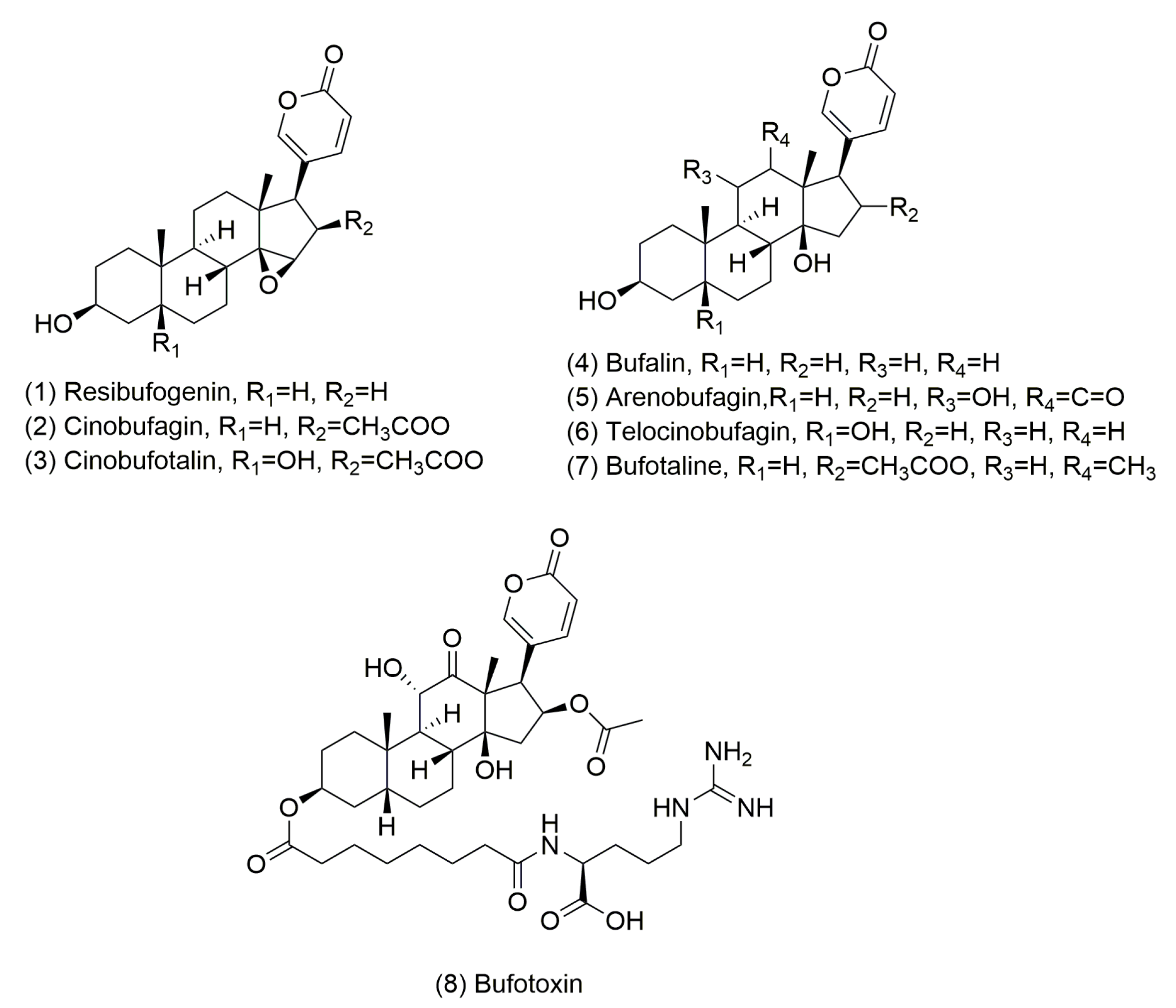
The Potential Mechanism of Bufadienolide-Like Chemicals on Breast Cancer via Bioinformatics Analysis

 ,
,
Abstract
1. Introduction
2. Results
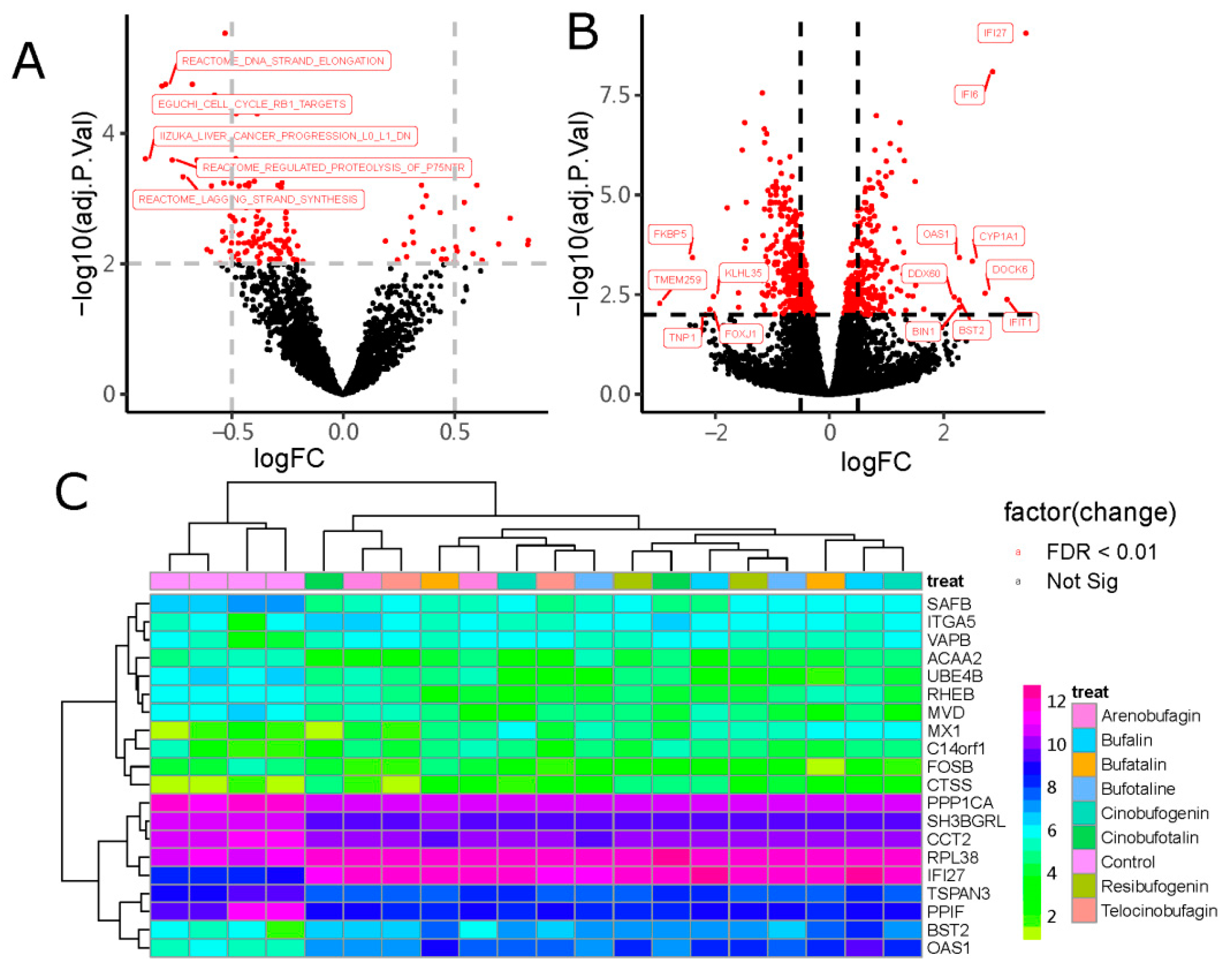
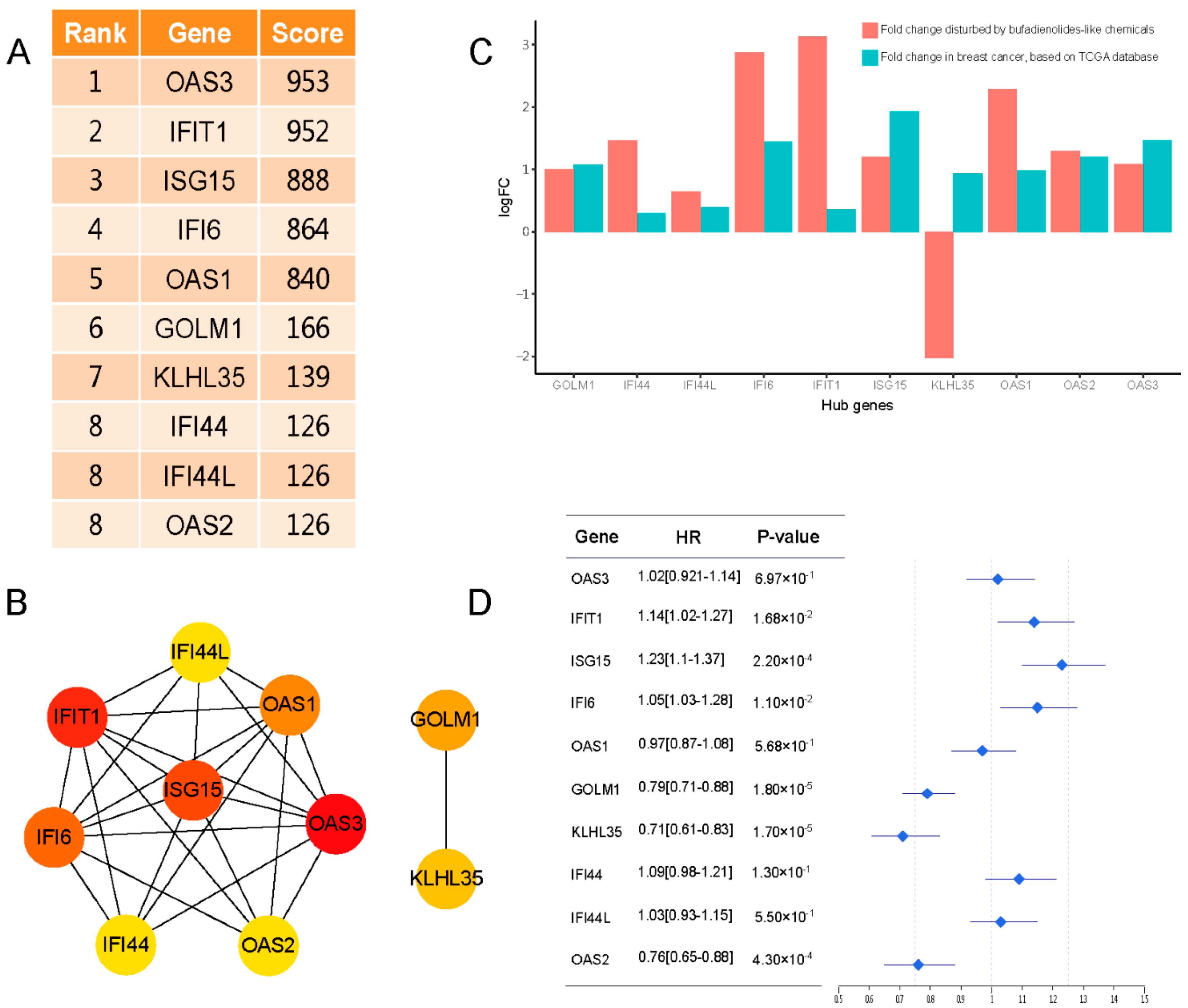
2.1. Identification of DEGs
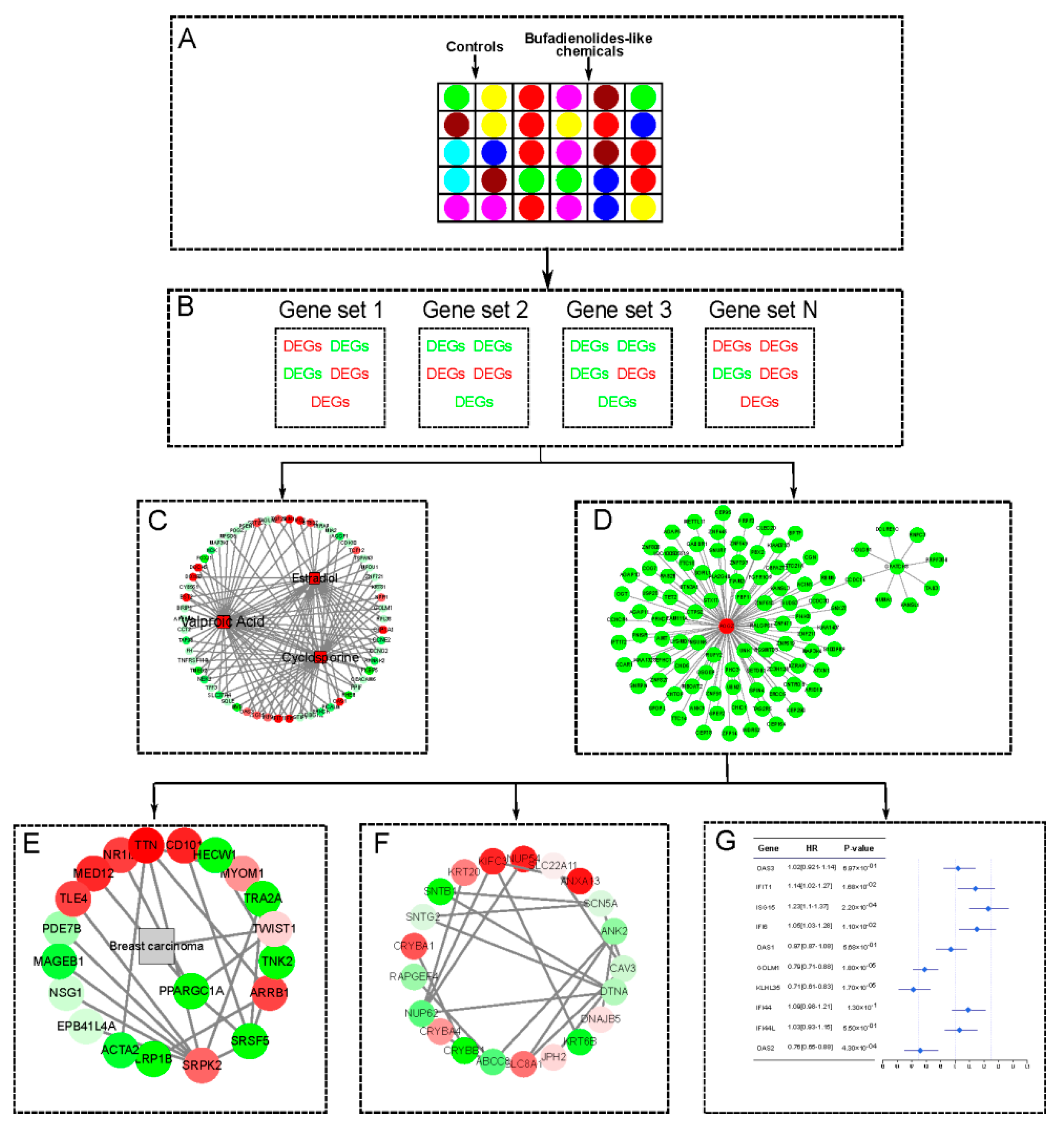
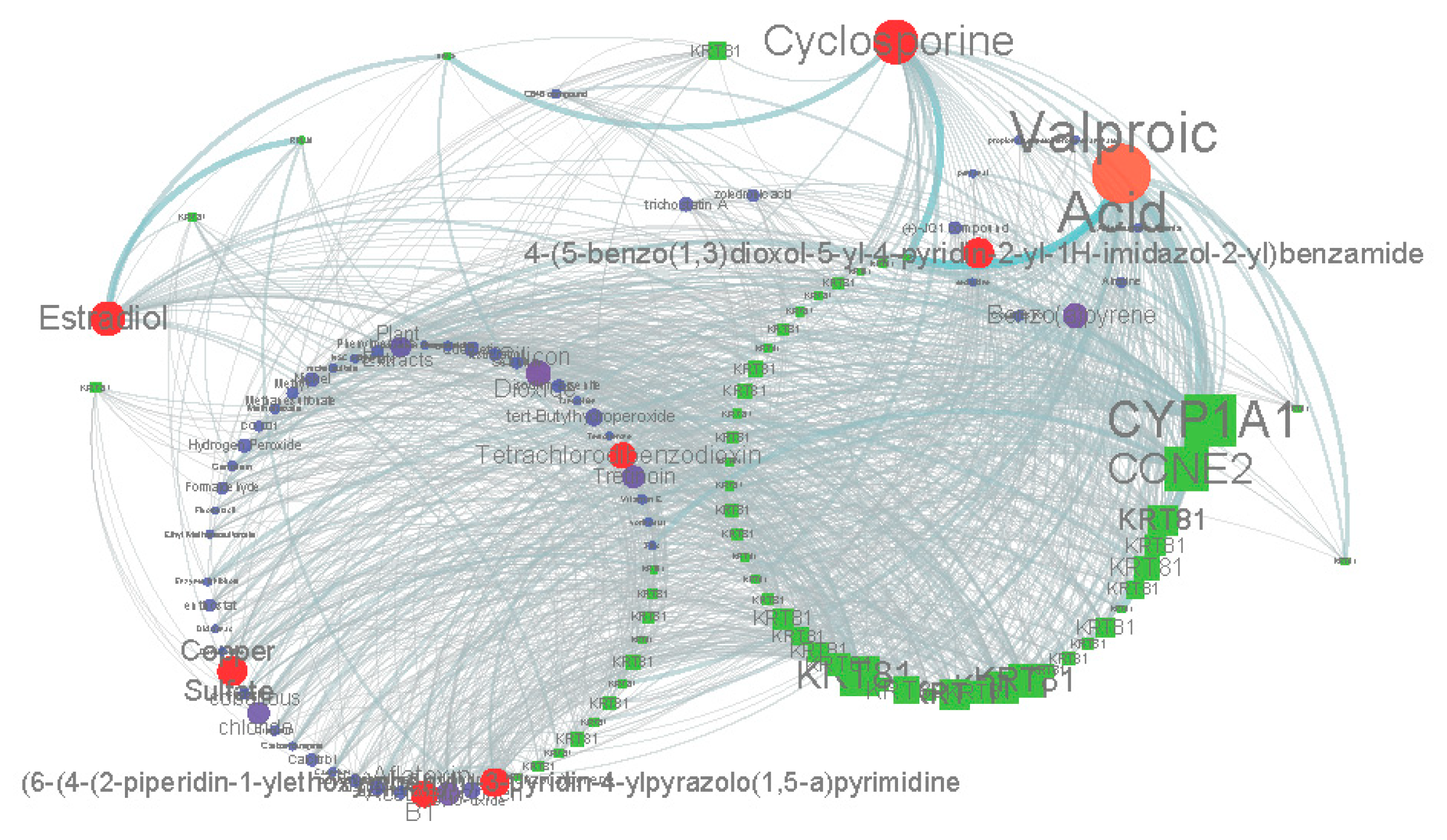
2.2. Similar Small Molecule Detection
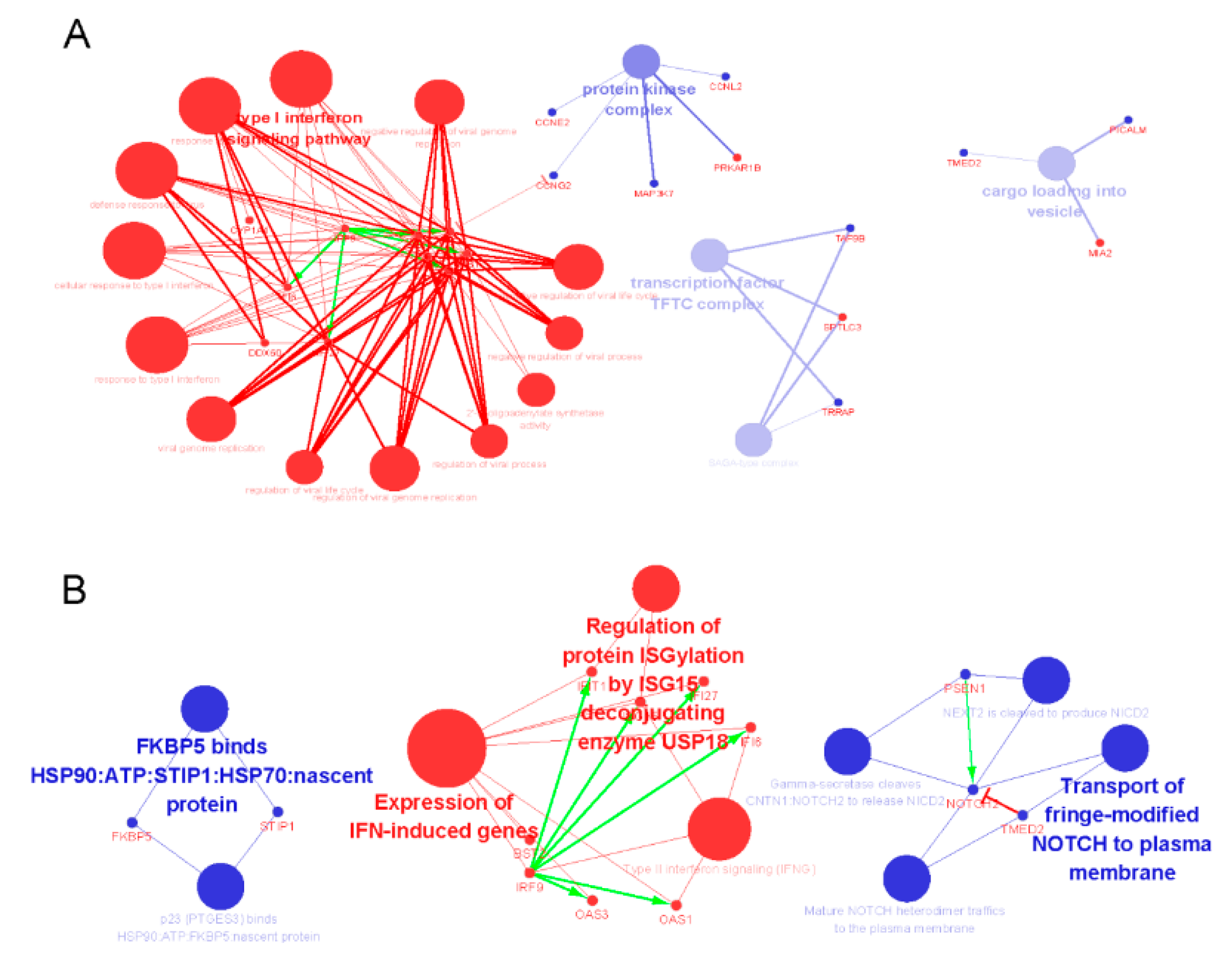
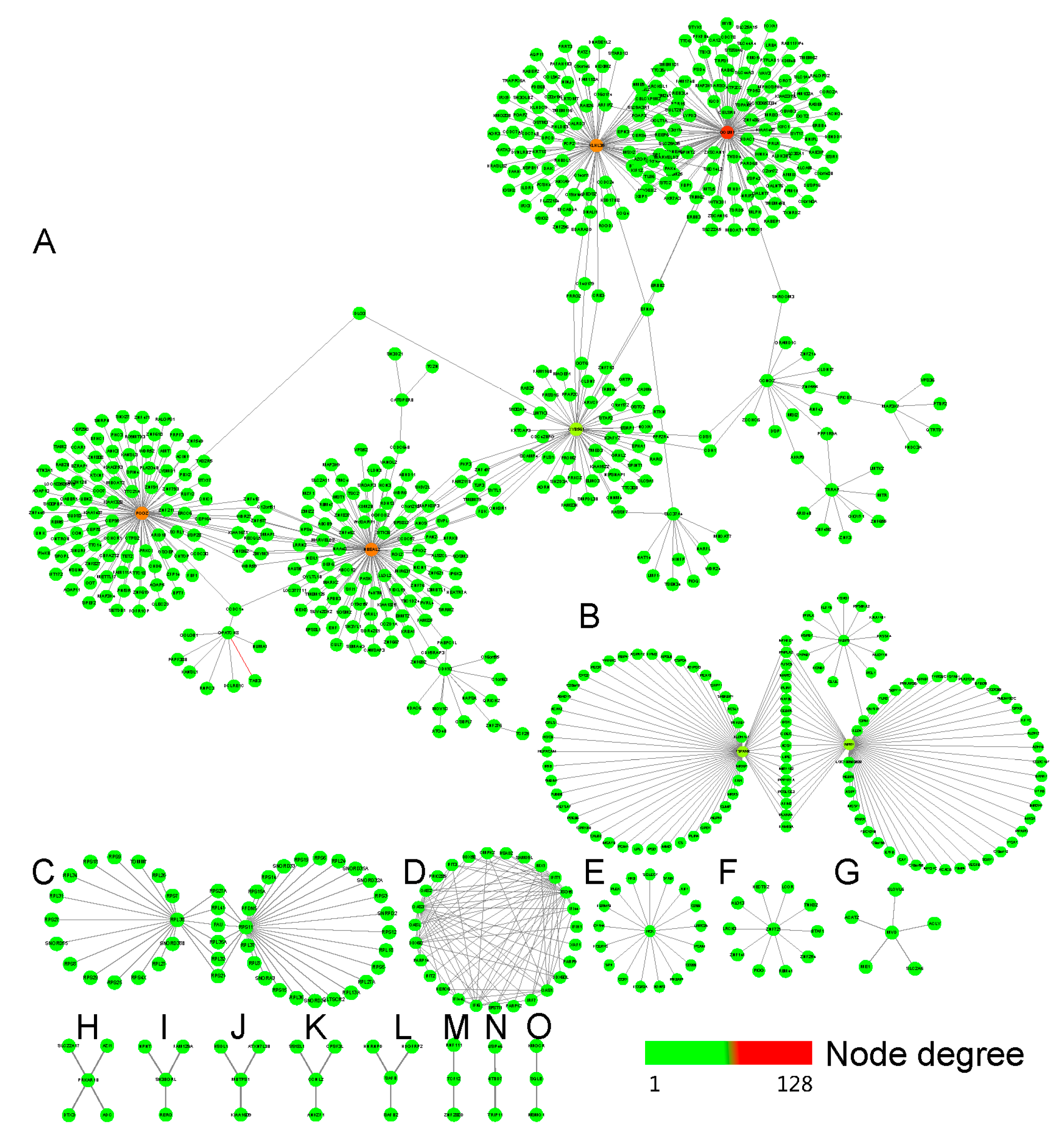
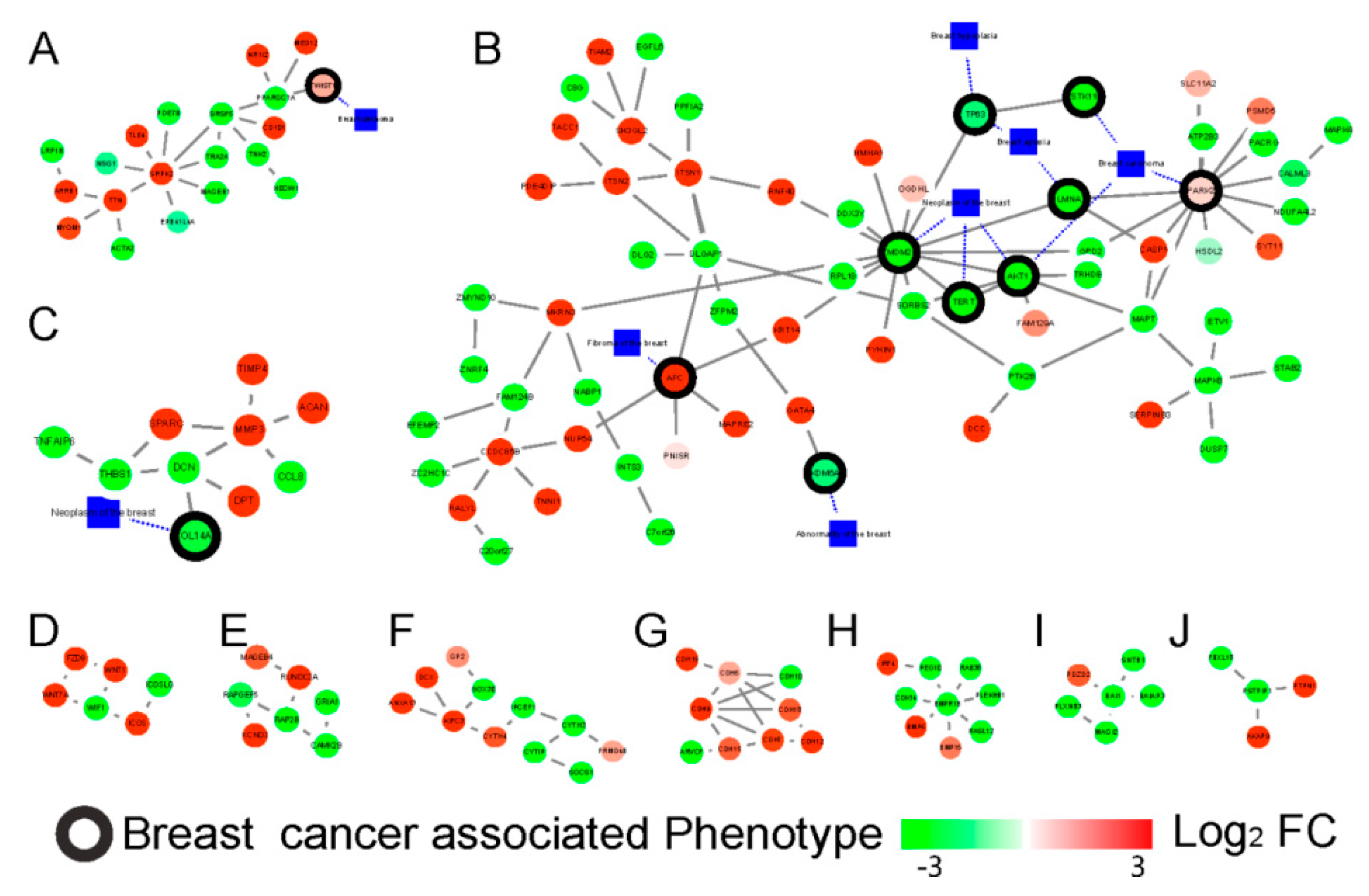
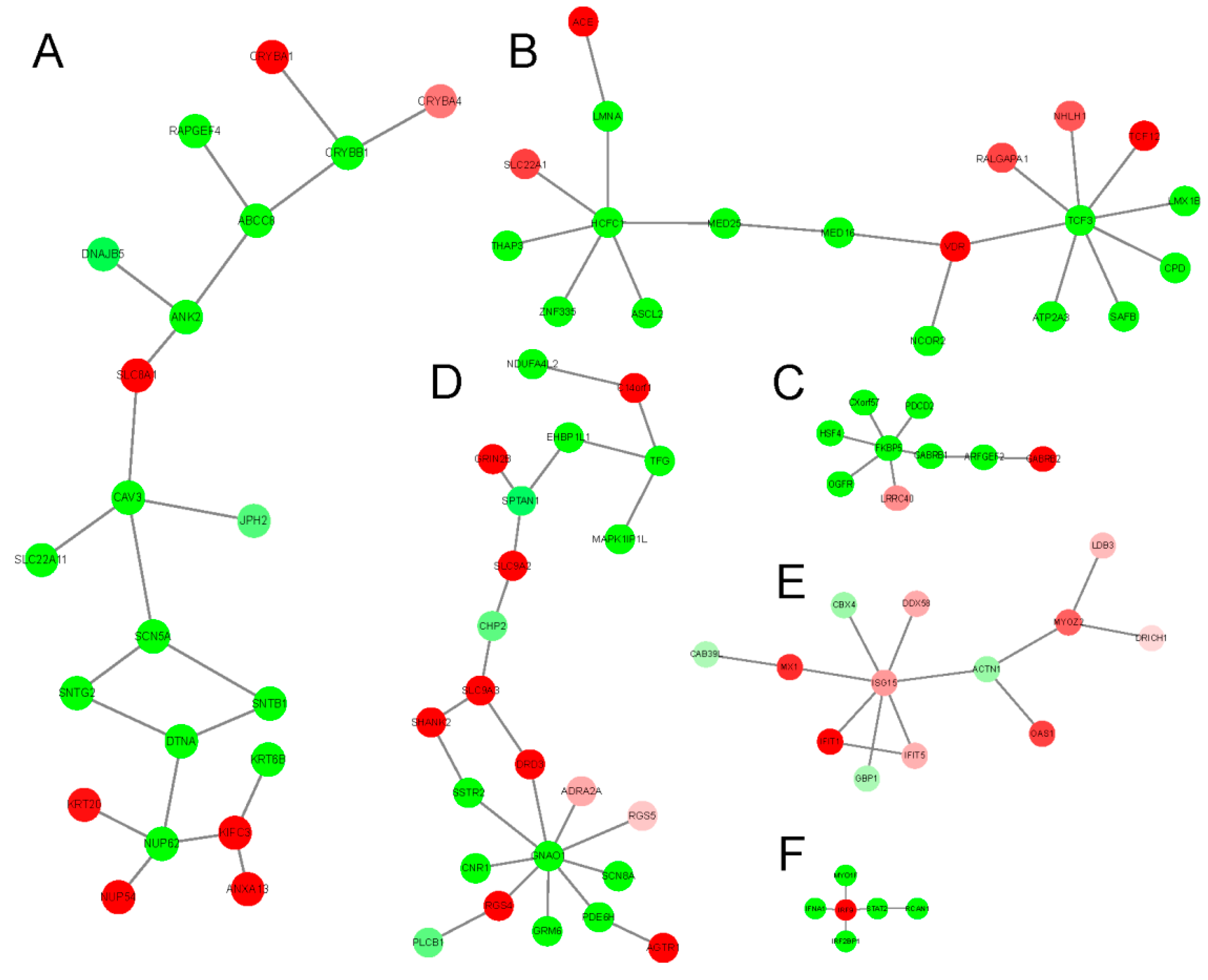
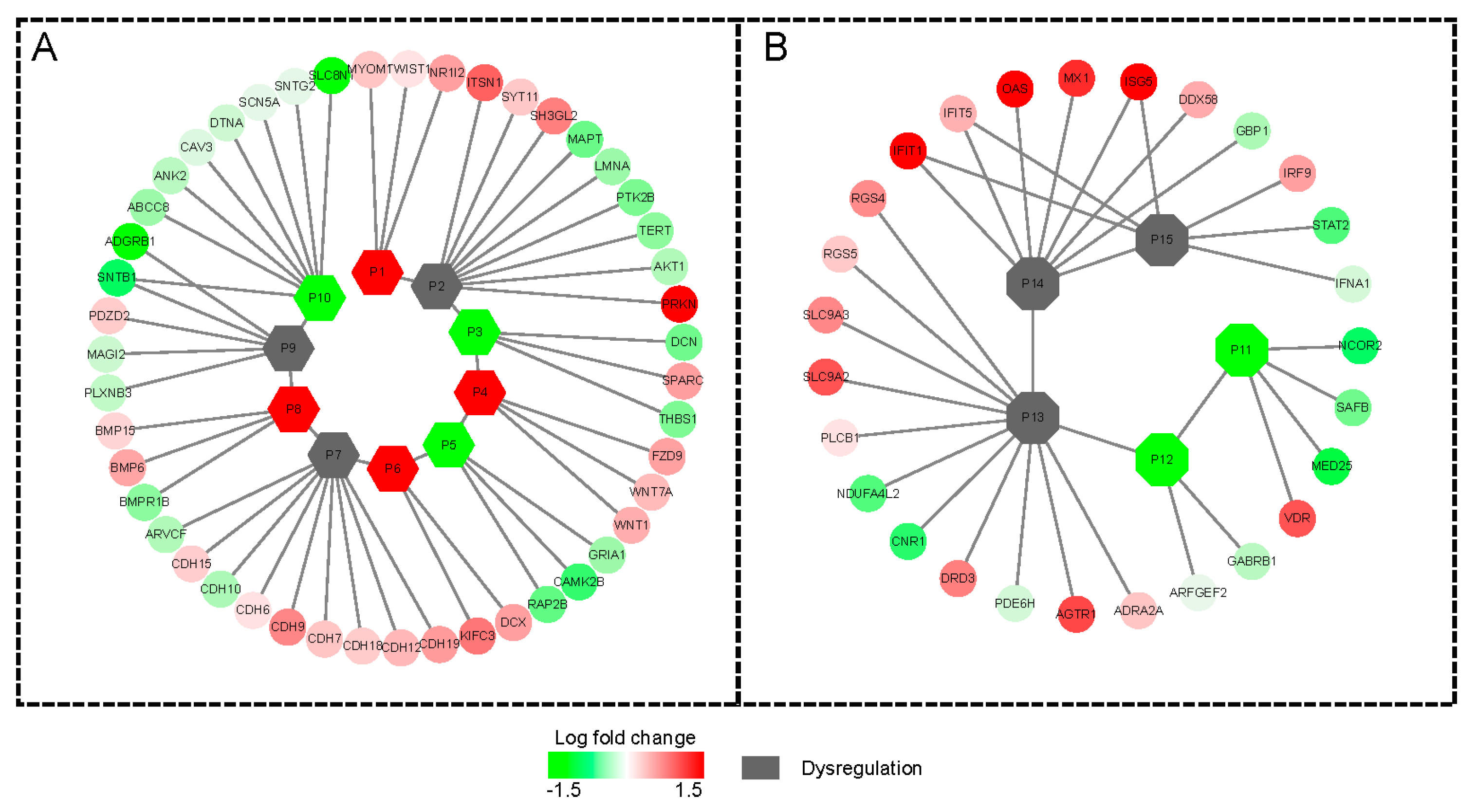
2.3. The Tissue Specific Co-Expression Network and Breast Cancer Associated Subnetwork Regulated by Bufadienolide-Like Chemicals
3. Discussion
4. Materials and Methods
4.1. Microarray Data Information
4.2. Identification of DEGs Associated with Relative Enrichment Pathways
4.3. Gene Enrichment Analysis
4.4. Similar Small Molecule Detection
4.5. Gene Co-Expression Network Analysis and Disease Phenotype Association
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhou, H.; Li, J.; Zhang, Z.; Ye, R.; Shao, N.; Cheang, T.; Wang, S. RING1 and YY1 binding protein suppresses breast cancer growth and metastasis. Int. J. Oncol. 2016, 49, 2442–2452. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wu, K.; Tian, Y.; Liu, Q.; Han, N.; Yuan, X.; Zhang, L.; Wu, G.S.; Wu, K. CD44 correlates with clinicopathological characteristics and is upregulated by EGFR in breast cancer. Int. J. Oncol. 2016, 49, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Jing, W.; He, K.; Zhang, L.; Long, X. SATB1 is Correlated with Progression and Metastasis of Breast Cancers: A Meta-Analysis. Cell. Physiol. Biochem. 2016, 38, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Mai, F.T.; Omar, H.A. Immunotherapy, an evolving approach for the management of triple negative breast cancer: Converting non-responders to responders. Crit. Rev. Oncol. Hematol. 2018, 122, 202–207. [Google Scholar]
- Akiko, E.; Mun-Chual, R.; Kanki, K.; Masahiko, H. Inhibitory effects of bufadienolides on interleukin-6 in MH-60 cells. J. Nat. Prod. 2004, 67, 2070–2072. [Google Scholar]
- Qin, T.-J.; Zhao, X.-H.; Yun, J.; Zhang, L.-X.; Ruan, Z.-P. Efficacy and safety of gemcitabine-oxaliplatin combined with huachansu in patients with advanced gallbladder carcinoma. World J. Gastroenterol. 2008, 14, 5210–5216. [Google Scholar] [CrossRef]
- Wang, J.; Jin, Y.; Xu, Z.; Zheng, Z.; Wan, S. Involvement of caspase-3 activity and survivin downregulation in cinobufocini-induced apoptosis in A 549 cells. Exp. Boil. Med. 2009, 234, 566–572. [Google Scholar] [CrossRef]
- Hong, Z.; Chan, K.; Yeung, H.W. Simultaneous determination of bufadienolides in the traditional Chinese medicine preparation, liu-shen-wan, by liquid chromatography. J. Pharm. Pharmacol. 2011, 44, 1023–1026. [Google Scholar] [CrossRef]
- Chun, L.; Hashimi, S.M.; Siyu, C.; Mellick, A.S.; Wei, D.; David, G.; Wei, M.Q. The mechanisms of chansu in inducing efficient apoptosis in colon cancer cells. Evid. Based Complement. Altern. Med. 2013, 2013, 849054. [Google Scholar] [CrossRef]
- Yeh, J.-Y.; Huang, W.J.; Kan, S.-F.; Wang, P.S. Effects of bufalin and cinobufagin on the proliferation of androgen dependent and independent prostate cancer cells. Prostate 2010, 54, 112–124. [Google Scholar] [CrossRef]
- Yu, C.H.; Kan, S.F.; Pu, H.F.; Chien, E.J.; Wang, P.S. Apoptotic signaling in bufalin- and cinobufagin-treated androgen-dependent and -independent human prostate cancer cells. Cancer Sci. 2010, 99, 2467–2476. [Google Scholar] [CrossRef]
- Dong, Y.Q.; Ma, W.L.; Gu, J.B.; Zheng, W.L. Effect of cinobufagin on nuclear factor-kappa B pathway in HepG2 cells. J. South. Med. Univ. 2010, 30, 137–139. [Google Scholar]
- Wang, L.; Jun, W.U.; Min, L.I.; Yang, X.W. Pilot study on the mechanisms of growth inhibitory effect of cinobufagin on HeLa cells. Chin. J. Oncol. 2005, 27, 717–720. [Google Scholar]
- Ko, W.S.; Park, T.Y.; Park, C.; Kim, Y.H.; Yoon, H.J.; Lee, S.Y.; Hong, S.H.; Choi, B.T.; Lee, Y.T.; Choi, Y.H. Induction of apoptosis by Chansu, a traditional Chinese medicine, in human bladder carcinoma T24 cells. Oncol. Rep. 2005, 14, 475–480. [Google Scholar] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; King, B.L.; McMorran, R.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. The Comparative Toxicogenomics Database: Update 2017. Nucleic Acids Res. 2017, 45, D972–D978. [Google Scholar] [CrossRef] [PubMed]
- Justin, L.; Crawford, E.D.; David, P.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Jim, L.; Jean-Philippe, B.; Aravind, S.; Ross, K.N. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- Lee, S.; Zhang, C.; Arif, M.; Liu, Z.; Benfeitas, R.; Bidkhori, G.; Deshmukh, S.; Al, S.M.; Lovric, A.; Boren, J. TCSBN: A database of tissue and cancer specific biological networks. Nucleic Acids Res. 2018, 46, D595–D600. [Google Scholar] [CrossRef]
- Soul, J.; Dunn, S.L.; Hardingham, T.E.; Boothandford, R.P.; Schwartz, J.M. PhenomeScape: A cytoscape app to identify differentially regulated sub-networks using known disease associations. Bioinformatics 2016, 32, 3847–3849. [Google Scholar] [CrossRef]
- Kosinski, M.; Biecek, P. RTCGA: The Cancer Genome Atlas Data Integration, R package version 1.12.0 [Software]; 2018. Available online: https://rdrr.io/bioc/RTCGA/ (accessed on 28 November 2018).
- Balazs, G.; Andras, L.; Eklund, A.C.; Carsten, D.; Jan, B.; Qiyuan, L.; Zoltan, S. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat 2010, 123, 725–731. [Google Scholar]
- Bindea, G.; Mlecnik, B.H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological network exploration with cytoscape 3. Curr. Protoc. Bioinform. 2014, 47, 8.13.1–8.13.24. [Google Scholar] [CrossRef] [PubMed]
- Minegaki, T.S.A.; Mori, M.; Tsuji, S.; Yamamoto, S.; Watanabe, A.; Tsuzuki, T.; Tsunoda, T.; Yamamoto, A.; Tsujimoto, M.; Nishiguchi, K. Histone deacetylase inhibitors sensitize 5-fluorouracil-resistant MDA-MB-468 breast cancer cells to 5-fluorouracil. Oncol. Lett. 2018, 16, 6202–6208. [Google Scholar] [CrossRef] [PubMed]
- Hankinson, S.E.; Willett, W.C.; Manson, J.E.; Colditz, G.A.; Hunter, D.J.; Spiegelman, D.; Barbieri, R.L.; Speizer, F.E. Plasma sex steroid hormone levels and risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 1998, 91, 1292–1299. [Google Scholar] [CrossRef]
- Barros-Oliveira, M.d.C.; Costa-Silva, D.R.; Andrade, D.B.d.; Borges, U.S.; Tavares, C.B.; Borges, R.S.; Silva, J.d.M.; Silva, B.B.d. Use of anastrozole in the chemoprevention and treatment of breast cancer: A literature review. Rev. Assoc. Medica Bras. 2017, 63. [Google Scholar] [CrossRef] [PubMed]
- Bennink, H.J.T.C.; Verhoeven, C.; Dutman, A.E.; Thijssen, J. The use of high-dose estrogens for the treatment of breast cancer. Maturitas 2017, 95, 11–23. [Google Scholar] [CrossRef]
- Barabási, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef]
- Lea, I.A.; Gong, H.; Paleja, A.; Rashid, A.; Fostel, J. CEBS: A comprehensive annotated database of toxicological data. Nucleic Acids Res. 2017, 45, D964–D971. [Google Scholar] [CrossRef]
- Pecoraro, M.; Sorrentino, R.; Franceschelli, S.; Pizzo, M.D.; Pinto, A.; Popolo, A. Doxorubicin-Mediated Cardiotoxicity: Role of Mitochondrial Connexin 43. Cardiovasc. Toxicol. 2015, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.V.; Peter, F.; Lucas, L.; Pál, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, 1453–1467. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Boil. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef] [PubMed]
- Soul, J.; Hardingham, T.E.; Boot-Handford, R.P.; Schwartz, J.M. PhenomeExpress: A refined network analysis of expression datasets by inclusion of known disease phenotype. Sci. Rep. 2015, 5, 8117. [Google Scholar] [CrossRef] [PubMed]
- Dunn, S.L.; Soul, J.; Anand, S.; Schwartz, J.M.; Boot-Handford, R.P.; Hardingham, T.E. Gene expression changes in damaged osteoarthritic cartilage identify a signature of non-chondrogenic and mechanical responses. Osteoarthr. Cartil. 2016, 24, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Chao, L.; Wu, X.; Xia, W.; Su, J.; Zeng, H.; Jing, Z.; Shan, L.; Liu, R.; Li, H.; Xuan, L. The gene expression profiles in response to 102 traditional Chinese medicine (TCM) components: A general template for research on TCMs. Sci. Rep. 2017, 7, 352. [Google Scholar] [CrossRef]
- Sean, D.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Brazma, A. Minimum Information About a Microarray Experiment (MIAME)—Successes, failures, challenges. Sci. World J. 2009, 9, 420–423. [Google Scholar] [CrossRef]
- Dondrup, M.; Albaum, S.P.; Griebel, T.; Henckel, K.; Jünemann, S.; Kahlke, T.; Kleindt, C.K.; Küster, H.; Linke, B.; Mertens, D. EMMA 2—A MAGE-compliant system for the collaborative analysis and integration of microarray data. BMC Bioinform. 2009, 10, 50. [Google Scholar] [CrossRef]
- Gautier, L.; Cope LBolstad, B.M.; Irizarry, R.A. Affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- Gharaibeh, R.Z.; Fodor, A.A.; Gibas, C.J. Background correction using dinucleotide affinities improves the performance of GCRMA. BMC Bioinform. 2008, 9, 452. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.V.; Huber, W.; Hahne, F. Genefilter: Genefilter: Methods for Filtering Genes from High-throughput Experiments, R package version 1.64.0 [Software]; 2018. Available online: https://rdrr.io/bioc/genefilter/ (accessed on 28 November 2018).
- Morgan, M.; Falcon, S.; Gentleman, R. GSEABase: Gene Set Enrichment Data Structures and Methods, R package version 1.44.0 [Software]; 2018. Available online: https://rdrr.io/bioc/GSEABase/ (accessed on 28 November 2018).
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Arthur, L.; Aravind, S.; Reid, P.; Helga, T.; Pablo, T.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Wolfgang, H.; Carey, V.J.; Robert, G.; Simon, A.; Marc, C.; Carvalho, B.S.; Hector Corrada, B.; Sean, D.; Laurent, G.; Thomas, G. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar]
- Castelo, R. GSVAdata: Data Employed in the Vignette of the GSVA Package, R package version 1.18.0 [Software]; 2018. Available online: https://www.bioconductor.org/packages/release/data/experiment/html/GSVAdata.html (accessed on 28 November 2018).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Kassambara, A. ggpubr: ‘ggplot2’ Based Publication Ready Plots, R package version 0.2 [Software]; 2018. Available online: https://rdrr.io/cran/ggpubr/ (accessed on 28 November 2018).
- Kolde, R. pheatmap: Pretty Heatmaps, R package version 1.0.12 [Software]; 2018. Available online: https://rdrr.io/cran/pheatmap/ (accessed on 28 November 2018).
- Wilke, C.O. cowplot: Streamlined Plot Theme and Plot Annotations for ‘ggplot2’, R package version 0.9.4 [Software]; 2018. Available online: https://rdrr.io/cran/cowplot/ (accessed on 28 November 2018).
- Berkopec, A. HyperQuick algorithm for discrete hypergeometric distribution. J. Discret. Algorithms 2007, 5, 341–347. [Google Scholar] [CrossRef]
- Xia, J.; Gill, E.E.; Hancock, R.E.W. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844. [Google Scholar] [CrossRef]
- Sebastian, K.H.; Schulz, M.H.; Peter, K.; Sebastian, B.; Sandra, D.L.; Ott, C.E.; Christine, M.; Denise, H.; Stefan, M.; Robinson, P.N. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am. J. Hum. Genet. 2009, 85, 457–464. [Google Scholar]
- Yassen, A.; Fidel, R.; Sven-Eric, S.; Thomas, L.; Mario, A. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | ATC Code | Mean Score | Enrichment | p-Value | Specificity |
|---|---|---|---|---|---|
| 1 | V03AF | −0.471 | −0.71 | 4.45 × 10−3 | 3.82 × 10−2 |
| 2 | G03GB | 0.449 | 0.655 | 3.29 × 10−2 | 7.47 × 10−2 |
| 3 | C05AX | 0.41 | 0.689 | 1.95 × 10−2 | 4.76 × 10−2 |
| 4 | C05CX | 0.41 | 0.689 | 1.95 × 10−2 | 4.76 × 10−2 |
| 5 | D07XC | −0.372 | −0.661 | 1.44 × 10−3 | 8.10 × 10−3 |
| 6 | N05BE | −0.359 | −0.719 | 1.26 × 10−2 | 1.22 × 10−2 |
| 7 | C08EA | 0.292 | 0.539 | 1.87 × 10−2 | 1.45 × 10−1 |
| 8 | N05AC | 0.259 | 0.365 | 2.32 × 10−3 | 3.90 × 10−1 |
| 9 | D06BB | −0.252 | −0.405 | 9.39 × 10−3 | 1.44 × 10−1 |
| 10 | D06BX | −0.249 | −0.72 | 3.74 × 10−3 | 1.38 × 10−2 |
| 11 | N02BB | 0.244 | 0.404 | 2.71 × 10−3 | 1.75 × 10−2 |
| 12 | N02CX | 0.189 | 0.481 | 3.16 × 10−2 | 4.43 × 10−2 |
| 13 | A07EA | −0.186 | −0.343 | 6.96 × 10−3 | 2.55 × 10−2 |
| 14 | S02BA | −0.167 | −0.383 | 5.03 × 10−3 | 1.31 × 10−2 |
| 15 | B01AC | 0.152 | 0.243 | 2.71 × 10−2 | 1.19 × 10−1 |
| 16 | S03BA | −0.144 | −0.366 | 2.02 × 10−2 | 4.80 × 10−2 |
| 17 | R03BA | −0.141 | −0.29 | 1.19 × 10−2 | 4.00 × 10−2 |
| 18 | S01CB | −0.136 | −0.326 | 1.21 × 10−2 | 2.61 × 10−2 |
| 19 | R01AD | −0.113 | −0.266 | 4.30 × 10−3 | 4.83 × 10−2 |
| 20 | C07AA | −0.109 | −0.262 | 1.14 × 10−2 | 2.22 × 10−1 |
| Subnetwork Number | Nodes | Edges | Seeds | KEGG Enrichment | GO Enrichment | ||
|---|---|---|---|---|---|---|---|
| KEGG Pathway | p-Value | BP Term | p-Value | ||||
| A | 492 | 558 | 13 | Tight junction | 4.19 × 10−4 | Establishment or maintenance of cell polarity | 2.83 × 10−4 |
| B | 113 | 128 | 3 | PPAR signaling pathway | 7.75 × 10−6 | Triglyceride metabolic process | 1.25 × 10−7 |
| C | 46 | 50 | 2 | mTOR signaling pathway | 9.62 × 10−3 | Protein targeting to membrane | 4.93 × 10−67 |
| D | 27 | 86 | 6 | Influenza A | 3.04 × 10−10 | Defense response to virus | 1.24 × 10−22 |
| E | 18 | 17 | 1 | Tuberculosis | 2.01 × 10−4 | Tuberculosis | 2.01 × 10−4 |
| F | 11 | 10 | 1 | N-Glycan biosynthesis | 9.19 × 10−3 | Post-translational protein modification | 6.33 × 10−3 |
| G | 6 | 5 | 1 | Terpenoid backbone biosynthesis | 1.72 × 10−4 | Coenzyme biosynthetic process | 1.55 × 10−5 |
| H | 5 | 4 | 1 | Notch signaling pathway | 2.98 × 10−2 | Gamete generation | 1.34 × 10−2 |
| I | 4 | 3 | 1 | Null | Null | Transcription, DNA-dependent | 1.31 × 10−2 |
| J | 4 | 3 | 1 | Null | Null | Positive regulation of translation | 1.17 × 10−2 |
| K | 4 | 3 | 1 | Null | Null | Endoplasmic reticulum unfolded protein response | 6.51 × 10−3 |
| L | 4 | 3 | 1 | Regulation of cyclin-dependent protein kinase activity | 1.24 × 10−2 | Regulation of cyclin-dependent protein kinase activity | 1.24 × 10−2 |
| M | 3 | 2 | 1 | Steroid biosynthesis | 7.68 × 10−3 | Steroid biosynthetic process | 2.07 × 10−6 |
| N | 3 | 2 | 1 | Null | Null | Regulation of transcription, DNA-dependent | 1.84 × 10−2 |
| O | 3 | 2 | 1 | Null | Null | Intra-Golgi vesicle-mediated transport | 4.47 × 10−3 |
| Subnetwork Number | No. of Nodes | GO-BP | Empirical p-Value |
|---|---|---|---|
| A | 21 | RNA splicing | 2.00 × 10−3 |
| B | 73 | apoptotic process | 2.00 × 10−3 |
| C | 11 | extracellular matrix organization | 1.00 × 10−3 |
| D | 6 | canonical Wnt signaling pathway | 2.20 × 10−2 |
| E | 7 | synaptic transmission | 1.40 × 10−2 |
| F | 11 | negative regulation of JAK-STAT cascade | 4.20 × 10−2 |
| G | 9 | adherens junction organization | 3.80 × 10−2 |
| H | 9 | BMP signaling pathway | 4.10 × 10−2 |
| I | 6 | negative regulation of cell migration | 1.30 × 10−2 |
| J | 4 | activation of signaling protein activity involved in unfolded protein response | 1.90 × 10−2 |
| K | 12 | drug metabolic process | 1.20 × 10−2 |
| L | 6 | negative regulation of lipid storage | 4.50 × 10−2 |
| M | 6 | xenobiotic metabolic process | 1.70 × 10−2 |
| N | 8 | relaxation of cardiac muscle | 4.80 × 10−2 |
| O | 5 | very long-chain fatty acid metabolic process | 1.70 × 10−2 |
| P | 4 | oligosaccharide metabolic process | 3.10 × 10−2 |
| Q | 4 | collagen catabolic process | 2.50 × 10−2 |
| R | 4 | response to cocaine | 2.70 × 10−2 |
| S | 4 | behavioral response to nicotine | 4.20 × 10−2 |
| Subnetwork Number | No. of Nodes | GO-BP | Empirical p-Value |
|---|---|---|---|
| A | 21 | Membrane depolarization during action potential | 3.70 × 10−2 |
| B | 19 | Retinoic acid receptor binding | 2.00 × 10−3 |
| C | 9 | GABA receptor binding | 3.00 × 10−3 |
| D | 23 | Positive regulation of nuclear division | 5.00 × 10−3 |
| E | 13 | Negative regulation of viral genome replication | 3.00 × 10−3 |
| F | 6 | Negative regulation of viral life cycle | 1.00 × 10−3 |
| Phenotype ID | Phenotype Description |
|---|---|
| HP:0100783 | Breast aplasia |
| HP:0100013 | Neoplasm of the breast |
| HP:0003002 | Breast carcionma |
| HP:0003187 | Breast hypoplasia |
| HP:0000769 | Abnormality of the breast |
| HP:0010619 | Fibroma of the breast |
| Phenotype ID | Phenotype Description |
|---|---|
| HP:0011675 | Arrhythmia |
| HP:0005110 | Atrial fibrillation |
| HP:0004749 | atrial flutter |
| HP:0011215 | Hemihypsarrhythmia |
| HP:0002521 | Hypsarrhythmia |
| HP:0040182 | Inappropriate sinus tachycardia |
| HP:0001962 | Palpitations |
| HP:0005115 | Supraventricular arrhythmia |
| HP:0004755 | Surpraventricular tachycardia |
| HP:0004308 | Ventricular arrhythmia |
| HP:0011841 | Ventricular flutter |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Tang, X.; Pang, Y.; Huang, L.; Wang, D.; Yuan, C.; Hu, X.; Qu, L. The Potential Mechanism of Bufadienolide-Like Chemicals on Breast Cancer via Bioinformatics Analysis. Cancers 2019, 11, 91. https://doi.org/10.3390/cancers11010091
Zhang Y, Tang X, Pang Y, Huang L, Wang D, Yuan C, Hu X, Qu L. The Potential Mechanism of Bufadienolide-Like Chemicals on Breast Cancer via Bioinformatics Analysis. Cancers. 2019; 11(1):91. https://doi.org/10.3390/cancers11010091
Chicago/Turabian StyleZhang, Yingbo, Xiaomin Tang, Yuxin Pang, Luqi Huang, Dan Wang, Chao Yuan, Xuan Hu, and Liping Qu. 2019. "The Potential Mechanism of Bufadienolide-Like Chemicals on Breast Cancer via Bioinformatics Analysis" Cancers 11, no. 1: 91. https://doi.org/10.3390/cancers11010091
APA StyleZhang, Y., Tang, X., Pang, Y., Huang, L., Wang, D., Yuan, C., Hu, X., & Qu, L. (2019). The Potential Mechanism of Bufadienolide-Like Chemicals on Breast Cancer via Bioinformatics Analysis. Cancers, 11(1), 91. https://doi.org/10.3390/cancers11010091