Analysis of the Whole-Exome Sequencing of Tumor and Circulating Tumor DNA in Metastatic Melanoma

 , , ,
, , ,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Patient Cohort
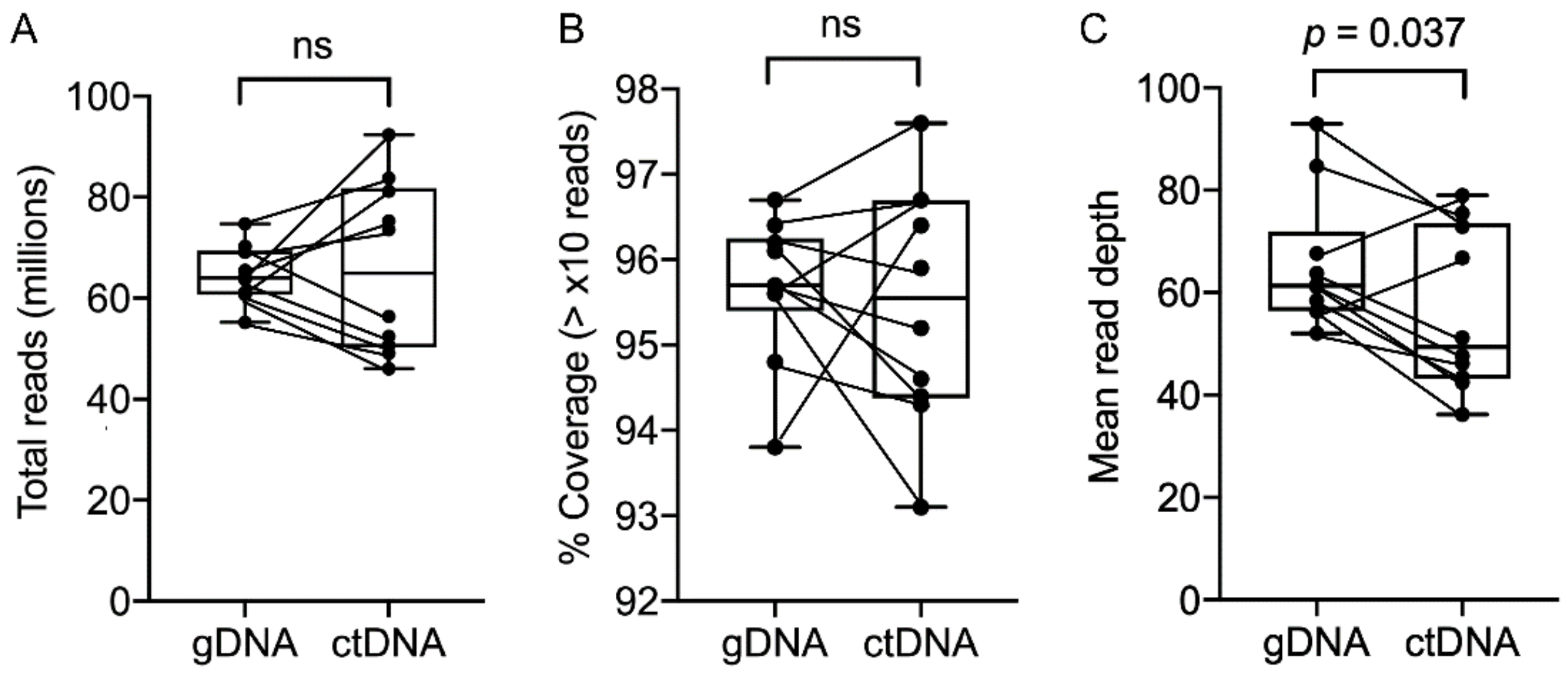
2.2. WES and Droplet Digital PCR
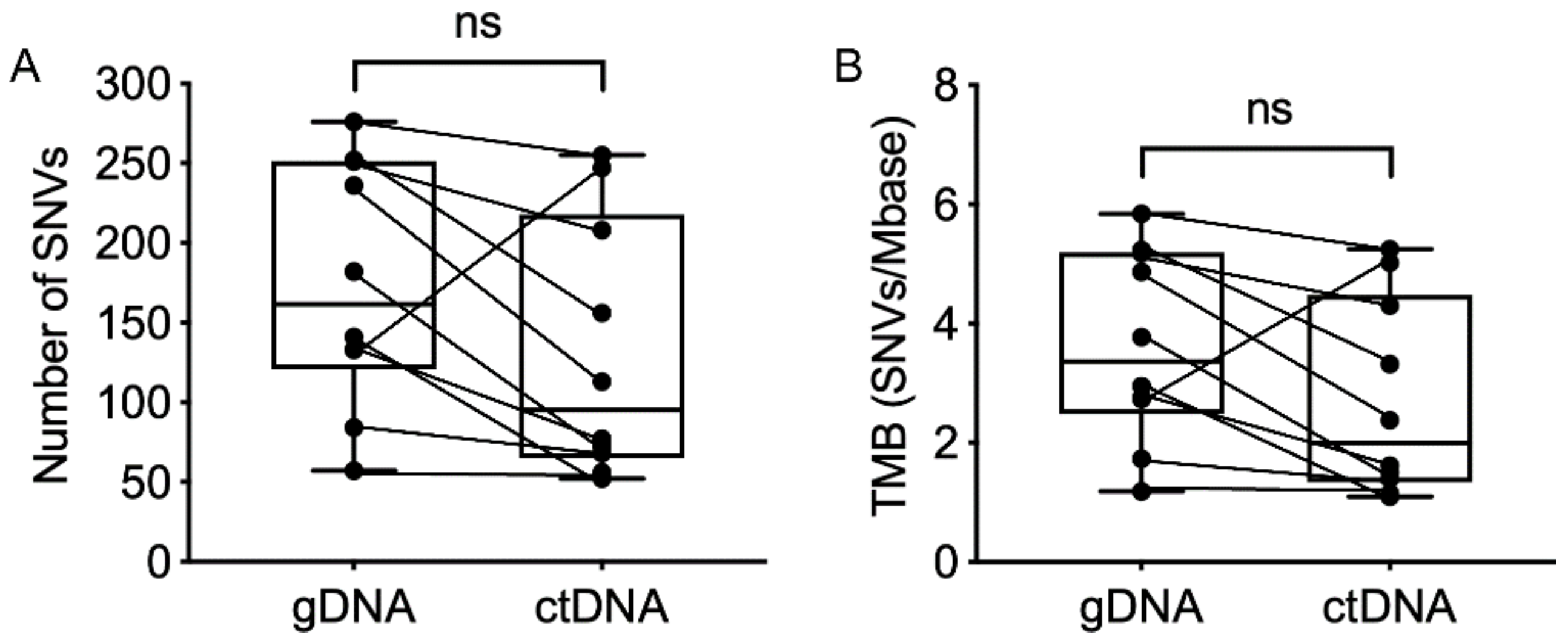
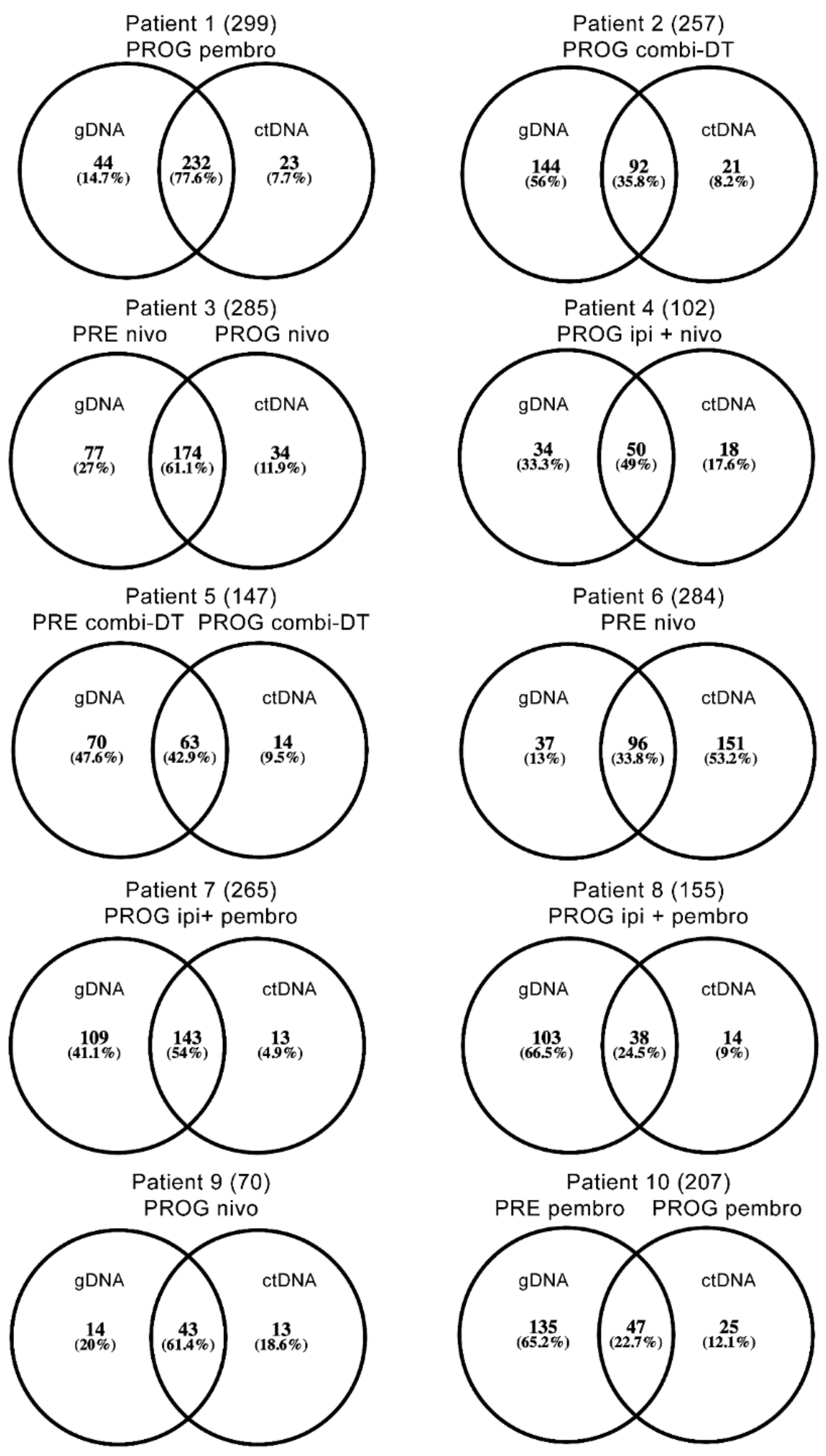
2.3. Single Nucleotide Variant Analysis
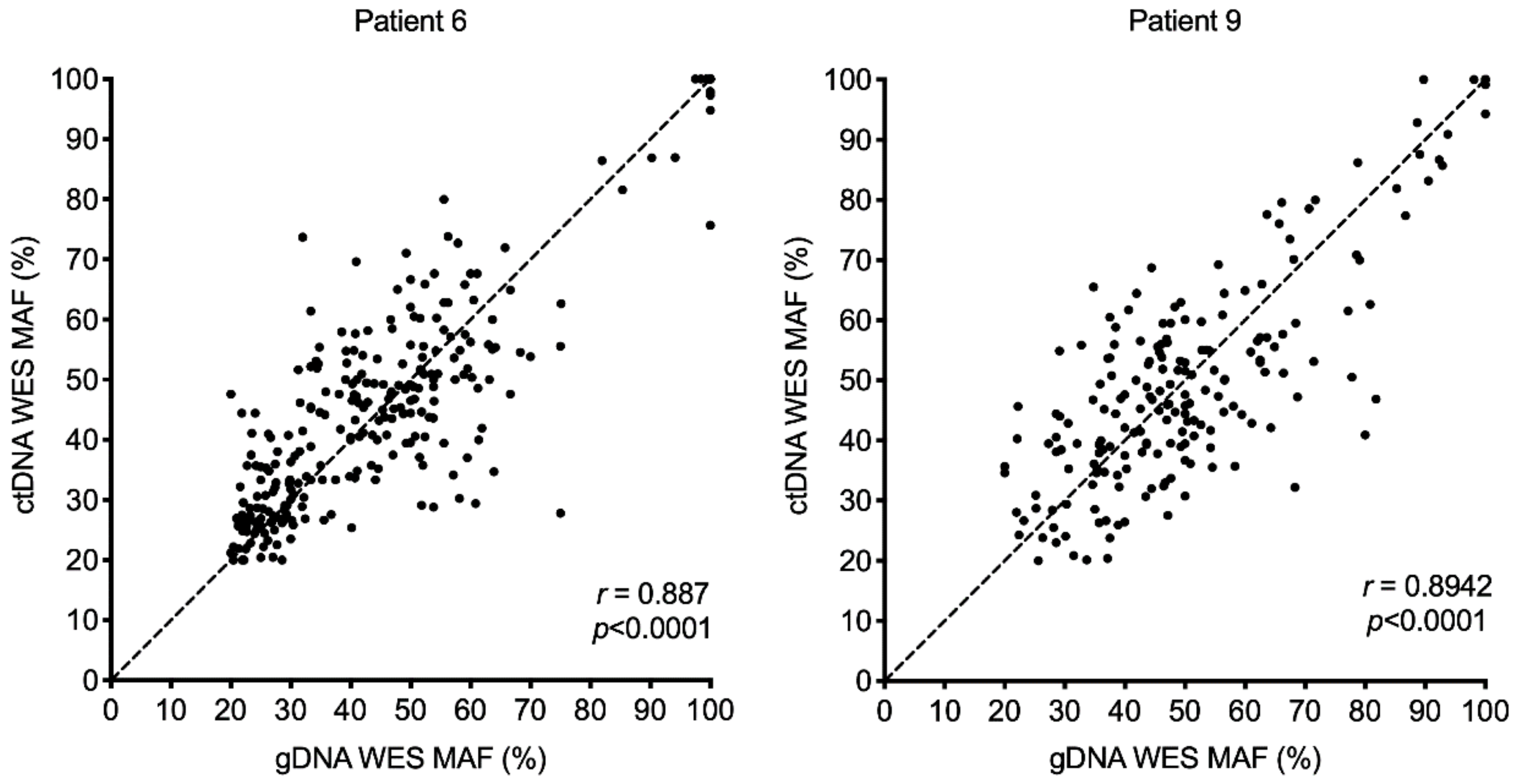
2.4. Mutant Allele Frequency (MAF) of Single Nucleotide Variants
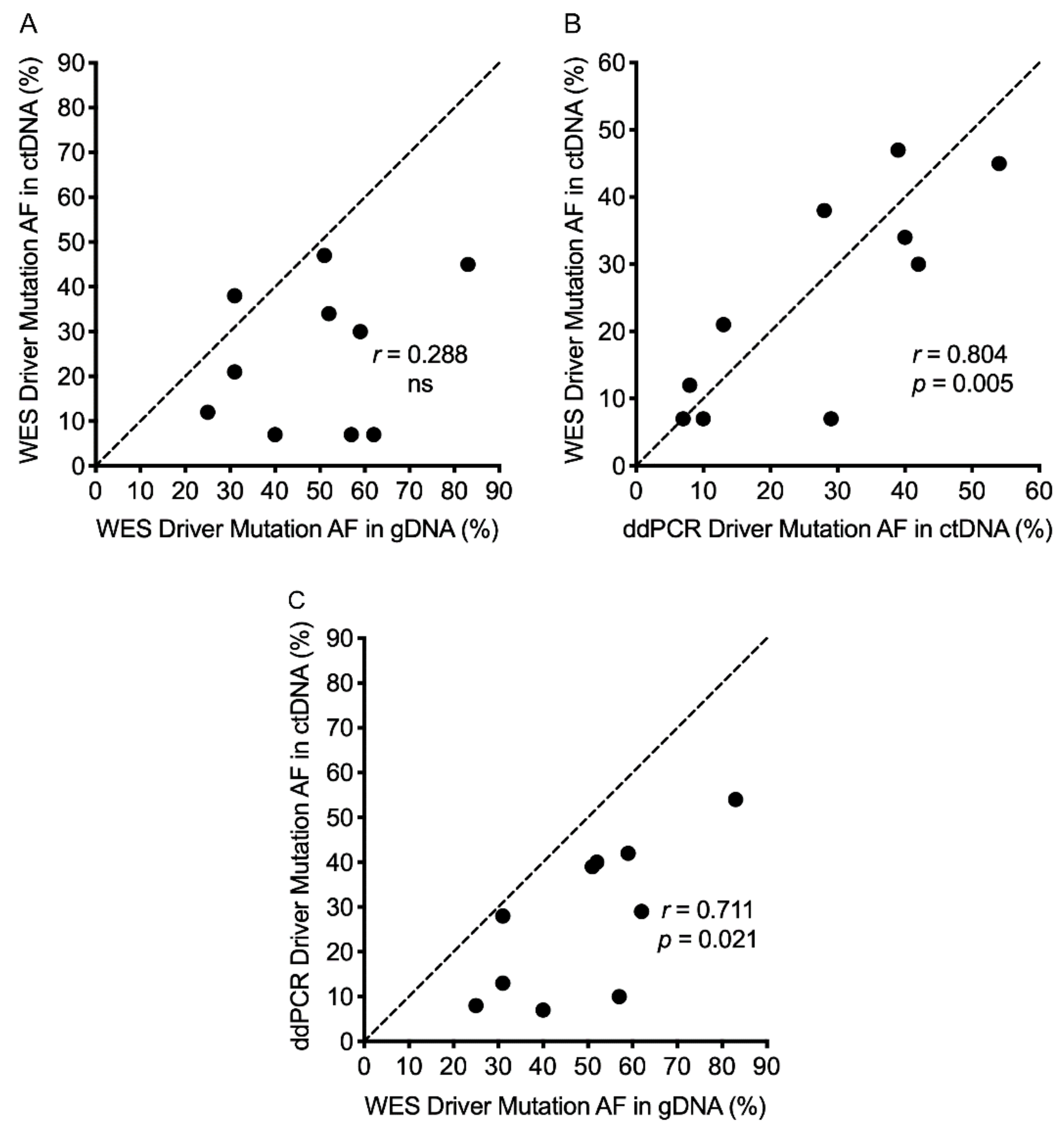
2.5. Melanoma Driver Mutations
2.6. Other Highlighted Mutations
3. Discussion
4. Materials and Methods
4.1. Human Melanoma Samples
4.2. DNA Extractions
4.3. Analysis of Purified ctDNA from Plasma
4.4. Whole Exome Sequencing
4.5. Filtered Single Nucleotide Variants
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yi, X.; Ma, J.; Guan, Y.; Chen, R.; Yang, L.; Xia, X. The feasibility of using mutation detection in ctDNA to assess tumor dynamics. Int. J. Cancer 2017, 140, 2642–2647. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- De Rubis, G.; Krishnan, S.R.; Bebawy, M. Circulating tumor DNA—Current state of play and future perspectives. Pharmacol. Res. 2018, 136, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, J.; Park, B.H. Circulating tumor DNA: Measurement and clinical utility. Annu. Rev. Med. 2018, 69, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Klevebring, D.; Neiman, M.; Sundling, S.; Eriksson, L.; Darai Ramqvist, E.; Celebioglu, F.; Czene, K.; Hall, P.; Egevad, L.; Gronberg, H.; et al. Evaluation of exome sequencing to estimate tumor burden in plasma. PLoS ONE 2014, 9, e104417. [Google Scholar] [CrossRef]
- Ahlborn, L.B.; Rohrberg, K.S.; Gabrielaite, M.; Tuxen, I.V.; Yde, C.W.; Spanggaard, I.; Santoni-Rugiu, E.; Nielsen, F.C.; Lassen, U.; Mau-Sorensen, M.; et al. Application of cell-free DNA for genomic tumor profiling: A feasibility study. Oncotarget 2019, 10, 1388–1398. [Google Scholar] [CrossRef]
- Butler, T.M.; Johnson-Camacho, K.; Peto, M.; Wang, N.J.; Macey, T.A.; Korkola, J.E.; Koppie, T.M.; Corless, C.L.; Gray, J.W.; Spellman, P.T. Exome sequencing of cell-free DNA from metastatic cancer patients identifies clinically actionable mutations distinct from primary disease. PLoS ONE 2015, 10, e0136407. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Chicard, M.; Colmet-Daage, L.; Clement, N.; Danzon, A.; Bohec, M.; Bernard, V.; Baulande, S.; Bellini, A.; Deveau, P.; Pierron, G.; et al. Whole-exome sequencing of cell-free DNA reveals temporo-spatial heterogeneity and identifies treatment-resistant clones in neuroblastoma. Clin. Cancer Res. 2018, 24, 939–949. [Google Scholar] [CrossRef]
- Dietz, S.; Schirmer, U.; Merce, C.; von Bubnoff, N.; Dahl, E.; Meister, M.; Muley, T.; Thomas, M.; Sultmann, H. Low input whole-exome sequencing to determine the representation of the tumor exome in circulating DNA of non-small cell lung cancer patients. PLoS ONE 2016, 11, e0161012. [Google Scholar] [CrossRef]
- Jimenez, I.; Chicard, M.; Colmet-Daage, L.; Clement, N.; Danzon, A.; Lapouble, E.; Pierron, G.; Bohec, M.; Baulande, S.; Berrebi, D.; et al. Circulating tumor DNA analysis enables molecular characterization of pediatric renal tumors at diagnosis. Int. J. Cancer 2018, 144, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Imperial, R.; Nazer, M.; Ahmed, Z.; Kam, A.E.; Pluard, T.J.; Bahaj, W.; Levy, M.; Kuzel, T.M.; Hayden, D.M.; Pappas, S.G.; et al. Matched whole-genome sequencing (WGS) and whole-exome sequencing (WES) of tumor tissue with circulating tumor DNA (ctDNA) analysis: Complementary modalities in clinical practice. Cancers 2019, 11, 1399. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Walker, A.; Gagnon, R.; Mazumdar, J.; Casey, M.; Long, G.V.; Schadendorf, D.; Flaherty, K.; Kefford, R.; Hauschild, A.; Hwu, P.; et al. Correlation of BRAF mutation status in circulating-free DNA and tumor and association with clinical outcome across four BRAFi and MEKi clinical trials. Clin. Cancer Res. 2016, 22, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.; Scolyer, R.A.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef]
- Lee, J.H.; Saw, R.P.; Thompson, J.F.; Lo, S.; Spillane, A.J.; Shannon, K.F.; Stretch, J.R.; Howle, J.; Menzies, A.M.; Carlino, M.S.; et al. Pre-operative ctDNA predicts survival in high-risk stage III cutaneous melanoma patients. Ann. Oncol. 2019, 30, 815–822. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Menzies, A.M.; Lo, S.; Guminski, A.; Whitbourne, K.; Peranec, M.; Scolyer, R.; Kefford, R.F.; Rizos, H.; et al. Association between circulating tumor DNA and pseudoprogression in patients with metastatic melanoma treated with anti-programmed cell death 1 antibodies. JAMA Oncol. 2018, 4, 717–721. [Google Scholar] [CrossRef]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Thompson, J.F.; Long, G.V.; Ross, M.I. Melanoma of the skin. In AJCC Cancer Staging Manual; Amin, M.B., Edge, S., Greene, F., Byrd, D.R., Brookland, R.K., Washington, M.K., Gershenwald, J.E., Compton, C.C., Hess, K.R., Sullivan, D.C., et al., Eds.; Springer: New York, NY, USA, 2017; pp. 563–585. [Google Scholar]
- Pires da Silva, I.; Lo, S.; Quek, C.; Gonzalez, M.; Carlino, M.S.; Long, G.V.; Menzies, A.M. Site-specific response patterns, pseudoprogression, and acquired resistance in patients with melanoma treated with ipilimumab combined with anti-PD-1 therapy. Cancer 2019. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Arafeh, R.; Qutob, N.; Emmanuel, R.; Keren-Paz, A.; Madore, J.; Elkahloun, A.; Wilmott, J.S.; Gartner, J.J.; Di Pizio, A.; Winograd-Katz, S.; et al. Recurrent inactivating RASA2 mutations in melanoma. Nat. Genet. 2015, 47, 1408–1410. [Google Scholar] [CrossRef] [PubMed]
- Newell, F.; Kong, Y.; Wilmott, J.S.; Johansson, P.A.; Ferguson, P.M.; Cui, C.; Li, Z.; Kazakoff, S.H.; Burke, H.; Dodds, T.J.; et al. Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat. Commun. 2019, 10, 3163. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Puntervoll, H.E.; Yang, X.R.; Vetti, H.H.; Bachmann, I.M.; Avril, M.F.; Benfodda, M.; Catricala, C.; Dalle, S.; Duval-Modeste, A.B.; Ghiorzo, P.; et al. Melanoma prone families with CDK4 germline mutation: Phenotypic profile and associations with MC1R variants. J. Med. Genet. 2013, 50, 264–270. [Google Scholar] [CrossRef]
- Loree, J.M.; Miron, B.; Holla, V.; Overman, M.J.; Pereira, A.A.L.; Lam, M.; Morris, V.K.; Raghav, K.P.S.; Routbort, M.; Shaw, K.R.; et al. Not all RAS mutations created equal: Functional and clinical characterization of 80 different KRAS and NRAS mutations. J. Clin. Oncol. 2017, 35, 3589. [Google Scholar] [CrossRef]
- Prickett, T.D.; Zerlanko, B.; Gartner, J.J.; Parker, S.C.J.; Dutton-Regester, K.; Lin, J.C.; Teer, J.K.; Wei, X.; Jiang, J.; Nisc Comparative Sequencing, P.; et al. Somatic mutations in MAP3K5 attenuate its proapoptotic function in melanoma through increased binding to thioredoxin. J. Invest. Dermatol. 2014, 134, 452–460. [Google Scholar] [CrossRef]
- Koeppel, F.; Blanchard, S.; Jovelet, C.; Genin, B.; Marcaillou, C.; Martin, E.; Rouleau, E.; Solary, E.; Soria, J.C.; Andre, F.; et al. Whole exome sequencing for determination of tumor mutation load in liquid biopsy from advanced cancer patients. PLoS ONE 2017, 12, e0188174. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martinez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.M.; Jalali, A.; Kircher, D.A.; Lee, W.C.; McQuade, J.L.; Haydu, L.E.; Joon, A.Y.; Reuben, A.; de Macedo, M.P.; Carapeto, F.C.L.; et al. Molecular profiling reveals unique immune and metabolic features of melanoma brain metastases. Cancer Discov. 2019, 9, 628–645. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Atkinson, V.; Lo, S.; Sandhu, S.; Guminski, A.D.; Brown, M.P.; Wilmott, J.S.; Edwards, J.; Gonzalez, M.; Scolyer, R.A.; et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: A multicentre randomised phase 2 study. Lancet Oncol. 2018, 19, 672–681. [Google Scholar] [CrossRef]
- Davies, M.A.; Saiag, P.; Robert, C.; Grob, J.J.; Flaherty, K.T.; Arance, A.; Chiarion-Sileni, V.; Thomas, L.; Lesimple, T.; Mortier, L.; et al. Dabrafenib plus trametinib in patients with BRAF(V600)-mutant melanoma brain metastases (COMBI-MB): A multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 863–873. [Google Scholar] [CrossRef]
- Long, G.V.; Trefzer, U.; Davies, M.A.; Kefford, R.F.; Ascierto, P.A.; Chapman, P.B.; Puzanov, I.; Hauschild, A.; Robert, C.; Algazi, A.; et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2012, 13, 1087–1095. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Forsyth, P.A.; Algazi, A.; Hamid, O.; Hodi, F.S.; Moschos, S.J.; Khushalani, N.I.; Lewis, K.; Lao, C.D.; Postow, M.A.; et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N. Engl. J. Med. 2018, 379, 722–730. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Calapre, L.; Warburton, L.; Millward, M.; Ziman, M.; Gray, E.S. Circulating tumour DNA (ctDNA) as a liquid biopsy for melanoma. Cancer Lett. 2017, 404, 62–69. [Google Scholar] [CrossRef]
- Johnson, D.B.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 2015, 51, 2792–2799. [Google Scholar] [CrossRef]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Lee, J.H.; Rizos, H. Monitoring melanoma using circulating free DNA. Am. J. Clin. Dermatol. 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Calapre, L.; Giardina, T.; Robinson, C.; Reid, A.L.; Al-Ogaili, Z.; Pereira, M.R.; McEvoy, A.C.; Warburton, L.; Hayward, N.K.; Khattak, M.A.; et al. Locus-specific concordance of genomic alterations between tissue and plasma circulating tumor DNA in metastatic melanoma. Mol. Oncol. 2019, 13, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Sukhai, M.A.; Misyura, M.; Thomas, M.; Garg, S.; Zhang, T.; Stickle, N.; Virtanen, C.; Bedard, P.L.; Siu, L.L.; Smets, T.; et al. Somatic tumor variant filtration strategies to optimize tumor-only molecular profiling using targeted next-generation sequencing panels. J. Mol. Diagn. 2019, 21, 261–273. [Google Scholar] [CrossRef]
- Fu, W.; O’Connor, T.D.; Jun, G.; Kang, H.M.; Abecasis, G.; Leal, S.M.; Gabriel, S.; Rieder, M.J.; Altshuler, D.; Shendure, J.; et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 2013, 493, 216–220. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age (Years) | Sex | Tumor Biopsy Site | Current Treatment When Tissue Biopsy Procured | Current Treatment When Liquid Biopsy Procured | Time Between Biopsies (Months) * | Mutation | Stage (AJCC 8th ed [19]) | Serum LDH ** | OS (Months) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 76 | M | Brain | PROG pembro | PROG pembro | Concurrent | BRAF V600E NRAS Q22K | M1d | NA | 4.8 |
| 2 | 61 | F | Inguinal LN | PROG combi-DT | PROG combi-DT | 6 | BRAF V600E | M1c | <ULN | Alive (33.7) |
| 3 | 58 | M | Brain | PRE nivo | PROG nivo | 2 | BRAF V600E | M1d | >2× ULN | 19.2 |
| 4 | 48 | M | Thigh SC | PROG ipi + nivo | PROG ipi + nivo | 1 | BRAF V600E | M1d | <ULN | 20.7 |
| 5 | 56 | M | Inguinal LN | PRE combi-DT | PROG combi-DT | 4 | BRAF V600K | M1c | >2× ULN | 10.4 |
| 6 | 70 | M | Liver | PRE nivo | PRE nivo | Concurrent | BRAF G466E | M1d | >2× ULN | 1.2 |
| 7 | 37 | F | Ovary | PROG ipi + pembro | PROG ipi + pembro | 4 | NRAS Q61K | M1c | >1× ULN | 25.7 |
| 8 | 65 | M | Abdominal LN | PROG ipi + pembro | PROG ipi + pembro | Concurrent | NRAS Q61K | M1c | <ULN | Alive (52.2) |
| 9 | 65 | M | Liver | PROG nivo | PROG nivo | 3 | NRAS Q61R | M1c | >1× ULN | 20.4 |
| 10 | 69 | M | Brain | PRE pembro | PROG pembro | 6 | NRAS Q61R | M1c | <ULN | 11.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diefenbach, R.J.; Lee, J.H.; Strbenac, D.; Yang, J.Y.H.; Menzies, A.M.; Carlino, M.S.; Long, G.V.; Spillane, A.J.; Stretch, J.R.; Saw, R.P.M.; et al. Analysis of the Whole-Exome Sequencing of Tumor and Circulating Tumor DNA in Metastatic Melanoma. Cancers 2019, 11, 1905. https://doi.org/10.3390/cancers11121905
Diefenbach RJ, Lee JH, Strbenac D, Yang JYH, Menzies AM, Carlino MS, Long GV, Spillane AJ, Stretch JR, Saw RPM, et al. Analysis of the Whole-Exome Sequencing of Tumor and Circulating Tumor DNA in Metastatic Melanoma. Cancers. 2019; 11(12):1905. https://doi.org/10.3390/cancers11121905
Chicago/Turabian StyleDiefenbach, Russell J., Jenny H. Lee, Dario Strbenac, Jean Y. H. Yang, Alexander M. Menzies, Matteo S. Carlino, Georgina V. Long, Andrew J. Spillane, Jonathan R. Stretch, Robyn P. M. Saw, and et al. 2019. "Analysis of the Whole-Exome Sequencing of Tumor and Circulating Tumor DNA in Metastatic Melanoma" Cancers 11, no. 12: 1905. https://doi.org/10.3390/cancers11121905
APA StyleDiefenbach, R. J., Lee, J. H., Strbenac, D., Yang, J. Y. H., Menzies, A. M., Carlino, M. S., Long, G. V., Spillane, A. J., Stretch, J. R., Saw, R. P. M., Thompson, J. F., Ch’ng, S., Scolyer, R. A., Kefford, R. F., & Rizos, H. (2019). Analysis of the Whole-Exome Sequencing of Tumor and Circulating Tumor DNA in Metastatic Melanoma. Cancers, 11(12), 1905. https://doi.org/10.3390/cancers11121905