Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy


Abstract
:1. Introduction
1.1. Repair of Single-Strand DNA Breaks (SSBs)
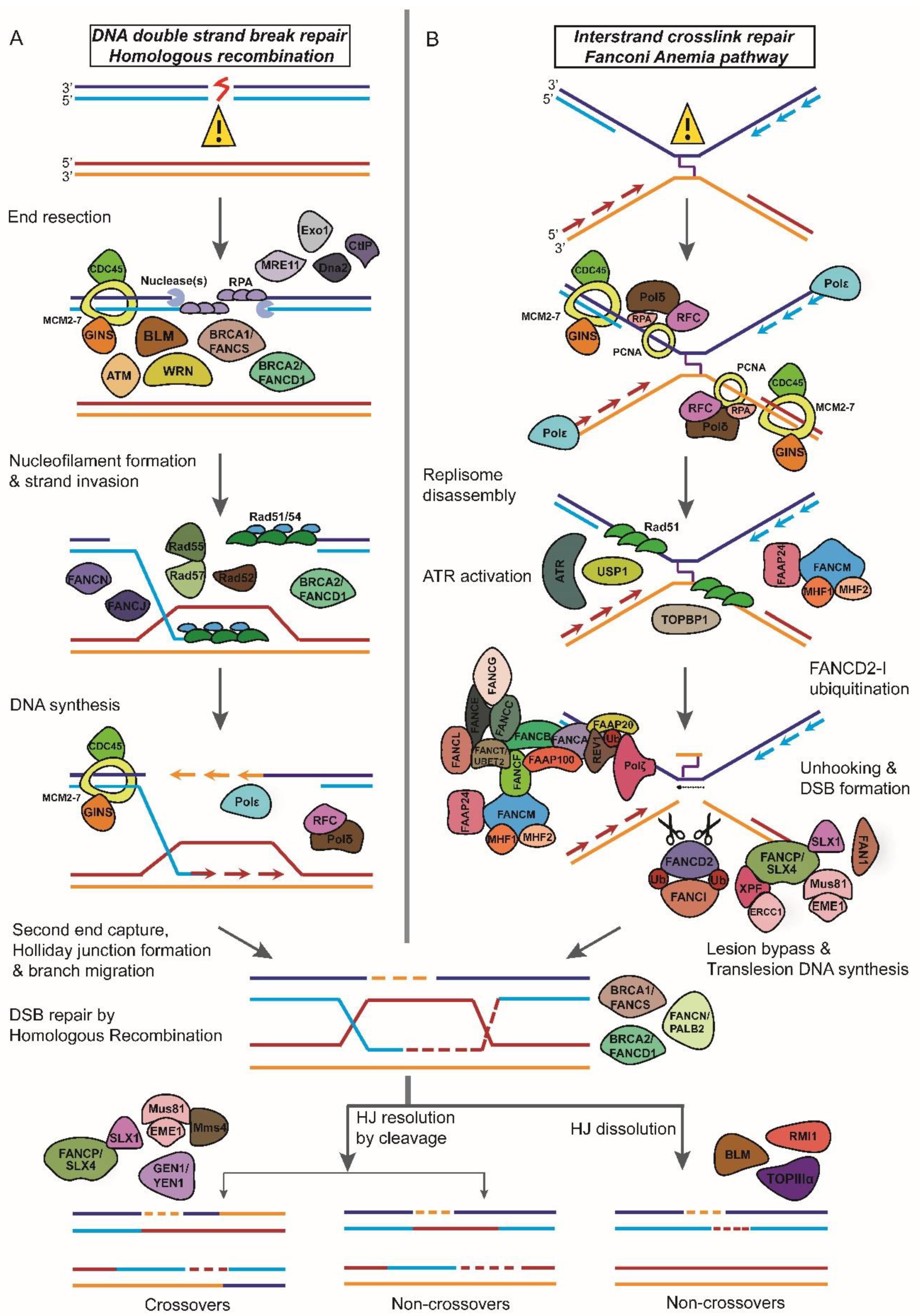
1.2. Repair of Double-Strand DNA Breaks (DSBR)
2. Leveraging Insights from the Biology of Rare Genetic Diseases for Targeted Cancer Therapy
3. Werner Syndrome
3.1. Werner Helicase in Non-Homologous End-Joining and Base Excision Repair
3.2. Small Molecule Inhibitors and Werner Helicase
4. Bloom Syndrome
4.1. Bloom Helicase and Homologous Recombination
4.2. Small Molecule Inhibitors and Bloom Helicase
5. Ataxia Telangiectasia
5.1. ATM and Double-Strand Break Repair
5.2. Small Molecule Inhibitors and ATM-CHK2 Helicases
6. Fanconi Anemia
6.1. Fanconi Anemia Proteins and DNA Interstrand Crosslink Repair
6.2. Small Molecule Inhibitors and Fanconi Anemia Proteins
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Alt-NHEJ | Alternative-NHEJ |
| ATTM | Ammonium tetrathiomolybdate |
| AT | Ataxia telangiectasia |
| BER | Base excision repair |
| BS | Bloom syndrome |
| CRC | Colorectal cancer |
| CPD | Cyclobutane pyrimidine dimers |
| D-loops | Displacement loops |
| DDR | DNA damage response |
| DSBs | Double-strand breaks |
| DSBR | Double-strand break repair |
| FA | Fanconi Anemia |
| OOPD | FDA Office of Orphan Products Development |
| HJ | Holliday junction |
| HR | Homologous recombination |
| ICLs | Interstrand crosslinks |
| LCS-1 | Lung Cancer Screen-1 |
| MMEJ | Microhomology-mediated end-joining |
| MMR | Mismatch repair |
| MRN | Mre11-Rad50-Nbs1 |
| NHEJ | Non-homologous end-joining |
| NER | Nucleotide excision repair |
| PARP | Poly(ADP ribose) polymerase |
| PRR | Post-replication repair |
| RPA | Replication protein A |
| SSBs | Single-strand breaks |
| SCE | Sister chromatid exchanges |
| SOD1 | Superoxide dismutase 1 |
| WS | Werner syndrome |
References
- Boycott, K.M.; Vanstone, M.R.; Bulman, D.E.; MacKenzie, A.E. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 2013, 14, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, S.; Nandi, S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life 2017, 69, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Fishel, R. Mismatch repair. J. Biol. Chem. 2015, 290, 26395–26403. [Google Scholar] [CrossRef] [PubMed]
- Hanawalt, P.C.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Scharer, O.D. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 2006, 106, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Castel, S.E.; Ren, J.; Bhattacharjee, S.; Chang, A.Y.; Sanchez, M.; Valbuena, A.; Antequera, F.; Martienssen, R.A. Dicer Promotes Transcription Termination at Sites of Replication Stress to Maintain Genome Stability. Cell 2014, 159, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.D.; Marsischky, G.T. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 1999, 9, 89–96. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Meers, C.; Keskin, H.; Storici, F. DNA repair by RNA: Templated, or not templated, that is the question. DNA Repair (Amst.) 2016, 44, 17–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decottignies, A. Alternative end-joining mechanisms: A historical perspective. Front. Genet. 2013, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell. Biol. 2003, 4, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.B. The origin of variation. Amer. Nat. 1922, 56, 51–63. [Google Scholar] [CrossRef]
- Biss, M.; Xiao, W. Selective tumor killing based on specific DNA-damage response deficiencies. Cancer Biol. Ther. 2012, 13, 239–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Furgason, J.M.; Bahassi El, M. Targeting DNA repair mechanisms in cancer. Pharmacol. Ther. 2013, 137, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Dietlein, F.; Thelen, L.; Reinhardt, H.C. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014, 30, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. Choices have consequences: The nexus between DNA repair pathways and genomic instability in cancer. Clin. Transl. Med. 2016, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic lethality and cancer therapy: Lessons learned from the development of PARP inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.T.; Yu, X.C. Functions of PARylation in DNA Damage Repair Pathways. Genom. Proteom. Bioinform. 2016, 14, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W.; Aoufouchi, S.; Johnson, P.; Shall, S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly(ADP-ribose) polymerase, and DNA ligase III is a novel molecular ‘nick-sensor’ in vitro. Nucleic Acids Res. 1996, 24, 4387–4394. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, M. Werner’s syndrome: From clinics to genetics. Clin. Exp. Rheumatol. 2000, 18, 760–766. [Google Scholar] [PubMed]
- O, W. On cataract in conjunction with scleroderma. Otto Werner, doctoral dissertation, 1904, Royal Ophthalmology Clinic, Royal Christian Albrecht University of Kiel. Adv. Exp. Med. Biol. 1985, 190, 1–14. [Google Scholar]
- Huang, S.R.; Li, B.M.; Gray, M.D.; Oshima, J.; Mian, S.I.; Campisi, J. The premature ageing syndrome protein, WRN, is a 3′→5′ exonuclease. Nat. Genet. 1998, 20, 114–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, C.J.; Martin, G.M.; Schultz, A.L.; Motulsky, A.G. Werner’s syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore) 1966, 45, 177–221. [Google Scholar] [CrossRef] [PubMed]
- Muftuoglu, M.; Oshima, J.; von Kobbe, C.; Cheng, W.H.; Leistritz, D.F.; Bohr, V.A. The clinical characteristics of Werner syndrome: Molecular and biochemical diagnosis. Hum. Genet. 2008, 124, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Masala, M.V.; Scapaticci, S.; Olivieri, C.; Pirodda, C.; Montesu, M.A.; Cuccuru, M.A.; Pruneddu, S.; Danesino, C.; Cerimele, D. Epidemiology and clinical aspects of Werner’s syndrome in North Sardinia: Description of a cluster. Eur J. Dermatol. 2007, 17, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Oshima, J. Werner Syndrome. J. Biomed. Biotechnol. 2002, 2, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamanna, R.A.; Lu, H.M.; de Freitas, J.K.; Tian, J.; Croteau, D.L.; Bohr, V.A. WRN regulates pathway choice between classical and alternative non-homologous end joining. Nat. Commun. 2016, 7, 13785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Zhang, T.; Ren, Y.; Wang, Z.; Ling, C.C.; He, F.; Li, G.C.; Wang, C.; Wen, B. Inhibiting DNA-PKcs in a non-homologous end-joining pathway in response to DNA double-strand breaks. Oncotarget 2017, 8, 22662–22673. [Google Scholar] [CrossRef] [PubMed]
- Li, B.M.; Comai, L. Functional interaction between Ku and the Werner syndrome protein in DNA end processing. J. Biol. Chem. 2000, 275, 39800. [Google Scholar] [CrossRef] [PubMed]
- Von Kobbe, C.; May, A.; Grandori, C.; Bohr, V.A. Werner syndrome cells escape hydrogen peroxide-induced cell proliferation arrest. FASEB J. 2004, 18, 1970–1972. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Wilson, D.M., 3rd; Prasad, R.; Opresko, P.L.; Beck, G.; May, A.; Wilson, S.H.; Bohr, V.A. The Werner syndrome protein operates in base excision repair and cooperates with DNA polymerase beta. Nucleic Acids Res. 2006, 34, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.L.; Ghosh, A.K.; Bohr, V.A. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst.) 2010, 9, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattler, U.; Frit, P.; Salles, B.; Calsou, P. Long-patch DNA repair synthesis during base excision repair in mammalian cells. EMBO Rep. 2003, 4, 363–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dianov, G.L.; Prasad, R.; Wilson, S.H.; Bohr, V.A. Role of DNA polymerase beta in the excision step of long patch mammalian base excision repair. J. Biol. Chem. 1999, 274, 13741–13743. [Google Scholar] [CrossRef] [PubMed]
- Sobol, R.W.; Horton, J.K.; Kuhn, R.; Gu, H.; Singhal, R.K.; Prasad, R.; Rajewsky, K.; Wilson, S.H. Requirement of mammalian DNA polymerase-beta in base-excision repair (vol 379, pg 183, 1996). Nature 1996, 383, 457. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Opresko, P.L.; von Kobbe, C.; Kedar, P.S.; Prasad, R.; Wilson, S.H.; Bohr, V.A. The Werner syndrome protein stimulates DNA polymerase beta strand displacement synthesis via its helicase activity. J. Biol. Chem. 2003, 278, 22686–22695. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Fan, J.; Momand, J.; Perrino, F.W.; Bohr, V.A.; Wilson, D.M. WRN exonuclease activity is blocked by DNA termini harboring 3′ obstructive groups. Mech. Ageing Dev. 2007, 128, 259–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosh, R.M.; Driscoll, H.C.; Dianov, G.L.; Sommers, J.A. Biochemical characterization of the WRN-FEN-1 functional interaction. Biochemistry 2002, 41, 12204–12216. [Google Scholar] [CrossRef] [PubMed]
- von Kobbe, C.; Harrigan, J.A.; May, A.; Opresko, P.L.; Dawut, L.; Cheng, W.H.; Bohr, V.A. Central role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damage. Mol. Cell. Biol. 2003, 23, 8601–8613. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.; Harrigan, J.A.; Indig, F.E.; Wilson, D.M.; Bohr, V.A. Regulation of WRN helicase activity in human base excision repair. J. Biol. Chem. 2004, 279, 53465–53474. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Banerjee, T.; Sommers, J.A.; Brosh, R.M., Jr. Targeting an Achilles’ heel of cancer with a WRN helicase inhibitor. Cell Cycle 2013, 12, 3329–3335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, M.; Sommers, J.A.; Shoemaker, R.H.; Brosh, R.M., Jr. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1525–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- German, J.; Sanz, M.M.; Ciocci, S.; Ye, T.Z.; Ellis, N.A. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum. Mutat. 2007, 28, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.H.; Dexheimer, T.S.; Rosenthal, A.S.; Chu, W.K.; Singh, D.K.; Mosedale, G.; Bachrati, C.Z.; Schultz, L.; Sakurai, M.; Savitsky, P.; et al. A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells. Chem. Biol. 2013, 20, 55–62. [Google Scholar] [CrossRef] [PubMed]
- German, J. Bloom’s syndrome. XX. The first 100 cancers. Cancer Genet. Cytogenet. 1997, 93, 100–106. [Google Scholar] [CrossRef]
- Roa, B.B.; Savino, C.V.; Richards, C.S. Ashkenazi Jewish population frequency of the Bloom syndrome gene 2281 delta 6ins7 mutation. Genet. Test. 1999, 3, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Eng, C.; Desnick, R.J.; German, J.; Ellis, N.A. Carrier frequency of the Bloom syndrome blmAsh mutation in the Ashkenazi Jewish population. Mol. Genet. Metab. 1998, 64, 286–290. [Google Scholar] [CrossRef] [PubMed]
- German, J.; Roe, A.M.; Leppert, M.F.; Ellis, N.A. Bloom syndrome: An analysis of consanguineous families assigns the locus mutated to chromosome band 15q26.1. Proc. Natl. Acad. Sci. USA 1994, 91, 6669–6673. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.K.; Hickson, I.D. RecQ helicases: Multifunctional genome caretakers. Nat. Rev. Cancer 2009, 9, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Cunniff, C.; Bassetti, J.A.; Ellis, N.A. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol. Syndromol. 2017, 8, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Mohaghegh, P.; Karow, J.K.; Brosh, R.M., Jr.; Bohr, V.A.; Hickson, I.D. The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001, 29, 2843–2849. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.M., Jr.; Li, J.L.; Kenny, M.K.; Karow, J.K.; Cooper, M.P.; Kureekattil, R.P.; Hickson, I.D.; Bohr, V.A. Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity. J. Biol. Chem. 2000, 275, 23500–23508. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Bellaoui, M.; Zhang, C.; Desai, R.; Morozov, P.; Delgado-Cruzata, L.; Rothstein, R.; Freyer, G.A.; Boone, C.; Brown, G.W. RMI1/NCE4, a suppressor of genome instability, encodes a member of the RecQ helicase/Topo III complex. EMBO J. 2005, 24, 2024–2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meetei, A.R.; Sechi, S.; Wallisch, M.; Yang, D.; Young, M.K.; Joenje, H.; Hoatlin, M.E.; Wang, W. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol. Cell. Biol. 2003, 23, 3417–3426. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Davies, S.L.; Levitt, N.C.; Hickson, I.D. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J. Biol. Chem. 2001, 276, 19375–19381. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Chan, K.L.; Ralf, C.; Bernstein, D.A.; Garcia, P.L.; Bohr, V.A.; Vindigni, A.; Janscak, P.; Keck, J.L.; Hickson, I.D. The HRDC domain of BLM is required for the dissolution of double Holliday junctions. EMBO J. 2005, 24, 2679–2687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Smith, K.; Waldman, B.C.; Waldman, A.S. Depletion of the bloom syndrome helicase stimulates homology-dependent repair at double-strand breaks in human chromosomes. DNA Repair (Amst.) 2011, 10, 416–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Salah, G.; Hadj Salem, I.; Masmoudi, A.; Kallabi, F.; Turki, H.; Fakhfakh, F.; Ayadi, H.; Kamoun, H. A novel frameshift mutation in BLM gene associated with high sister chromatid exchanges (SCE) in heterozygous family members. Mol. Biol. Rep. 2014, 41, 7373–7380. [Google Scholar] [CrossRef] [PubMed]
- Chaganti, R.S.; Schonberg, S.; German, J. A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc. Natl. Acad. Sci. USA 1974, 71, 4508–4512. [Google Scholar] [CrossRef] [PubMed]
- Reddi, A.R.; Culotta, V.C. SOD1 integrates signals from oxygen and glucose to repress respiration. Cell 2013, 152, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Sajesh, B.V.; McManus, K.J. Targeting SOD1 induces synthetic lethal killing in BLM- and CHEK2-deficient colorectal cancer cells. Oncotarget 2015, 6, 27907–27922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, A.M.R.; Lam, Z.; Last, J.I.; Byrd, P.J. Ataxia telangiectasia: More variation at clinical and cellular levels. Clin. Genet. 2015, 87, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Lempiainen, H.; Halazonetis, T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009, 28, 3067–3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovejoy, C.A.; Cortez, D. Common mechanisms of PIKK regulation. DNA Repair (Amst.) 2009, 8, 1004–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bednarski, J.J.; Sleckman, B.P. Integrated signaling in developing lymphocytes The role of DNA damage responses. Cell Cycle 2012, 11, 4129–4134. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Smolka, M.B.; Albuquerque, C.P.; Chen, S.H.; Zhou, H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 10364–10369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beli, P.; Lukashchuk, N.; Wagner, S.A.; Weinert, B.T.; Olsen, J.V.; Baskcomb, L.; Mann, M.; Jackson, S.P.; Choudhary, C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell. 2012, 46, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, A.; Schmidt, A.; Ziv, Y.; Elkon, R.; Wang, S.Y.; Chen, D.J.; Aebersold, R.; Shiloh, Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal. 2010, 3, rs3. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007, 11, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Andegeko, Y.; Moyal, L.; Mittelman, L.; Tsarfaty, I.; Shiloh, Y.; Rotman, G. Nuclear retention of ATM at sites of DNA double strand breaks. J. Biol. Chem. 2001, 276, 38224–38230. [Google Scholar] [PubMed]
- Bekker-Jensen, S.; Lukas, C.; Kitagawa, R.; Melander, F.; Kastan, M.B.; Bartek, J.; Lukas, J. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J. Cell Biol. 2006, 173, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.P.; Maser, R.S.; Olivares, H.; Davis, E.M.; Le Beau, M.; Yates, J.R.; Hays, L.; Morgan, W.F.; Petrini, J.H.J. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell 1998, 93, 477–486. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Wang, B.; Bignell, C.R.; Taylor, A.M.; Elledge, S.J. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 2003, 421, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Edwards, R.A.; Thede, G.L.; Glover, J.N. Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the gamma-H2AX histone tail. J. Biol. Chem. 2005, 280, 32053–32056. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Minter-Dykhouse, K.; Franco, S.; Gostissa, M.; Rivera, M.A.; Celeste, A.; Manis, J.P.; van Deursen, J.; Nussenzweig, A.; Paull, T.T.; et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell. 2006, 21, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Jackson, S.P. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008, 9, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melander, F.; Bekker-Jensen, S.; Falck, J.; Bartek, J.; Mailand, N.; Lukas, J. Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J. Cell Biol. 2008, 181, 213–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spycher, C.; Miller, E.S.; Townsend, K.; Pavic, L.; Morrice, N.A.; Janscak, P.; Stewart, G.S.; Stucki, M. Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J. Cell Biol. 2008, 181, 227–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, R.; Xie, A. Double strand break repair functions of histone H2AX. Mutat. Res. 2013, 750, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huen, M.S.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Huang, J.; Chen, J.J. CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nat. Struct. Mol. Biol. 2007, 14, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Chen, J.; Yu, X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 2007, 316, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Elledge, S.J. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl. Acad. Sci. USA 2007, 104, 20759–20763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Sobhian, B.; Shao, G.; Lilli, D.R.; Culhane, A.C.; Moreau, L.A.; Xia, B.; Livingston, D.M.; Greenberg, R.A. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 2007, 316, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Polo, S.; Miller, K.M.; Jackson, S.P. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 2009, 462, 935–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, G.S. Solving the RIDDLE of 53BP1 recruitment to sites of damage. Cell Cycle 2009, 8, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, S.; Rendtlew Danielsen, J.; Fugger, K.; Gromova, I.; Nerstedt, A.; Lukas, C.; Bartek, J.; Lukas, J.; Mailand, N. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell. Biol. 2010, 12, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Meerang, M.; Ritz, D.; Paliwal, S.; Garajova, Z.; Bosshard, M.; Mailand, N.; Janscak, P.; Hubscher, U.; Meyer, H.; Ramadan, K. The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat. Cell Biol. 2011, 13, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Mattiroli, F.; Vissers, J.H.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Riballo, E.; Kuhne, M.; Rief, N.; Doherty, A.; Smith, G.C.; Recio, M.J.; Reis, C.; Dahm, K.; Fricke, A.; Krempler, A.; et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell. 2004, 16, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.P.; Uematsu, N.; Kobayashi, J.; Lerenthal, Y.; Krempler, A.; Yajima, H.; Lobrich, M.; Shiloh, Y.; Chen, D.J. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J. Biol. Chem. 2007, 282, 6582–6587. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Yu, Y.; Riballo, E.; Douglas, P.; Walker, S.A.; Ye, R.; Harer, C.; Marchetti, C.; Morrice, N.; Jeggo, P.A.; et al. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J. 2006, 25, 3880–3889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.L.; Shi, L.Z.; Wong, C.C.L.; Han, X.M.; Hwang, P.Y.H.; Truong, L.N.; Zhu, Q.Y.; Shao, Z.P.; Chen, D.J.; Berns, M.W.; et al. The Interaction of CtIP and Nbs1 Connects CDK and ATM to Regulate HR-Mediated Double-Strand Break Repair. PLoS Genet. 2013, 9, e1003277. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Lobrich, M.; et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, U509-U506. [Google Scholar] [CrossRef] [PubMed]
- Chanut, P.; Britton, S.; Coates, J.; Jackson, S.P.; Calsou, P. Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks. Nat. Commun. 2016, 7, 12889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zuo, B.; Wang, H.; Ren, L.; Yang, P.; Zeng, M.; Duan, D.; Liu, C.; Li, M. CGK733 enhances multinucleated cell formation and cytotoxicity induced by taxol in Chk1-deficient HBV-positive hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2012, 422, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Rainey, M.D.; Charlton, M.E.; Stanton, R.V.; Kastan, M.B. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008, 68, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Alao, J.P.; Sunnerhagen, P. The ATM and ATR inhibitors CGK733 and caffeine suppress cyclin D1 levels and inhibit cell proliferation. Radiat. Oncol. 2009, 4, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batey, M.A.; Zhao, Y.; Kyle, S.; Richardson, C.; Slade, A.; Martin, N.M.; Lau, A.; Newell, D.R.; Curtin, N.J. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol. Cancer Ther. 2013, 12, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Cmielova, J.; Havelek, R.; Kohlerova, R.; Soukup, T.; Bruckova, L.; Suchanek, J.; Vavrova, J.; Mokry, J.; Rezacova, M. The effect of ATM kinase inhibition on the initial response of human dental pulp and periodontal ligament mesenchymal stem cells to ionizing radiation. Int. J. Radiat. Biol. 2013, 89, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Jo, Y.H.; Cho, C.H.; Choe, W.; Kang, I.; Baik, H.H.; Yoon, K.S. ATM-deficient human fibroblast cells are resistant to low levels of DNA double-strand break induced apoptosis and subsequently undergo drug-induced premature senescence. Biochem. Biophys. Res. Commun. 2013, 430, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Mao, C.; Wu, J.; Li, S.; Ma, R.; Cao, H.; Ji, M.; Jing, C.; Tang, J. Improved ataxia telangiectasia mutated kinase inhibitor KU60019 provides a promising treatment strategy for non-invasive breast cancer. Oncol. Lett. 2014, 8, 2043–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobson, A.G.; Lountos, G.T.; Lorenzi, P.L.; Llamas, J.; Connelly, J.; Cerna, D.; Tropea, J.E.; Onda, A.; Zoppoli, G.; Kondapaka, S.; et al. Cellular inhibition of checkpoint kinase 2 (Chk2) and potentiation of camptothecins and radiation by the novel Chk2 inhibitor PV1019 [7-nitro-1H-indole-2-carboxylic acid {4-[1-(guanidinohydrazone)-ethyl]-phenyl}-amide]. J. Pharmacol. Exp. Ther. 2009, 331, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.E.; Walton, M.I.; Eve, P.D.; Boxall, K.J.; Antoni, L.; Caldwell, J.J.; Aherne, W.; Pearl, L.H.; Oliver, A.W.; Collins, I.; et al. CCT241533 is a potent and selective inhibitor of CHK2 that potentiates the cytotoxicity of PARP inhibitors. Cancer Res. 2011, 71, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Riesterer, O.; Matsumoto, F.; Wang, L.; Pickett, J.; Molkentine, D.; Giri, U.; Milas, L.; Raju, U. A novel Chk inhibitor, XL-844, increases human cancer cell radiosensitivity through promotion of mitotic catastrophe. Investig. New Drugs 2011, 29, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.J.; Yakes, F.M.; Chen, J.; Tadano, M.; Bornheim, L.; Clary, D.O.; Tai, A.; Wagner, J.M.; Miller, N.; Kim, Y.D.; et al. Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell Cycle 2007, 6, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Fanconi, G. Familial constitutional panmyelocytopathy, Fanconi’s anemia (F.A.). I. Clinical aspects. Semin. Hematol. 1967, 4, 233–240. [Google Scholar] [PubMed]
- Lobitz, S.; Velleuer, E. Guido Fanconi (1892–1979): A jack of all trades. Nat. Rev. Cancer 2006, 6, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Mathew, C.G. Fanconi anaemia genes and susceptibility to cancer. Oncogene 2006, 25, 5875–5884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meetei, A.R.; Levitus, M.; Xue, Y.T.; Medhurst, A.L.; Zwaan, M.; Ling, C.; Rooimans, M.A.; Bier, P.; Hoatlin, M.; Pals, G.; et al. X-linked inheritance of Fanconi anemia complementation group B. Nat. Genet. 2004, 36, 1219–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantinou, A. Rescue of replication failure by Fanconi anaemia proteins. Chromosoma 2012, 121, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P. Fanconi anemia and the development of leukemia. Best Pract. Res. Clin. Haematol. 2014, 27, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, P.S.; Tamary, H.; Alter, B.P. How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi Anemia in the United States and Israel. Am. J. Med. Genet. 2011, 155A, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Daly, M.; Arends, M.J.; Patel, K.J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 2012, 489, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Parmar, K.; Mouly, E.; Delord, M.; Kim, J.M.; Regairaz, M.; Pla, M.; Vasquez, N.; Zhang, Q.S.; Pondarre, C.; et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. 2012, 11, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway. Cell Commun. Signal. 2017, 15. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, S.; Wu, Z. Fanconi anemia pathway defects in inherited and sporadic cancers. Transl. Pediatr. 2014, 3, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell. Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Cheung, R.S.; Castella, M.; Abeyta, A.; Gafken, P.R.; Tucker, N.; Taniguchi, T. Ubiquitination-Linked Phosphorylation of the FANCI S/TQ Cluster Contributes to Activation of the Fanconi Anemia I/D2 Complex. Cell Rep. 2017, 19, 2432–2440. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Pradhan, A.; Singh, T.R.; Du, C.H.; Li, J.; Wahengbam, K.; Grassman, E.; Auerbach, A.D.; Pang, Q.S.; Meetei, A.R. FAAP20: A novel ubiquitin-binding FA nuclear core-complex protein required for functional integrity of the FA-BRCA DNA repair pathway. Blood 2012, 119, 3285–3294. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Osman, F.; Feeney, L.; Lorenz, A.; Bryer, C.; Whitby, M.C. MHF1-2/CENP-S-X performs distinct roles in centromere metabolism and genetic recombination (Retracted article. See vol. 8, art no. 180010, 2018). Open Biol. 2013, 3, 130102. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Ling, C.; Coulthard, R.; Yan, Z.J.; Xue, Y.T.; Meetei, A.R.; Laghmani, E.H.; Joenje, H.; McDonald, N.; de Winter, J.P.; et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol. Cell 2007, 25, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.B.; Nebert, D.W.; Bruford, E.A.; Thompson, D.C.; Joenje, H.; Vasiliou, V. Update of the human and mouse Fanconi anemia genes. Hum. Genomics 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yang, K.L.; Dejsuphong, D.; D’Andrea, A.D. Regulation of Rev1 by the Fanconi anemia core complex. Nat. Struct. Mol. Biol. 2012, 19, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Ishiai, M.; Ali, A.M.; Medhurst, A.L.; Neveling, K.; Kalb, R.; Yan, Z.J.; Xue, Y.T.; Oostra, A.B.; Auerbach, A.D.; et al. FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. EMBO J. 2007, 26, 2104–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandi, S.; Whitby, M.C. The ATPase activity of Fml1 is essential for its roles in homologous recombination and DNA repair. Nucleic Acids Res. 2012, 40, 9584–9595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Nandi, S.; Osman, F.; Ahn, J.S.; Jakovleska, J.; Lorenz, A.; Whitby, M.C. The FANCM Ortholog Fml1 Promotes Recombination at Stalled Replication Forks and Limits Crossing Over during DNA Double-Strand Break Repair. Mol. Cell 2008, 32, 118–128. [Google Scholar] [CrossRef] [PubMed]
- de Oca, R.M.; Andreassen, P.R.; Margossian, S.P.; Gregory, R.C.; Taniguchi, T.; Wang, X.Z.; Houghtaling, S.; Grompe, M.; D’Andrea, A.D. Regulated interaction of the Fanconi anemia protein, FANCD2, with chromatin. Blood 2005, 105, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Dorsman, J.C.; Levitus, M.; Rockx, D.; Rooimans, M.A.; Oostra, A.B.; Haitjema, A.; Bakker, S.T.; Steltenpool, J.; Schuler, D.; Mohan, S.; et al. Identification of the Fanconi anemia complementation group I gene, FANCI. Cell. Oncol. 2007, 29, 211–218. [Google Scholar] [PubMed]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P.; Hofmann, K.; D’Andrea, A.D.; et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007, 129, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Mamrak, N.E.; Shimamura, A.; Howlett, N.G. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev. 2017, 31, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.J.; Huang, T.T. The Fanconi anemia pathway in replication stress and DNA crosslink repair. Cell. Mol. Life Sci. 2012, 69, 3963–3974. [Google Scholar] [CrossRef] [PubMed]
- Palovcak, A.; Liu, W.; Yuan, F.; Zhang, Y. Maintenance of genome stability by Fanconi anemia proteins. Cell Biosci. 2017, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Hira, A.; Yoshida, K.; Sato, K.; Okuno, Y.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Miyano, S.; Shimamoto, A.; Tahara, H.; et al. Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. Am. J. Hum. Genet. 2015, 96, 1001–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, J.A.; Frost, M.G.; Carroll, E.; Rowe, M.L.; Howard, M.J.; Sidhu, A.; Chaugule, V.K.; Alpi, A.F.; Walden, H. The Fanconi Anemia DNA Repair Pathway Is Regulated by an Interaction between Ubiquitin and the E2-like Fold Domain of FANCL. J. Biol. Chem. 2015, 290, 20995–21006. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Zhan, B.; Yoshikawa, Y.; Haas, W.; Gygi, S.P.; Cohn, M.A. UHRF1 is a sensor for DNA interstrand crosslinks and recruits FANCD2 to initiate the Fanconi anemia pathway. Cell Rep. 2015, 10, 1947–1956. [Google Scholar] [CrossRef] [PubMed]
- Knipscheer, P.; Raschle, M.; Smogorzewska, A.; Enoiu, M.; Ho, T.V.; Scharer, O.D.; Elledge, S.J.; Walter, J.C. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 2009, 326, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.N.; Kobayashi, S.; Tsuda, M.; Kurumizaka, H.; Takata, M.; Kono, K.; Jiricny, J.; Takeda, S.; Hirota, K. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 6492–6496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Spitz, G.S.; Veturi, U.; Lach, F.P.; Auerbach, A.D.; Smogorzewska, A. Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood 2013, 121, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Klein Douwel, D.; Boonen, R.A.; Long, D.T.; Szypowska, A.A.; Raschle, M.; Walter, J.C.; Knipscheer, P. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell. 2014, 54, 460–471. [Google Scholar] [CrossRef] [PubMed]
- McMahon, L.W.; Sangerman, J.; Goodman, S.R.; Kumaresan, K.; Lambert, M.W. Human α Spectrin II and the FANCA, FANCC, and FANCG Proteins Bind to DNA Containing Psoralen Interstrand Cross-Links†. Biochemistry 2001, 40, 7025–7034. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.W. Nuclear alpha spectrin: Critical roles in DNA interstrand cross-link repair and genomic stability. Exp. Biol. Med. (Maywood) 2016, 241, 1621–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 2009, 73, 134–154. [Google Scholar] [CrossRef] [PubMed]
- Murakumo, Y. The property of DNA polymerase ζ: REV7 is a putative protein involved in translesion DNA synthesis and cell cycle control. Mutat. Res. 2002, 510, 37–44. [Google Scholar] [CrossRef]
- Hanada, K.; Budzowska, M.; Davies, S.L.; van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007, 14, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Clauson, C.; Scharer, O.D.; Niedernhofer, L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012732. [Google Scholar] [CrossRef] [PubMed]
- Chirnomas, D.; Taniguchi, T.; de la Vega, M.; Vaidya, A.P.; Vasserman, M.; Hartman, A.R.; Kennedy, R.; Foster, R.; Mahoney, J.; Seiden, M.V.; et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol. Cancer Ther. 2006, 5, 952–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkitt, K.; Ljungman, M. Phenylbutyrate interferes with the Fanconi anemia and BRCA pathway and sensitizes head and neck cancer cells to cisplatin. Mol. Cancer 2008, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Dexheimer, T.S.; Ai, Y.; Liang, Q.; Villamil, M.A.; Inglese, J.; Maloney, D.J.; Jadhav, A.; Simeonov, A.; Zhuang, Z. Selective and cell-active inhibitors of the USP1/UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem. Biol. 2011, 18, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Dexheimer, T.S.; Zhang, P.; Rosenthal, A.S.; Villamil, M.A.; You, C.; Zhang, Q.; Chen, J.; Ott, C.A.; Sun, H.; et al. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat. Chem. Biol. 2014, 10, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Hsieh, G.; Buhrlage, S.J.; Huang, M.; Park, E.; Cuny, G.D.; Galinsky, I.; Stone, R.M.; Gray, N.S.; D’Andrea, A.D.; et al. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol. Cancer Ther. 2013, 12, 2651–2662. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, P.R.; D’Andrea, A.D.; Taniguchi, T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004, 18, 1958–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Target | Molecule | Stage of Testing | Tumor Type | Reference/ ClinicalTrial.gov Identifier |
|---|---|---|---|---|
| CHK1/CHK2 | AZD7762 | Phase 1 Administered as single intravenous unit and in combination with gemcitabine | Solid Tumors | NCT00413686 |
| CHK1/CHK2 | CBP501 | Phase 1 Administered in combination with cisplatin/nivolumab | Advanced Solid Tumors | NCT03113188 |
| CHK1/CHK2 | LY2606368 | Phase 1 Administered in combination with cisplatin/cetuximab/pemetrexed/fluorouracil | Neoplasm Metastasis Colorectal Neoplasm Breast Cancer | NCT02124148 |
| CHK1/CHK2 | LY2606368 | Phase 2 Single agent | Ovarian Cancer Breast Cancer Prostate Cancer | NCT02203513 |
| ATM/ATR | CGK733 | Preclinical testing using Chk1-deficient HBV-positive hepatocellular carcinoma cells | Hepatocellular carcinoma | [115] |
| ATM | CP466722 | Preclinical testing using multiple cell lines in combination with infrared radiation (IR) | Cervical cancer Fibroblasts | [116] |
| ATM/ATR | Caffeine | Preclinical testing as single agent using human cancer cells and non-transformed mouse fibroblast cell lines | Breast Cancer Prostate cancer | [117] |
| ATM | KU59403 | Preclinical testing using p53 functional and dysfunctional models of human cancer in combination with camptothecin, doxorubicin or etoposide | Osteosarcoma | [118] |
| ATM | KU55933 | Preclinical testing using human mesenchymal stem cells in combination with IR | Mesenchymal stem cells | [119] |
| ATM | KU55933 | Preclinical testing using ATM-defective and normal human fibroblast cells in combination with doxorubicin | Fibroblast | [120] |
| ATM | KU-60019 | Preclinical testing using human glioma cells in combination with IR | Glioma | [121] |
| ATM | KU-60019 | Preclinical testing using non-invasive breast cancer cells in combination with doxorubicin | Breast Cancer | [122] |
| CHK2 | PV1019 | Preclinical testing using human tumor cell lines in combination with topotecan, camptothecin or radiation | Ovarian Carcinoma | [123] |
| CHK2 | CCT241533 | Preclinical testing using human tumor cell lines in combination with PARP inhibitors | Colon Cancer Breast Cancer Glioblastoma Osteosarcoma Lung Cancer Cervical Cancer | [124] |
| CHK1/CHK2 | XL-844 | Preclinical testing using HT-29 cell line in combination with IR | Colon Cancer | [125] |
| CHK1/CHK2 | XL-844 | Preclinical testing using multiple cell lines in combination with gemcitabine | Pancreatic Cancer Cervical Cancer Ovarian Cancer | [126] |
| Target | Molecule | Stage of Testing | Tumor Type | Reference/ ClinicalTrial.gov Identifier |
|---|---|---|---|---|
| 26S proteasome | Bortezomib | Phase 3 Administered in combination with Daratumumab/bortezomib/dexamethasone | Relapsed or Refractory Multiple Myeloma | NCT03234972 |
| 26S proteasome | Bortezomib | Phase 3 Administered in combination with daratumumab/cyclophosphamide bortezomib/dexamethasone | Amyloidosis | NCT03201965 |
| 26S proteasome | Bortezomib | Phase 2 As single agent | Multiple Myeloma | NCT00153920 |
| 26S proteasome | Bortezomib | Phase 2 As single agent | Multiple Myeloma Stage I, II, and III | NCT00075881 |
| 26S proteasome | Bortezomib | Phase 2 As single agent | Primary Peritoneal Cavity Cancer and Recurrent Ovarian Epithelial Cancer | NCT00023712 |
| 26S proteasome | Bortezomib | Phase 2 Administered in combination with bortezomib/vorinostat | Acute Lymphoblastic Leukemia | NCT02553460 |
| FANCF | Curcumin | Preclinical testing using cell lines in combination with cisplatin | Ovarian and Breast Cancer | [167] |
| FANCS | Phenylbutyrate | Preclinical testing using cell lines in combination with cisplatin | Head and Neck Cancer | [168] |
| USP1 | GW7647 | Preclinical testing using cell lines in combination with cisplatin | Non-small-cell Lung Cancer | [169] |
| USP1 | Pimozide | Preclinical testing using cell lines in combination with cisplatin | Non-small-cell Lung Cancer | [169] |
| USP1-UAF1 | ML323 | Preclinical testing using cell lines in combination with cisplatin | Non-small-cell Lung Cancer and Osteosarcoma | [170] |
| USP1 | C527 | Preclinical testing using cell lines in combination with MMC and camptothecin | Leukemia | [171] |
| ATR | Wortmannin | Preclinical testing using HeLa cell lines in combination with IR or mitomycin C (MMC) | Cervical Cancer | [172] |
| PKA PKC PKG | H-9 | Preclinical testing using HeLa cell lines in combination with IR | Cervical Cancer | [167] |
| CDK GSK3 | Alsterpaullone | Preclinical testing using HeLa cell lines in combination with IR | Cervical Cancer | [167] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharjee, S.; Nandi, S. Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy. Cancers 2018, 10, 298. https://doi.org/10.3390/cancers10090298
Bhattacharjee S, Nandi S. Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy. Cancers. 2018; 10(9):298. https://doi.org/10.3390/cancers10090298
Chicago/Turabian StyleBhattacharjee, Sonali, and Saikat Nandi. 2018. "Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy" Cancers 10, no. 9: 298. https://doi.org/10.3390/cancers10090298
APA StyleBhattacharjee, S., & Nandi, S. (2018). Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy. Cancers, 10(9), 298. https://doi.org/10.3390/cancers10090298