The Role of Activator Protein-1 (AP-1) Family Members in CD30-Positive Lymphomas

 ,
,  ,
,
Abstract
:1. Introduction
1.1. AP-1 Transcription Factors
1.2. Transcriptional and Translational Regulation of AP-1
1.3. AP-1 Functions in Tumourigenesis
2. AP-1 TFs in Classical Hodgkin Lymphoma (CHL)
2.1. Deregulated Signalling Pathways and AP-1 TFs in CHL
2.1.1. The NF-κB/AP-1 Signalling Axis in CHL
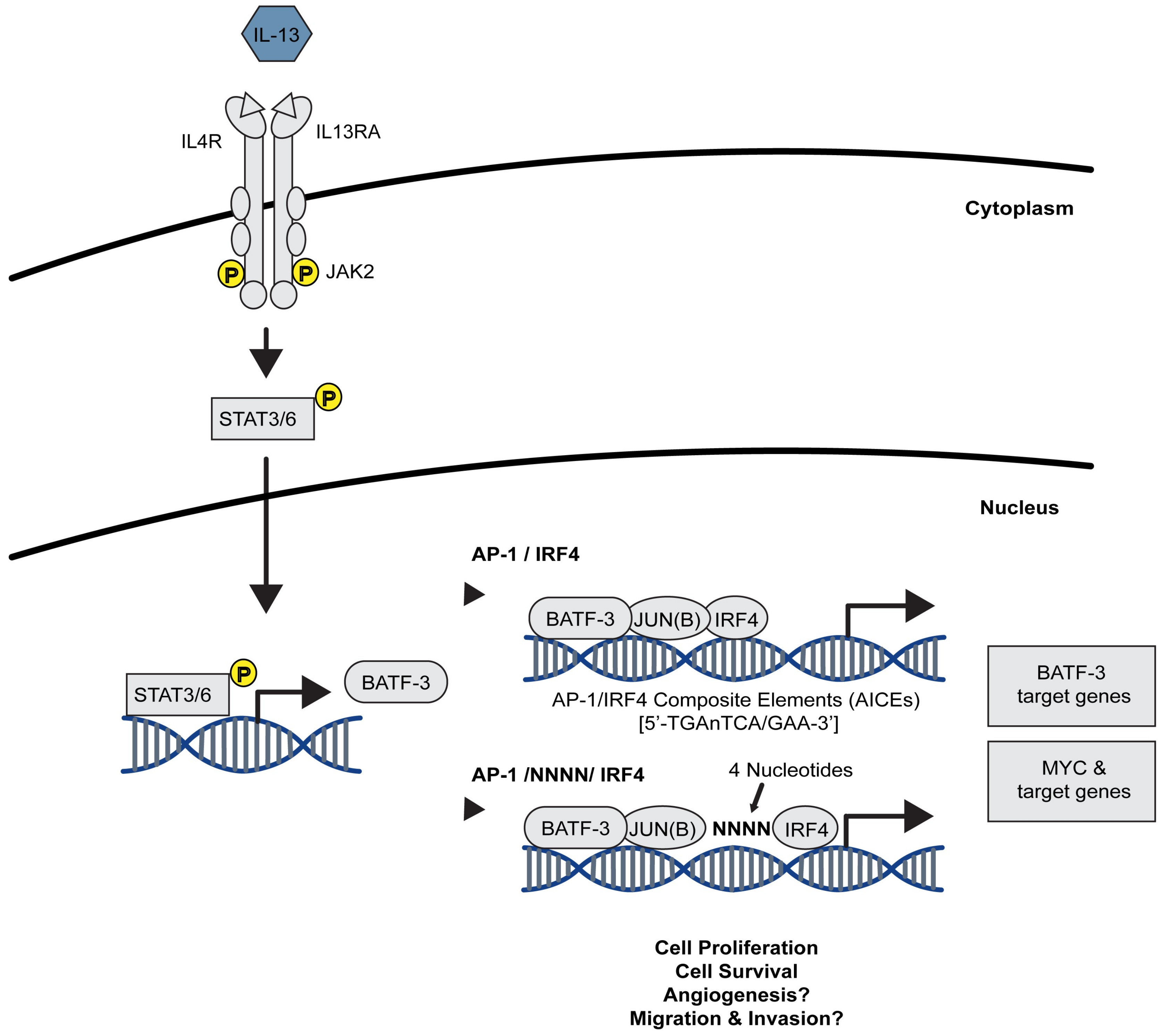
2.1.2. The STAT/AP-1 Signalling Axis in CHL
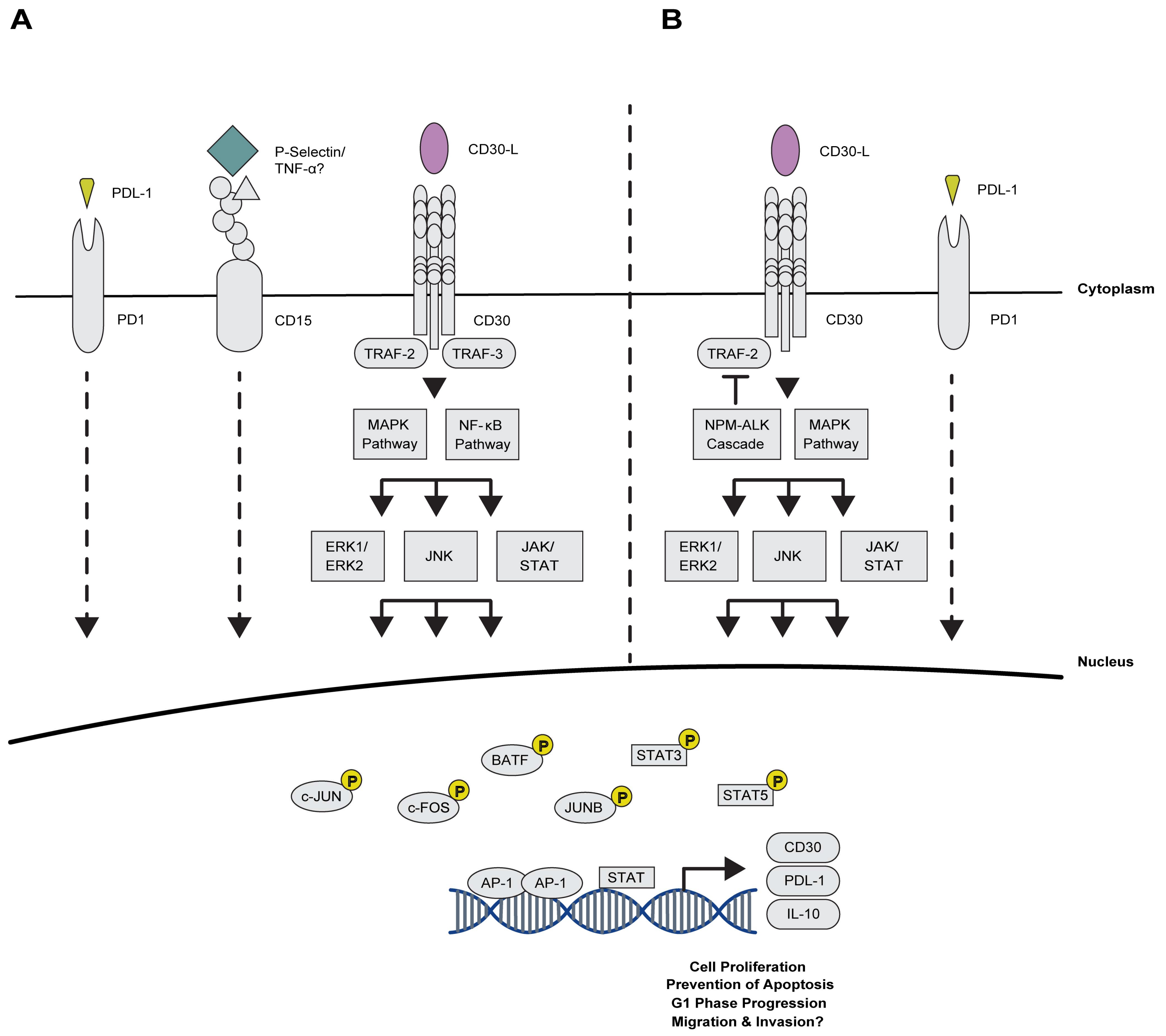
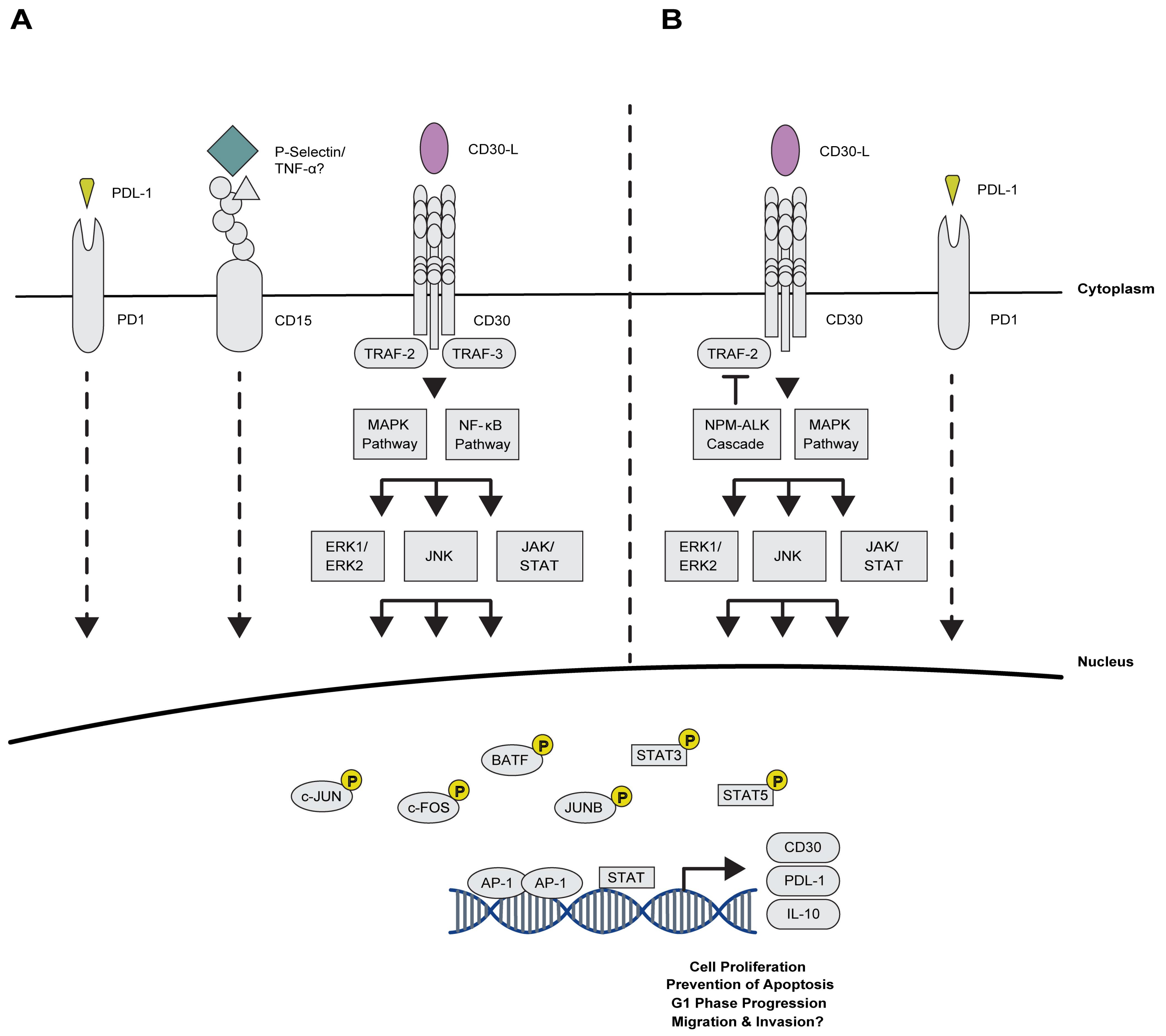
2.2. Deciphering the Cross-Talk between AP-1 and Cell Surface Proteins in CHL
2.2.1. Maintaining Proliferation via AP-1 and the TNFRS Family Members
2.2.2. Novel Association of AP-1 with Immuno-Surveillance and -Suppression Mechanisms
3. Involvement of AP-1 TFs in the Pathogenesis of CD30+ Peripheral T-Cell Lymphomas (PTCLs)
3.1. Anaplastic Large Cell Lymphoma (ALCL)
3.1.1. AP-1 TFs Are Expressed in ALCL, Mediate Key Signalling Pathways and Account for Typical Features of this Malignancy
3.1.2. AP-1 TFs Provide Therapeutic Targets for the Treatment of ALCL
3.2. Involvement of AP-1 TFs in PTCL-Not Otherwise Specified (PTCL-NOS)
3.3. Implication of AP-1 TFs in Extranodal, Cutaneous T-Cell Lymphomas (CTCLs)
3.4. AP-1 in Other PTCLs and Diffuse Large B-Cell Lymphoma (DLBCL)
4. AP-1 and Future Treatment Options
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Angel, P.; Imagawa, M.; Chiu, R.; Stein, B.; Imbra, R.J.; Rahmsdorf, H.J.; Jonat, C.; Herrlich, P.; Karin, M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell 1987, 49, 729–739. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Glover, J.N.; Harrison, S.C. Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature 1995, 373, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973. [Google Scholar] [CrossRef] [PubMed]
- Haslinger, A.; Karin, M. Upstream promoter element of the human metallothionein-IIA gene can act like an enhancer element. Proc. Natl. Acad. Sci. USA 1985, 82, 8572–8576. [Google Scholar] [CrossRef] [PubMed]
- Chinenov, Y.; Kerppola, T.K. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 2001, 20, 2438–2452. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, H.; Motohashi, H.; Sueno, S.; Kimura, M.; Takagawa, H.; Kanno, Y.; Yamamoto, M.; Tanaka, T. Structural basis of alternative DNA recognition by Maf transcription factors. Mol. Cell. Biol. 2009, 29, 6232–6244. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Georgopoulos, K.; Greenberg, M.E.; Leder, P. c-Jun dimerizes with itself and with c-Fos, forming complexes of different DNA binding affinities. Cell 1988, 55, 917–924. [Google Scholar] [CrossRef]
- O’Shea, E.K.; Rutkowski, R.; Kim, P.S. Mechanism of specificity in the Fos-Jun oncoprotein heterodimer. Cell 1992, 68, 699–708. [Google Scholar] [CrossRef]
- Bakiri, L.; Matsuo, K.; Wisniewska, M.; Wagner, E.F.; Yaniv, M. Promoter specificity and biological activity of tethered AP-1 dimers. Mol. Cell. Biol. 2002, 22, 4952–4964. [Google Scholar] [CrossRef] [PubMed]
- Grondin, B.; Lefrancois, M.; Tremblay, M.; Saint-Denis, M.; Haman, A.; Waga, K.; Bédard, A.; Tenen, D.G.; Hoang, T. c-Jun homodimers can function as a context-specific coactivator. Mol. Cell. Biol. 2007, 27, 2919–2933. [Google Scholar] [CrossRef] [PubMed]
- Chiu, R.; Angel, P.; Karin, M. Jun-B differs in its biological properties from, and is a negative regulator of, c-Jun. Cell 1989, 59, 979–986. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Guru, S.C.; Heppner, C.; Erdos, M.R.; Collins, R.M.; Park, S.Y.; Saggar, S.; Chandrasekharappa, S.C.; Collins, F.S.; Spiegel, A.M.; et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 1999, 96, 143–152. [Google Scholar] [CrossRef]
- Szabowski, A.; Maas-Szabowski, N.; Andrecht, S.; Kolbus, A.; Schorpp-Kistner, M.; Fusenig, N.E.; Angel, P. c-Jun and JunB antagonistically control cytokine-regulated mesenchymal-epidermal interaction in skin. Cell 2000, 103, 745–755. [Google Scholar] [CrossRef]
- Johnston, I.M.; Spence, H.J.; Winnie, J.N.; McGarry, L.; Vass, J.K.; Meagher, L.; Stapleton, G.; Ozanne, B.W. Regulation of a multigenic invasion programme by the transcription factor, AP-1: Re-expression of a down-regulated gene, TSC-36, inhibits invasion. Oncogene 2000, 19, 5348–5358. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, M.; Kolbus, A.; Piu, F.; Szabowski, A.; Möhle-Steinlein, U.; Tian, J.; Karin, M.; Angel, P.; Wagner, E.F. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999, 13, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Passegué, E.; Wagner, E.F. JunB suppresses cell proliferation by transcriptional activation of p16(INK4a) expression. EMBO J. 2000, 19, 2969–2979. [Google Scholar] [CrossRef] [PubMed]
- Bakiri, L.; Lallemand, D.; Bossy-Wetzel, E.; Yaniv, M. Cell cycle-dependent variations in c-Jun and JunB phosphorylation: A role in the control of cyclin D1 expression. EMBO J. 2000, 19, 2056–2068. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Schreiber, M.; Piu, F.; Beeche, M.; Wagner, E.F.; Karin, M. The mammalian UV response: C-Jun induction is required for exit from p53-imposed growth arrest. Cell 2000, 103, 897–907. [Google Scholar] [CrossRef]
- MacLaren, A.; Black, E.J.; Clark, W.; Gillespie, D.A.F. c-Jun-deficient cells undergo premature senescence as a result of spontaneous DNA damage accumulation. Mol. Cell. Biol. 2004, 24, 9006–9018. [Google Scholar] [CrossRef] [PubMed]
- Angel, P.; Hattori, K.; Smeal, T.; Karin, M. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell 1988, 55, 875–885. [Google Scholar] [CrossRef]
- Staber, P.B.; Vesely, P.; Haq, N.; Ott, R.G.; Funato, K.; Bambach, I.; Fuchs, C.; Schauer, S.; Linkesch, W.; Hrzenjak, A.; et al. The oncoprotein NPM-ALK of anaplastic large-cell lymphoma induces JUNB transcription via ERK1/2 and JunB translation via mTOR signaling. Blood 2007, 110, 3374–3383. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yang, W.; Bu, W.; Ji, H.; Zhao, X.; Zheng, Y.; Lin, X.; Li, Y.; Lu, Z. Differential regulation of c-Jun protein plays an instrumental role in chemoresistance of cancer cells. J. Biol. Chem. 2013, 288, 19321–19329. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995, 270, 16483–16486. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Kallunki, T.; Su, B.; Tsigelny, I.; Sluss, H.K.; Derijard, B.; Moore, G.; Davis, R.; Karin, M. JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 1994, 8, 2996–3007. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Dérijard, B.; Davis, R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [PubMed]
- Boyle, W.J.; Smeal, T.; Defize, L.H.; Angel, P.; Woodgett, J.R.; Karin, M.; Hunter, T. Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell 1991, 64, 573–584. [Google Scholar] [CrossRef]
- Baker, S.J.; Kerppola, T.K.; Luk, D.; Vandenberg, M.T.; Marshak, D.R.; Curran, T.; Abate, C. Jun is phosphorylated by several protein kinases at the same sites that are modified in serum-stimulated fibroblasts. Mol. Cell. Biol. 1992, 12, 4694–4705. [Google Scholar] [CrossRef] [PubMed]
- Smeal, T.; Binetruy, B.; Mercola, D.A.; Birrer, M.; Karin, M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature 1991, 354, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.N.; Ku, T.K.S.; Salib, N.K.; Crowe, D.L. Extracellular Signals Regulate Rapid Coactivator Recruitment at AP-1 Sites by Altered Phosphorylation of both CREB Binding Protein and c-jun. Mol. Cell. Biol. 2008, 28, 4240–4250. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Lozach, J.; Jepsen, K.; Sawka-Verhelle, D.; Perissi, V.; Sasik, R.; Rose, D.W.; Johnson, R.S.; Rosenfeld, M.G.; Glass, C.K. A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation. Proc. Natl. Acad. Sci. USA 2004, 101, 14461–14466. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.Y.; Dolan, L.; Davis, R.J.; Ronai, Z. Phosphorylation-dependent targeting of c-Jun ubiquitination by Jun N-kinase. Oncogene 1996, 13, 1531–1535. [Google Scholar] [PubMed]
- Salvat, C.; Jariel-Encontre, I.; Acquaviva, C.; Omura, S.; Piechaczyk, M. Differential directing of c-Fos and c-Jun proteins to the proteasome in serum-stimulated mouse embryo fibroblasts. Oncogene 1998, 17, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, P.; Andermarcher, E.; Bossis, G.; Acquaviva, C.; Brockly, F.; Jariel-Encontre, I.; Piechaczyk, M. The structural determinants responsible for c-Fos protein proteasomal degradation differ according to the conditions of expression. Oncogene 2003, 22, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Bossis, G.; Ferrara, P.; Acquaviva, C.; Jariel-Encontre, I.; Piechaczyk, M. c-Fos proto-oncoprotein is degraded by the proteasome independently of its own ubiquitinylation in vivo. Mol. Cell. Biol. 2003, 23, 7425–7436. [Google Scholar] [CrossRef] [PubMed]
- Treier, M.; Staszewski, L.M.; Bohmann, D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell 1994, 78, 787–798. [Google Scholar] [CrossRef]
- Muller, S.; Berger, M.; Lehembre, F.; Seeler, J.S.; Haupt, Y.; Dejean, A. c-Jun and p53 activity is modulated by SUMO-1 modification. J. Biol. Chem. 2000, 275, 13321–13329. [Google Scholar] [CrossRef] [PubMed]
- Bossis, G.; Malnou, C.E.; Farras, R.; Andermarcher, E.; Hipskind, R.; Rodriguez, M.; Schmidt, D.; Muller, S.; Jariel-Encontre, I.; Piechaczyk, M. Down-regulation of c-Fos/c-Jun AP-1 dimer activity by sumoylation. Mol. Cell. Biol. 2005, 25, 6964–6979. [Google Scholar] [CrossRef] [PubMed]
- Monje, P.; Marinissen, M.J.; Gutkind, J.S. Phosphorylation of the carboxyl-terminal transactivation domain of c-Fos by extracellular signal-regulated kinase mediates the transcriptional activation of AP-1 and cellular transformation induced by platelet-derived growth factor. Mol. Cell. Biol. 2003, 23, 7030–7043. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wang, J.; Liu, T.-J.; Yung, W.K.A.; Hunter, T.; Lu, Z. c-Jun downregulation by HDAC3-dependent transcriptional repression promotes osmotic stress-induced cell apoptosis. Mol. Cell 2007, 25, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Lantowski, A.; Dannenberg, A.J.; Subbaramaiah, K. Histone deacetylase inhibitors suppress the induction of c-Jun and its target genes including COX-2. J. Biol. Chem. 2005, 280, 32569–32577. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wu, Y.; Tang, X.; Xia, Y.; He, G.; Min, Z.; Li, C.; Xiong, S.; Shi, Z.; Lu, Y.; et al. HDAC inhibitors suppress c-Jun/Fra-1-mediated proliferation through transcriptionally downregulating MKK7 and Raf1 in neuroblastoma cells. Oncotarget 2016, 7, 6727–6747. [Google Scholar] [CrossRef] [PubMed]
- Tili, E.; Michaille, J.-J.; Croce, C.M. MicroRNAs play a central role in molecular dysfunctions linking inflammation with cancer. Immunol. Rev. 2013, 253, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, L.; Zhang, R.; Liu, X.; Ren, Y.; Liu, Z.; Zhang, X.; Cheng, W.; Hua, Z.-C. Regulation of T lymphocyte activation by microRNA-21. Mol. Immunol. 2014, 59, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Ito, T.; Mizutani, T.; Minoguchi, S.; Yamamichi, N.; Sakurai, K.; Iba, H. miR-21 Gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J. Mol. Biol. 2008, 378, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Wang, X.; McBride, J.; Fewell, C.; Flemington, E. B-cell receptor activation induces BIC/miR-155 expression through a conserved AP-1 element. J. Biol. Chem. 2008, 283, 2654–2662. [Google Scholar] [CrossRef] [PubMed]
- Maki, Y.; Bos, T.J.; Davis, C.; Starbuck, M.; Vogt, P.K. Avian sarcoma virus 17 carries the jun oncogene. Proc. Natl. Acad. Sci. USA 1987, 84, 2848–2852. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Saitoh, K.; Kameda, T.; Murakami, M.; Niikura, Y.; Okazaki, S.; Morishita, Y.; Mori, S.; Yokouchi, Y.; Kuroiwa, A.; et al. Chondrocytes as a specific target of ectopic Fos expression in early development. Proc. Natl. Acad. Sci. USA 1997, 94, 3994–3999. [Google Scholar] [CrossRef] [PubMed]
- Jochum, W.; Passegué, E.; Wagner, E.F. AP-1 in mouse development and tumorigenesis. Oncogene 2001, 20, 2401–2412. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-T.; Tsou, H.-K.; Chang, C.-H.; Tang, C.-H. Hepatocyte Growth Factor Increases Osteopontin Expression in Human Osteoblasts through PI3K, Akt, c-Src, and AP-1 Signaling Pathway. PLoS ONE 2012, 7, e38378. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.A.; O’Hara, M.; Angel, P.; Chojkier, M.; Karin, M. Prolonged activation of jun and collagenase genes by tumour necrosis factor-α. Nature 1989, 337, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Goldgaber, D.; Harris, H.W.; Hla, T.; Maciag, T.; Donnelly, R.J.; Jacobsen, J.S.; Vitek, M.P.; Gajdusek, D.C. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc. Natl. Acad. Sci. USA 1989, 86, 7606–7610. [Google Scholar] [CrossRef] [PubMed]
- Laderoute, K.R. The interaction between HIF-1 and AP-1 transcription factors in response to low oxygen. Semin. Cell Dev. Biol. 2005, 16, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Piccaluga, P.P.; Tabanelli, V.; Pileri, S.A. Molecular genetics of peripheral T-cell lymphomas. Int. J. Hematol. 2014, 99, 219–226. [Google Scholar] [CrossRef] [PubMed]
- De Leval, L.; Gaulard, P. Pathology and biology of peripheral T-cell lymphomas. Histopathology 2011, 58, 49–68. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.S.; Nicolae, A.; Pittaluga, S. Peripheral T-cell and NK-cell lymphomas in the WHO classification: Pearls and pitfalls. Mod. Pathol. 2013, 26, 71–87. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, H.; Savage, K.J. The spectrum of peripheral T-cell lymphomas. Curr. Opin. Hematol. 2009, 16, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Costello, R.; Sanchez, C.; Le Treut, T.; Rihet, P.; Imbert, J.; Sébahoun, G. Peripheral T-cell lymphoma gene expression profiling and potential therapeutic exploitations. Br. J. Haematol. 2010, 150, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Foss, F.M.; Zinzani, P.L.; Vose, J.M.; Gascoyne, R.D.; Rosen, S.T.; Tobinai, K. Peripheral T-cell lymphoma. Blood 2011, 117, 6756–6767. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.; Pan, L.; Diss, T.; Isaacson, P.G. Expression of B-cell antigens by Hodgkin’s and Reed-Sternberg cells. Am. J. Pathol. 1991, 139, 701–707. [Google Scholar] [PubMed]
- Amini, R.-M.; Enblad, G. Relationship between Hodgkin’s and non-Hodgkin’s lymphomas. Med. Oncol. 2003, 20, 211–220. [Google Scholar] [CrossRef]
- Kreher, S.; Bouhlel, M.A.; Cauchy, P.; Lamprecht, B.; Li, S.; Grau, M.; Hummel, F.; Köchert, K.; Anagnostopoulos, I.; Jöhrens, K.; et al. Mapping of transcription factor motifs in active chromatin identifies IRF5 as key regulator in classical Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E4513–E4522. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R. The biology of Hodgkin’s lymphoma. Nat. Rev. Cancer 2009, 9, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Zandi, E.; Rothwarf, D.M.; Delhase, M.; Hayakawa, M.; Karin, M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 1997, 91, 243–252. [Google Scholar] [CrossRef]
- Bargou, R.C.; Emmerich, F.; Krappmann, D.; Bommert, K.; Mapara, M.Y.; Arnold, W.; Royer, H.D.; Grinstein, E.; Greiner, A.; Scheidereit, C.; et al. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J. Clin. Invest. 1997, 100, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Weniger, M.A.; Küppers, R. NF-κB deregulation in Hodgkin lymphoma. Semin. Cancer Biol. 2016, 39, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Telenius, A.; Shah, S.P.; Farinha, P.; Barclay, L.; Boyle, M.; Connors, J.M.; Horsman, D.E.; Gascoyne, R.D. Genome-wide copy number analysis of Hodgkin Reed-Sternberg cells identifies recurrent imbalances with correlations to treatment outcome. Blood 2010, 116, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Mathas, S.; Hinz, M.; Anagnostopoulos, I.; Krappmann, D.; Lietz, A.; Jundt, F.; Bommert, K.; Mechta-Grigoriou, F.; Stein, H.; Dörken, B.; et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. EMBO J. 2002, 21, 4104–4113. [Google Scholar] [CrossRef] [PubMed]
- Burger, R. Impact of Interleukin-6 in hematological malignancies. Transfus. Med. Hemother. 2013, 40, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Debnath, B.; Neamati, N. Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Hirano, T. IL-6 in autoimmune disease and chronic inflammatory proliferative disease. Cytokine Growth Factor Rev. 2002, 13, 357–368. [Google Scholar] [CrossRef]
- Khalaf, H.; Jass, J.; Olsson, P.E. Differential cytokine regulation by NF-κB and AP-1 in Jurkat T-cells. BMC Immunol. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Pares-Matos, E.I.; Tesmer, V.M.; Dai, C.; Ashworth, S.; Huai, J.; Bina, M. Organization of the promoter region of the human NF-IL6 gene. Biochim. Biophys. Acta Gene Struct. Expr. 2002, 1577, 102–108. [Google Scholar] [CrossRef]
- Matsusaka, T.; Fujikawa, K.; Nishio, Y.; Mukaida, N.; Matsushima, K.; Kishimoto, T.; Akira, S. Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc. Natl. Acad. Sci. USA 1993, 90, 10193–10197. [Google Scholar] [CrossRef] [PubMed]
- Abate, C.; Patel, L.; Rauscher, F.; Curran, T. Redox regulation of fos and jun DNA-binding activity in vitro. Science 1990, 249, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Han, S.-I.; Kim, H.-H.; Lee, Z.H. TAK1-dependent activation of AP-1 and c-Jun N-terminal kinase by receptor activator of NF-kappaB. J. Biochem. Mol. Biol. 2002, 35, 371–376. [Google Scholar] [PubMed]
- Verma, I.M.; Stevenson, J.K.; Schwarz, E.M.; Van Antwerp, D.; Miyamoto, S. Rel/NF-kappa B/I kappa B family: Intimate tales of association and dissociation. Genes Dev. 1995, 9, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lin, Y.; Guo, Z.; Cheng, J.; Huang, J.; Deng, L.; Liao, W.; Chen, Z.; Liu, Z.; Su, B. The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat. Immunol. 2001, 2, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Franzoso, G.; Carlson, L.; Brown, K.; Daucher, M.B.; Bressler, P.; Siebenlist, U. Activation of the serum response factor by p65/NF-kappaB. EMBO J. 1996, 15, 3403–3412. [Google Scholar] [PubMed]
- Eckerle, S.; Brune, V.; Döring, C.; Tiacci, E.; Bohle, V.; Sundström, C.; Kodet, R.; Paulli, M.; Falini, B.; Klapper, W.; et al. Gene expression profiling of isolated tumour cells from anaplastic large cell lymphomas: Insights into its cellular origin, pathogenesis and relation to Hodgkin lymphoma. Leukemia 2009, 23, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Joos, S.; Küpper, M.; Ohl, S.; Von Bonin, F.; Mechtersheimer, G.; Bentz, M.; Marynen, P.; Möller, P.; Pfreundschuh, M.; Trümper, L.; et al. Genomic imbalances including amplification of the tyrosine kinase gene JAK2 in CD30+Hodgkin cells. Cancer Res. 2000, 60, 549–552. [Google Scholar] [PubMed]
- Scheeren, F.A.; Diehl, S.A.; Smit, L.A.; Beaumont, T.; Naspetti, M.; Bende, R.J.; Blom, B.; Karube, K.; Ohshima, K.; van Noesel, C.J.M.; et al. IL-21 is expressed in Hodgkin lymphoma and activates STAT5: Evidence that activated STAT5 is required for Hodgkin lymphomagenesis. Blood 2008, 111, 4706–4715. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Lemke, P.; Anagnostopoulos, I.; Hacker, C.; Krappmann, D.; Mathas, S.; Dörken, B.; Zenke, M.; Stein, H.; Scheidereit, C. Nuclear Factor KappaB Dependent Gene Expression Profiling of Hodgkin’s Disease Tumor Cells, Pathogenetic Significance, and Link to Constitutive Signal Transducer and Activator of Transcription 5a Activity. J. Exp. Med. 2002, 196, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, B.F. Signal transducer and activator of transcription 6 is frequently activated in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 2002, 99, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Lollies, A.; Hartmann, S.; Schneider, M.; Bracht, T.; Weiß, A.L.; Arnolds, J.; Klein-Hitpass, L.; Sitek, B.; Hansmann, M.-L.; Küppers, R.; et al. An oncogenic axis of STAT-mediated BATF3 upregulation causing MYC activity in classical Hodgkin lymphoma and anaplastic large cell lymphoma. Leukemia 2018, 32, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Wright, G.; Leroy, K.; Yu, X.; Gaulard, P.; Gascoyne, R.D.; Chan, W.C.; Zhao, T.; Haioun, C.; Greiner, T.C.; et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med. 2003, 198, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Schwering, I.; Bräuninger, A.; Distler, V.; Jesdinsky, J.; Diehl, V.; Hansmann, M.-L.; Rajewsky, K.; Küppers, R. Profiling of Hodgkin’s lymphoma cell line L1236 and germinal center B cells: Identification of Hodgkin’s lymphoma-specific genes. Mol. Med. 2003, 9, 85–95. [Google Scholar] [PubMed]
- Murphy, T.L.; Tussiwand, R.; Murphy, K.M. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat. Rev. Immunol. 2013, 13, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Schleussner, N.; Merkel, O.; Costanza, M.; Liang, H.-C.; Hummel, F.; Romagnani, C.; Durek, P.; Anagnostopoulos, I.; Hummel, M.; Jöhrens, K.; et al. The AP-1-BATF and -BATF3 module is essential for growth, survival and TH17/ILC3 skewing of anaplastic large cell lymphoma. Leukemia 2018. [Google Scholar] [CrossRef]
- Clark, G.; Stockinger, H.; Balderas, R.; van Zelm, M.C.; Zola, H.; Hart, D.; Engel, P. Nomenclature of CD molecules from the Tenth Human Leucocyte Differentiation Antigen Workshop. Clin. Transl. Immunol. 2016, 5, e57. [Google Scholar] [CrossRef] [PubMed]
- Horie, R.; Watanabe, T.; Morishita, Y.; Ito, K.; Ishida, T.; Kanegae, Y.; Saito, I.; Higashihara, M.; Mori, S.; Kadin, M.E.; et al. Ligand-independent signaling by overexpressed CD30 drives NF-kappaB activation in Hodgkin-Reed-Sternberg cells. Oncogene 2002, 21, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Buchan, S.L.; Al-Shamkhani, A. Distinct motifs in the intracellular domain of human CD30 differentially activate canonical and alternative transcription factor NF-κB signaling. PLoS ONE 2012, 7, e45244. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Sasaki, M.; Itoh, K.; Higashihara, M.; Umezawa, K.; Kadin, M.E.; Abraham, L.J.; Watanabe, T.; Horie, R. JunB induced by constitutive CD30-extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase signaling activates the CD30 promoter in anaplastic large cell lymphoma and reed-sternberg cells of Hodgkin lymphoma. Cancer Res. 2005, 65, 7628–7634. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Nakano, K.; Togano, T.; Nakashima, M.; Higashihara, M.; Kadin, M.E.; Watanabe, T.; Horie, R. Targeted repression of overexpressed CD30 downregulates NF-kappaB and ERK1/2 pathway in Hodgkin lymphoma cell lines. Oncol. Res. 2011, 19, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Croager, E.J.; Muir, T.M.; Abraham, L.J. Analysis of the human and mouse promoter region of the non-Hodgkin’s lymphoma-associated CD30 gene. J. Interferon Cytokine Res. 1998, 18, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Croager, E.J.; Gout, A.M.; Abraham, L.J. Involvement of Sp1 and microsatellite repressor sequences in the transcriptional control of the human CD30 gene. Am. J. Pathol. 2000, 156, 1723–1731. [Google Scholar] [CrossRef]
- Watanabe, M.; Ogawa, Y.; Ito, K.; Higashihara, M.; Kadin, M.E.; Abraham, L.J.; Watanabe, T.; Horie, R. AP-1 mediated relief of repressive activity of the CD30 promoter microsatellite in Hodgkin and Reed-Sternberg cells. Am. J. Pathol. 2003, 163, 633–641. [Google Scholar] [CrossRef]
- Watanabe, M.; Itoh, K.; Togano, T.; Kadin, M.E.; Watanabe, T.; Higashihara, M.; Horie, R. Ets-1 activates overexpression of JunB and CD30 in Hodgkin’s lymphoma and anaplastic large-cell lymphoma. Am. J. Pathol. 2012, 180, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Von Wasielewski, R.; Mengel, M.; Fischer, R.; Hansmann, M.L.; Hübner, K.; Franklin, J.; Tesch, H.; Paulus, U.; Werner, M.; Diehl, V.; et al. Classical Hodgkin’s disease. Clinical impact of the immunophenotype. Am. J. Pathol. 1997, 151, 1123–1130. [Google Scholar] [PubMed]
- Benharroch, D.; Dima, E.; Levy, A.; Ohana-Malka, O.; Ariad, S.; Prinsloo, I.; Mejirovsky, E.; Sacks, M.; Gopas, J. Differential expression of sialyl and non-sialyl-CD15 antigens on Hodgkin-Reed-Sternberg cells: Significance in Hodgkin’s disease. Leuk. Lymphoma 2000, 39, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Ohana, O.M.; Ozer, J.; Prinsloo, I.; Benharroch, D.; Gopas, J. Hodgkin lymphoma cell lines bind to platelets. Incubation with platelets induces CD15 and P-selectin dependent adhesion of the cell lines to Human Umbilical Vein Endothelial cells (HUVEC). Cancer Biol. Ther. 2015, 16, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Jeffrey Medeiros, L.; Young, K.H. Signaling pathway and dysregulation of PD1 and its ligands in lymphoid malignancies. Biochim. Biophys. Acta Rev. Cancer 2016, 1865, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.M.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 genetic alterations define classical hodgkin lymphoma and predict outcome. J. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.R.; Shipp, M.A. Signaling pathways and immune evasion mechanisms in classical Hodgkin lymphoma. Blood 2017, 130, 2265–2270. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-l1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Vose, J.; Armitage, J.; Weisenburger, D. International T-Cell Lymphoma Project International Peripheral T-Cell and Natural Killer/T-Cell Lymphoma Study: Pathology Findings and Clinical Outcomes. J. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar] [CrossRef] [PubMed]
- Damm-Welk, C.; Klapper, W.; Oschlies, I.; Gesk, S.; Röttgers, S.; Bradtke, J.; Siebert, R.; Reiter, A.; Woessmann, W. Distribution of NPM1-ALK and X-ALK fusion transcripts in paediatric anaplastic large cell lymphoma: A molecular-histological correlation. Br. J. Haematol. 2009, 146, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Martelli, M.P. Anaplastic Large Cell Lymphoma: changes in the World Health Organization classification and perspectives for targeted therapy. Haematologica; 2009, 94, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Stein, H.; Foss, H.D.; Dürkop, H.; Marafioti, T.; Delsol, G.; Pulford, K.; Pileri, S.; Falini, B. CD30(+) anaplastic large cell lymphoma: A review of its histopathologic, genetic, and clinical features. Blood 2000, 96, 3681–3695. [Google Scholar] [PubMed]
- Tort, F.; Pinyol, M.; Pulford, K.; Roncador, G.; Hernandez, L.; Nayach, I.; Kluin-Nelemans, H.C.; Kluin, P.; Touriol, C.; Delsol, G.; et al. Molecular characterization of a new ALK translocation involving moesin (MSN-ALK) in anaplastic large cell lymphoma. Lab. Investig. 2001, 81, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. European T-Cell Lymphoma Study Group, T-Cell Project: Prospective Collection of Data in Patients with Peripheral T-Cell Lymphoma and the AIRC 5xMille Consortium “Genetics-Driven Targeted Management of Lymphoid Malignancies” Convergent Mutations and Kinase Fusions Lead to Oncogenic STAT3 Activation in Anaplastic Large Cell Lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Mereu, E.; Pellegrino, E.; Scarfò, I.; Inghirami, G.; Piva, R. The heterogeneous landscape of ALK negative ALCL. Oncotarget 2017, 8, 18525–18536. [Google Scholar] [CrossRef] [PubMed]
- Mathas, S.; Kreher, S.; Meaburn, K.J.; Jöhrens, K.; Lamprecht, B.; Assaf, C.; Sterry, W.; Kadin, M.E.; Daibata, M.; Joos, S.; et al. Gene deregulation and spatial genome reorganization near breakpoints prior to formation of translocations in anaplastic large cell lymphoma. Proc. Natl. Acad. Sci. USA 2009, 106, 5831–5836. [Google Scholar] [CrossRef] [PubMed]
- Janz, M.; Hummel, M.; Truss, M.; Wollert-Wulf, B.; Mathas, S.; Jöhrens, K.; Hagemeier, C.; Bommert, K.; Stein, H.; Dörken, B.; et al. Classical Hodgkin lymphoma is characterized by high constitutive expression of activating transcription factor 3 (ATF3), which promotes viability of Hodgkin/Reed-Sternberg cells. Blood 2006, 107, 2536–2539. [Google Scholar] [CrossRef] [PubMed]
- Agnelli, L.; Mereu, E.; Pellegrino, E.; Limongi, T.; Kwee, I.; Bergaggio, E.; Ponzoni, M.; Zamò, A.; Iqbal, J.; Piccaluga, P.P.; et al. European T-Cell Lymphoma Study Group Identification of a 3-gene model as a powerful diagnostic tool for the recognition of ALK-negative anaplastic large-cell lymphoma. Blood 2012, 120, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Orchard, G.; Lillington, D.M.; Russell-Jones, R.; Young, B.D.; Whittaker, S.J. Amplification and overexpression of JUNB is associated with primary cutaneous T-cell lymphomas. Blood 2003, 101, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Higuchi, T.; Oiso, N.; Kawada, A.; Yoshie, O. Expression and function of FRA2/JUND in cutaneous T-cell lymphomas. Anticancer Res. 2012, 32, 1367–1373. [Google Scholar] [PubMed]
- Leventaki, V.; Drakos, E.; Medeiros, L.J.; Lim, M.S.; Elenitoba-Johnson, K.S.; Claret, F.X.; Rassidakis, G.Z. NPM-ALK oncogenic kinase promotes cell-cycle progression through activation of JNK/cJun signaling in anaplastic large-cell lymphoma. Blood 2007, 110, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D.; Yeung, D.; Hadfield, K.; Cook, S.J.; Alexander, D.R. The NPM-ALK tyrosine kinase mimics TCR signalling pathways, inducing NFAT and AP-1 by RAS-dependent mechanisms. Cell. Signal. 2007, 19, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Atsaves, V.; Lekakis, L.; Drakos, E.; Leventaki, V.; Ghaderi, M.; Baltatzis, G.E.; Chioureas, D.; Jones, D.; Feretzaki, M.; Liakou, C.; et al. The oncogenic JUNB/CD30 axis contributes to cell cycle deregulation in ALK+ anaplastic large cell lymphoma. Br. J. Haematol. 2014, 167, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Weilemann, A.; Grau, M.; Erdmann, T.; Merkel, O.; Sobhiafshar, U.; Anagnostopoulos, I.; Hummel, M.; Siegert, A.; Hayford, C.; Madle, H.; et al. Essential role of IRF4 and MYC signaling for survival of anaplastic large cell lymphoma. Blood 2015, 125, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.Y.-Y.; Johnston, P.B.; Burke, K.A.; Zhao, Y. The expression of CD30 in anaplastic large cell lymphoma is regulated by nucleophosmin-anaplastic lymphoma kinase-mediated JunB level in a cell type-specific manner. Cancer Res. 2006, 66, 9002–9008. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, T.I.M.; Villarese, P.; Fairbairn, C.J.; Lamant, L.; Trinquand, A.; Hook, C.E.; Burke, G.A.A.; Brugières, L.; Hughes, K.; Payet, D.; et al. Anaplastic large cell lymphoma arises in thymocytes and requires transient TCR expression for thymic egress. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Ambrogio, C.; Martinengo, C.; Voena, C.; Tondat, F.; Riera, L.; Di Celle, P.F.; Inghirami, G.; Chiarle, R. NPM-ALK oncogenic tyrosine kinase controls T-cell identity by transcriptional regulation and epigenetic silencing in lymphoma cells. Cancer Res. 2009, 69, 8611–8619. [Google Scholar] [CrossRef] [PubMed]
- Hassler, M.R.; Pulverer, W.; Lakshminarasimhan, R.; Redl, E.; Hacker, J.; Garland, G.D.; Merkel, O.; Schiefer, A.I.; Simonitsch-Klupp, I.; Kenner, L.; et al. Insights into the Pathogenesis of Anaplastic Large-Cell Lymphoma through Genome-wide DNA Methylation Profiling. Cell Rep. 2016, 17, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Laimer, D.; Dolznig, H.; Kollmann, K.; Vesely, P.W.; Schlederer, M.; Merkel, O.; Schiefer, A.-I.; Hassler, M.R.; Heider, S.; Amenitsch, L.; et al. PDGFR blockade is a rational and effective therapy for NPM-ALK-driven lymphomas. Nat. Med. 2012, 18, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Wright, G.; Wang, C.; Rosenwald, A.; Gascoyne, R.D.; Weisenburger, D.D.; Greiner, T.C.; Smith, L.; Guo, S.; Wilcox, R.A.; et al. Lymphoma Leukemia Molecular Profiling Project and the International Peripheral T-cell Lymphoma Project Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood 2014, 123, 2915–2923. [Google Scholar] [CrossRef] [PubMed]
- Onaindia, A.; Martínez, N.; Montes-Moreno, S.; Almaraz, C.; Rodríguez-Pinilla, S.M.; Cereceda, L.; Revert, J.B.; Ortega, C.; Tardio, A.; González, L.; et al. CD30 expression by B and T cells: A frequent finding in angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma-not otherwise specified. Am. J. Surg. Pathol. 2016, 40, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Sabattini, E.; Pizzi, M.; Tabanelli, V.; Baldin, P.; Sagramoso Sacchetti, C.; Agostinelli, C.; Luigi Zinzani, P.; Pileri, S.A. CD30 expression in peripheral T-cell lymphomas. Haematologica 2013, 98, e81–e82. [Google Scholar] [CrossRef] [PubMed]
- Bisig, B.; de Reyniès, A.; Bonnet, C.; Sujobert, P.; Rickman, D.S.; Marafioti, T.; Delsol, G.; Lamant, L.; Gaulard, P.; de Leval, L. CD30-positive peripheral T-cell lymphomas share molecular and phenotypic features. Haematologica 2013, 98, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Drakos, E.; Leventaki, V.; Schlette, E.J.; Jones, D.; Lin, P.; Medeiros, L.J.; Rassidakis, G.Z. c-Jun expression and activation are restricted to CD30+ lymphoproliferative disorders. Am. J. Surg. Pathol. 2007, 31, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Rassidakis, G.Z.; Thomaides, A.; Atwell, C.; Ford, R.; Jones, D.; Claret, F.X.; Medeiros, L.J. JunB expression is a common feature of CD30+ lymphomas and lymphomatoid papulosis. Mod. Pathol. 2005, 18, 1365–1370. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Orchard, G.; Mitchell, T.J.; Oyama, N.; Russell-Jones, R.; Vermeer, M.H.; Willemze, R.; Van Doorn, R.; Tensen, C.P.; Young, B.D.; et al. A genomic and expression study of AP-1 in primary cutaneous T-cell lymphoma: Evidence for dysregulated expression of JUNB and JUND in MF and SS. J. Cutan. Pathol. 2008, 35, 899–910. [Google Scholar] [CrossRef] [PubMed]
- De Leval, L.; Gisselbrecht, C.; Gaulard, P. Advances in the understanding and management of angioimmunoblastic T-cell lymphoma. Br. J. Haematol. 2010, 148, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Vallois, D.; Dobay, M.P.D.; Morin, R.D.; Lemonnier, F.; Missiaglia, E.; Juilland, M.; Iwaszkiewicz, J.; Fataccioli, V.; Bisig, B.; Roberti, A.; et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood 2016, 128, 1490–1502. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Fujii, M.; Ikeda, S.; Yamada, Y.; Tomonaga, M.; Ballard, D.W.; Yamamoto, N. Constitutive activation of NF-kappaB in primary adult T-cell leukemia cells. Blood 1999, 93, 2360–2368. [Google Scholar] [PubMed]
- Mori, N.; Fujii, M.; Iwai, K.; Ikeda, S.; Yamasaki, Y.; Hata, T.; Yamada, Y.; Tanaka, Y.; Tomonaga, M.; Yamamoto, N. Constitutive activation of transcription factor AP-1 in primary adult T-cell leukemia cells. Blood 2000, 95, 3915–3921. [Google Scholar] [PubMed]
- Ishida, T.; Utsunomiya, A.; Iida, S.; Inagaki, H.; Takatsuka, Y.; Kusumoto, S.; Takeuchi, G.; Shimizu, S.; Ito, M.; Komatsu, H.; et al. Clinical significance of CCR4 expression in adult T-cell leukemia/lymphoma: Its close association with skin involvement and unfavorable outcome. Clin. Cancer Res. 2003, 9, 3625–3634. [Google Scholar] [PubMed]
- Nakayama, T.; Hieshima, K.; Arao, T.; Jin, Z.; Nagakubo, D.; Shirakawa, A.-K.; Yamada, Y.; Fujii, M.; Oiso, N.; Kawada, A.; et al. Aberrant expression of Fra-2 promotes CCR4 expression and cell proliferation in adult T-cell leukemia. Oncogene 2008, 27, 3221–3232. [Google Scholar] [CrossRef] [PubMed]
- Yoshie, O.; Fujisawa, R.; Nakayama, T.; Harasawa, H.; Tago, H.; Izawa, D.; Hieshima, K.; Tatsumi, Y.; Matsushima, K.; Hasegawa, H.; et al. Frequent expression of CCR4 in adult T-cell leukemia and human T-cell leukemia virus type 1-transformed T cells. Blood 2002, 99, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Nagakubo, D.; Jin, Z.; Hieshima, K.; Nakayama, T.; Shirakawa, A.-K.; Tanaka, Y.; Hasegawa, H.; Hayashi, T.; Tsukasaki, K.; Yamada, Y.; et al. Expression of CCR9 in HTLV-1+ T cells and ATL cells expressing Tax. Int. J. Cancer 2007, 120, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Hess, S.; Richardson, S.K.; Newton, S.; Showe, L.C.; Benoit, B.M.; Ubriani, R.; Vittorio, C.C.; Junkins-Hopkins, J.M.; Wysocka, M.; et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J. Clin. Investig. 2005, 115, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Kamioka, M.; Imamura, J.; Komatsu, N.; Daibata, M.; Sugiura, T. Testican 3 expression in adult T-cell leukemia. Leuk. Res. 2009, 33, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Hagiya, K.; Yasunaga, J.-I.; Satou, Y.; Ohshima, K.; Matsuoka, M. ATF3, an HTLV-1 bZip factor binding protein, promotes proliferation of adult T-cell leukemia cells. Retrovirology 2011, 8, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deleeuw, R.J.; Zettl, A.; Klinker, E.; Haralambieva, E.; Trottier, M.; Chari, R.; Ge, Y.; Gascoyne, R.D.; Chott, A.; Müller-Hermelink, H.-K.; et al. Whole-genome analysis and HLA genotyping of enteropathy-type T-cell lymphoma reveals 2 distinct lymphoma subtypes. Gastroenterology 2007, 132, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Yamaguchi, M.; Imai, H.; Kobayashi, T.; Tamaru, S.; Nishii, K.; Yuda, M.; Shiku, H.; Katayama, N. Gene expression profiling of peripheral T-cell lymphoma including gammadelta T-cell lymphoma. Blood 2009, 113, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Juilland, M.; Gonzalez, M.; Erdmann, T.; Banz, Y.; Jevnikar, Z.; Hailfinger, S.; Tzankov, A.; Grau, M.; Lenz, G.; Novak, U.; et al. CARMA1- and MyD88-dependent activation of Jun/ATF-type AP-1 complexes is a hallmark of ABC diffuse large B-cell lymphomas. Blood 2016, 127, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Papoudou-Bai, A.; Goussia, A.; Batistatou, A.; Stefanou, D.; Malamou-Mitsi, V.; Kanavaros, P. The expression levels of JunB, JunD and p-c-Jun are positively correlated with tumor cell proliferation in diffuse large B-cell lymphomas. Leuk. Lymphoma 2016, 57, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Meijer, C.A.; Le Haen, P.A.A.; van Dijk, R.A.; Hira, M.; Hamming, J.F.; van Bockel, J.H.; Lindeman, J.H. Activator protein-1 (AP-1) signalling in human atherosclerosis: Results of a systematic evaluation and intervention study. Clin. Sci. (Lond.) 2012, 122, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Muslin, A.J. MAPK signalling in cardiovascular health and disease: Molecular mechanisms and therapeutic targets. Clin. Sci. (Lond.) 2008, 115, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Uchihashi, S.; Fukumoto, H.; Onoda, M.; Hayakawa, H.; Ikushiro, S.; Sakaki, T. Metabolism of the c-Fos/activator protein-1 inhibitor T-5224 by multiple human UDP-glucuronosyltransferase isoforms. Drug Metab. Dispos. 2011, 39, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Ball, D.P.; Lewis, A.M.; Williams, D.; Resetca, D.; Wilson, D.J.; Gunning, P.T. Signal transducer and activator of transcription 3 (STAT3) inhibitor, S3I-201, acts as a potent and non-selective alkylating agent. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Cumaraswamy, A.A.; Lewis, A.M.; Geletu, M.; Todic, A.; Diaz, D.B.; Cheng, X.R.; Brown, C.E.; Laister, R.C.; Muench, D.; Kerman, K.; et al. Nanomolar-potency small molecule inhibitor of STAT5 protein. ACS Med. Chem. Lett. 2014, 5, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Rezaei Araghi, R.; Bird, G.H.; Ryan, J.A.; Jenson, J.M.; Godes, M.; Pritz, J.R.; Grant, R.A.; Letai, A.; Walensky, L.D.; Keating, A.E. Iterative optimization yields Mcl-1–targeting stapled peptides with selective cytotoxicity to Mcl-1–dependent cancer cells. Proc. Natl. Acad. Sci. USA 2018, 201712952. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Jaume, M.A.; Bergman, M.R.; Mahimkar, R.; Cheng, S.; Jin, Z.Q.; Karliner, J.S.; Lovett, D.H. Cardiac ischemia-reperfusion injury induces matrix metalloproteinase-2 expression through the AP-1 components FosB and JunB. Am. J. Physiol. Circ. Physiol. 2006, 291, H1838–H1846. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.W.; Bigam, C.G.; Erdman, P.E.; Palanki, M.S.; Anderson, D.W.; Goldman, M.E.; Ransone, L.J.; Suto, M.J. 2-Chloro-4-(trifluoromethyl)pyrimidine-5-N-(3′,5′- bis(trifluoromethyl)phenyl)-carboxamide: A potent inhibitor of NF-kappa B- and AP-1-mediated gene expression identified using solution-phase combinatorial chemistry. J. Med. Chem. 1998, 41, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Uray, I.P.; Li, Y.; Krisko, T.I.; Strecker, T.E.; Kim, H.T.; Brown, P.H. The AP-1 transcription factor regulates breast cancer cell growth via cyclins and E2F factors. Oncogene 2008, 27, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.S.; Thaker, H.M.; Giordano, T.; Williams, J.; Rogers, D.; Sudersanam, V.; Vasu, K.K. Design, synthesis and characterization of novel 2-(2,4-disubstituted-thiazole-5-yl)-3-aryl-3H-quinazoline-4-one derivatives as inhibitors of NF-kappaB and AP-1 mediated transcription activation and as potential anti-inflammatory agents. Eur. J. Med. Chem. 2009, 44, 2184–2189. [Google Scholar] [CrossRef] [PubMed]
- Palanki, M.S.S.; Manning, A.M.; Erdman, P.E.; Gayo-Fung, L.M.; Shevlin, G.I.; Sullivan, R.W.; Suto, M.J.; Goldman, M.E.; Ransone, L.J.; Bennett, B.L. Inhibitors of NF-κB and AP-1 gene expression: SAR studies on the pyrimidine portion of 2-chloro-4-trifluoromethylpyrimidine-5-[N-(3′,5′-bis(trifluoromethyl)-phenyl) carboxamide]. J. Med. Chem. 2000, 43, 3995–4004. [Google Scholar] [CrossRef] [PubMed]
- Palanki, M.S.; Erdman, P.E.; Manning, A.M.; Ow, A.; Ransone, L.J.; Spooner, C.; Suto, C.; Suto, M. Novel inhibitors of AP-1 and NF-κB mediated gene expression: Structure-activity relationship studies of ethyl 4-[(3-Methyl-2,5-dioxo(3-pyrrolinyl))amino]-2-(trifluoromethyl)pyrimidine-5-carboxylate. Bioorg. Med. Chem. Lett. 2000, 10, 1645–1648. [Google Scholar] [CrossRef]
- Aikawa, Y.; Morimoto, K.; Yamamoto, T.; Chaki, H.; Hashiramoto, A.; Narita, H.; Hirono, S.; Shiozawa, S. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat. Biotechnol. 2008, 26, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Kamide, D.; Yamashita, T.; Araki, K.; Tomifuji, M.; Tanaka, Y.; Tanaka, S.; Shiozawa, S.; Shiotani, A. Selective activator protein-1 inhibitor T-5224 prevents lymph node metastasis in an oral cancer model. Cancer Sci. 2016, 107, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wu, M.; Li, Y.; Chang, I.; Yuan, Q.; Ekimyan-Salvo, M.; Deng, P.; Yu, B.; Yu, Y.; Dong, J.; et al. Targeting BMI1+Cancer Stem Cells Overcomes Chemoresistance and Inhibits Metastases in Squamous Cell Carcinoma. Cell Stem Cell 2017, 20, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.S.; Thaker, H.M.; Giordano, T.; Chen, B.; Nuthalapaty, S.; Vasu, K.K.; Sudarsanam, V. Synthesis and evaluation of quinazolinone derivatives as inhibitors of NF-κB, AP-1 mediated transcription and eIF-4E mediated translational activation: Inhibitors of multi-pathways involve in cancer. Eur. J. Med. Chem. 2010, 45, 3558–3563. [Google Scholar] [CrossRef] [PubMed]
- Atsaves, V.; Zhang, R.; Ruder, D.; Pan, Y.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. Constitutive control of AKT1 gene expression by JUNB/CJUN in ALK+ anaplastic large-cell lymphoma: A novel crosstalk mechanism. Leukemia 2015, 29, 2162–2172. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Family | Neoplasms | Major Groups | Sub-Groups |
|---|---|---|---|
| Lymphoproliferative Disorders [62] | Hodgkin Lymphoma (HL) [63,64] | Classical Hodgkin Lymphoma (CHL) [64] | |
| Peripheral T-cell Lymphoma/Non-Hodgkin Lymphoma (NHL) [56,57,58,59,60,61,62] | Nodal Lymphoma [56,57,62] | Anaplastic Large Cell Lymphoma (ALCL) | |
| Angioimmunoblastic T-cell Lymphoma (AITL) | |||
| PTCL-Not Otherwise Specified (PTCL-NOS) | |||
| Extranodal Lymphoma [58,59,62] | Enteropathy-associated T-cell Lymphoma (EATL) | ||
| Hepatosplenic Υδ T-cell Lymphoma | |||
| Natural Killer (NK)/NK—like T-cell Lymphoma | |||
| Extranodal-cutaneous Lymphoma [58,60,62] | B-cell Cutaneous Lymphoma | ||
| T-cell Cutaneous Lymphoma | |||
| NK-cell Cutaneous Lymphoma | |||
| Leukaemic and Disseminated Disease [61,62] | Adult T-cell Leukaemia/Lymphoma (ATLL) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garces de los Fayos Alonso, I.; Liang, H.-C.; Turner, S.D.; Lagger, S.; Merkel, O.; Kenner, L. The Role of Activator Protein-1 (AP-1) Family Members in CD30-Positive Lymphomas. Cancers 2018, 10, 93. https://doi.org/10.3390/cancers10040093
Garces de los Fayos Alonso I, Liang H-C, Turner SD, Lagger S, Merkel O, Kenner L. The Role of Activator Protein-1 (AP-1) Family Members in CD30-Positive Lymphomas. Cancers. 2018; 10(4):93. https://doi.org/10.3390/cancers10040093
Chicago/Turabian StyleGarces de los Fayos Alonso, Ines, Huan-Chang Liang, Suzanne D. Turner, Sabine Lagger, Olaf Merkel, and Lukas Kenner. 2018. "The Role of Activator Protein-1 (AP-1) Family Members in CD30-Positive Lymphomas" Cancers 10, no. 4: 93. https://doi.org/10.3390/cancers10040093