Arrestin domain-containing protein 3 recruits the NEDD4 E3 ligase to mediate ubiquitination of the beta2-adrenergic receptor. EMBO Rep. 2010, 11, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Draheim, K.M.; Chen, H.-B.; Tao, Q.; Moore, N.; Roche, M.; Lyle, S. ARRDC3 suppresses breast cancer progression by negatively regulating integrin β4. Oncogene 2010, 29, 5032–5047. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; O’Hayre, M.; Gutkind, J.S.; Hurley, J.H. Structural and Biochemical Basis for Ubiquitin Ligase Recruitment by Arrestin-related Domain-containing Protein-3 (ARRDC3). J. Biol. Chem. 2014, 289, 4743–4752. [Google Scholar] [CrossRef] [PubMed]
- Soung, Y.H.; Pruitt, K.; Chung, J. Epigenetic silencing of ARRDC3 expression in basal-like breast cancer cells. Sci. Rep. 2014, 4, 3846. [Google Scholar] [CrossRef] [PubMed]
- Soung, Y.H.; Kashyap, T.; Nguyen, T.; Yadav, G.; Chang, H.; Landesman, Y.; Chung, J. Selective Inhibitors of Nuclear Export (SINE) compounds block proliferation and migration of triple negative breast cancer cells by restoring expression of ARRDC3. Oncotarget 2017, 8, 52935–52947. [Google Scholar] [CrossRef] [PubMed]
- Arancibia-Cárcamo, I.L.; Fairfax, B.P.; Moss, S.J.; Kittler, J.T. Studying the Localization, Surface Stability and Endocytosis of Neurotransmitter Receptors by Antibody Labeling and Biotinylation Approaches. In The Dynamic Synapse: Molecular Methods in Ionotropic Receptor Biology; Frontiers in Neuroscience; Kittler, J.T., Moss, S.J., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2006; ISBN 978-0-8493-1891-7. [Google Scholar]
- Le, T.L.; Yap, A.S.; Stow, J.L. Recycling of E-cadherin: A potential mechanism for regulating cadherin dynamics. J. Cell Biol. 1999, 146, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Dutta, U.; Shaw, L.M. A key tyrosine (Y1494) in the β4 integrin regulates multiple signaling pathways important for tumor development and progression. Cancer Res. 2008, 68, 8779–8787. [Google Scholar] [CrossRef]












© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soung, Y.H.; Ford, S.; Yan, C.; Chung, J. The Role of Arrestin Domain-Containing 3 in Regulating Endocytic Recycling and Extracellular Vesicle Sorting of Integrin β4 in Breast Cancer. Cancers 2018, 10, 507. https://doi.org/10.3390/cancers10120507
Soung YH, Ford S, Yan C, Chung J. The Role of Arrestin Domain-Containing 3 in Regulating Endocytic Recycling and Extracellular Vesicle Sorting of Integrin β4 in Breast Cancer. Cancers. 2018; 10(12):507. https://doi.org/10.3390/cancers10120507
Chicago/Turabian StyleSoung, Young Hwa, Shane Ford, Cecilia Yan, and Jun Chung. 2018. "The Role of Arrestin Domain-Containing 3 in Regulating Endocytic Recycling and Extracellular Vesicle Sorting of Integrin β4 in Breast Cancer" Cancers 10, no. 12: 507. https://doi.org/10.3390/cancers10120507