Potent Antitumor Activity of Liposomal Irinotecan in an Organoid- and CRISPR-Cas9-Based Murine Model of Gallbladder Cancer

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
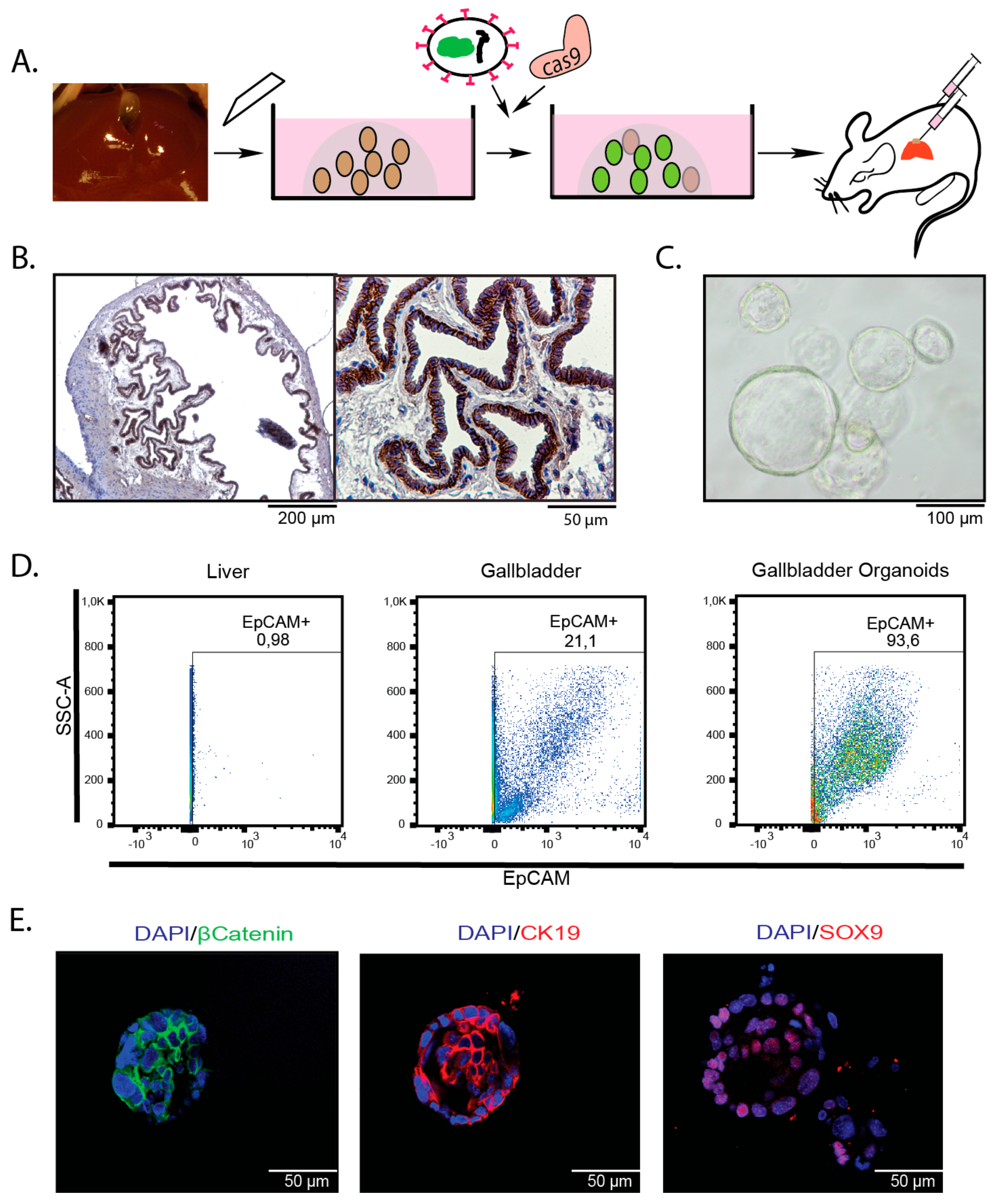
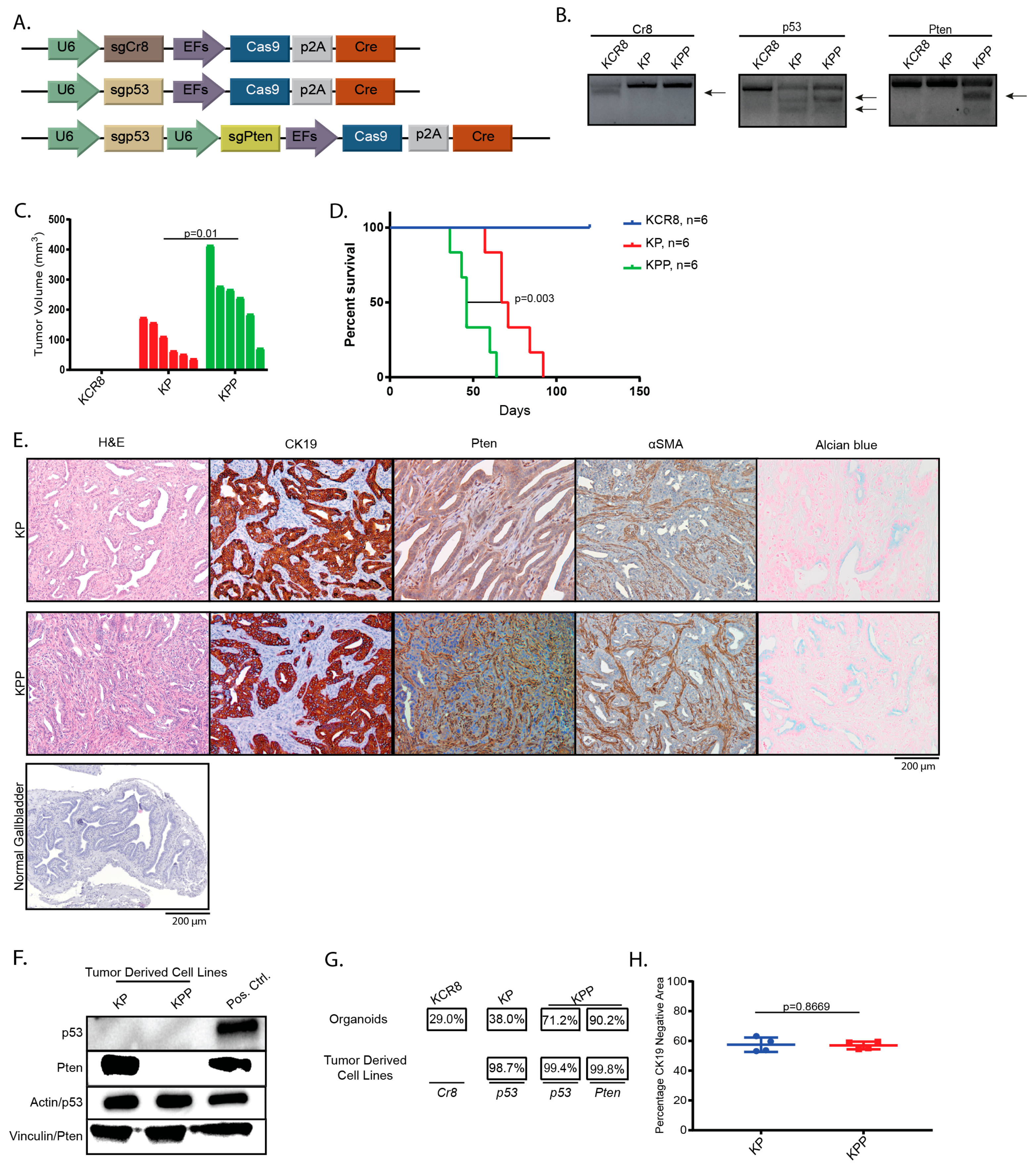
2.1. Introduction of Cancer Drivers into GB orGanoids Leads to Tumor Formation in Mice
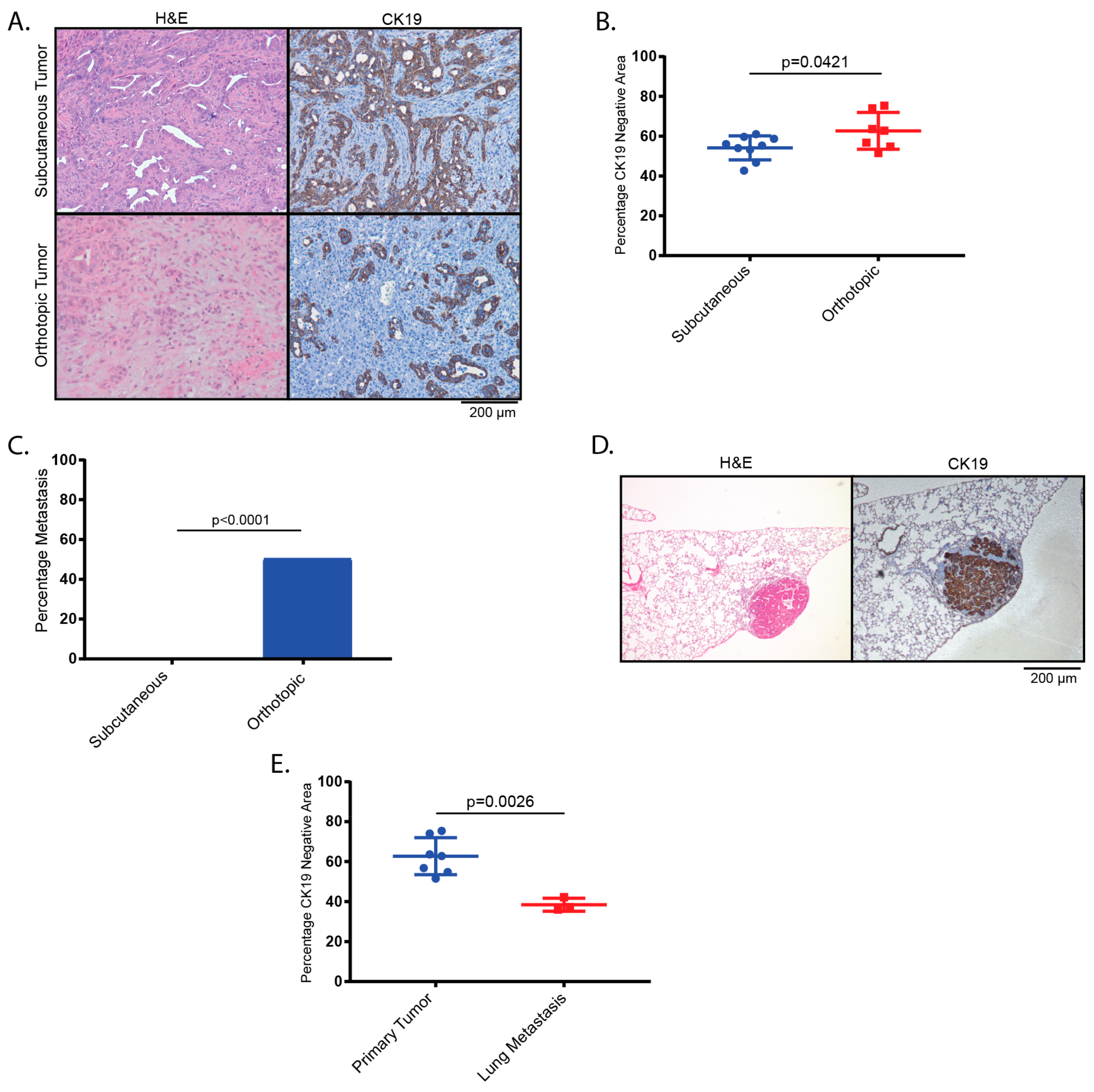
2.2. Tumors Derived from Orthotopic Transplantation of Genetically Altered orGanoids Frequently Metastasize to the Lung
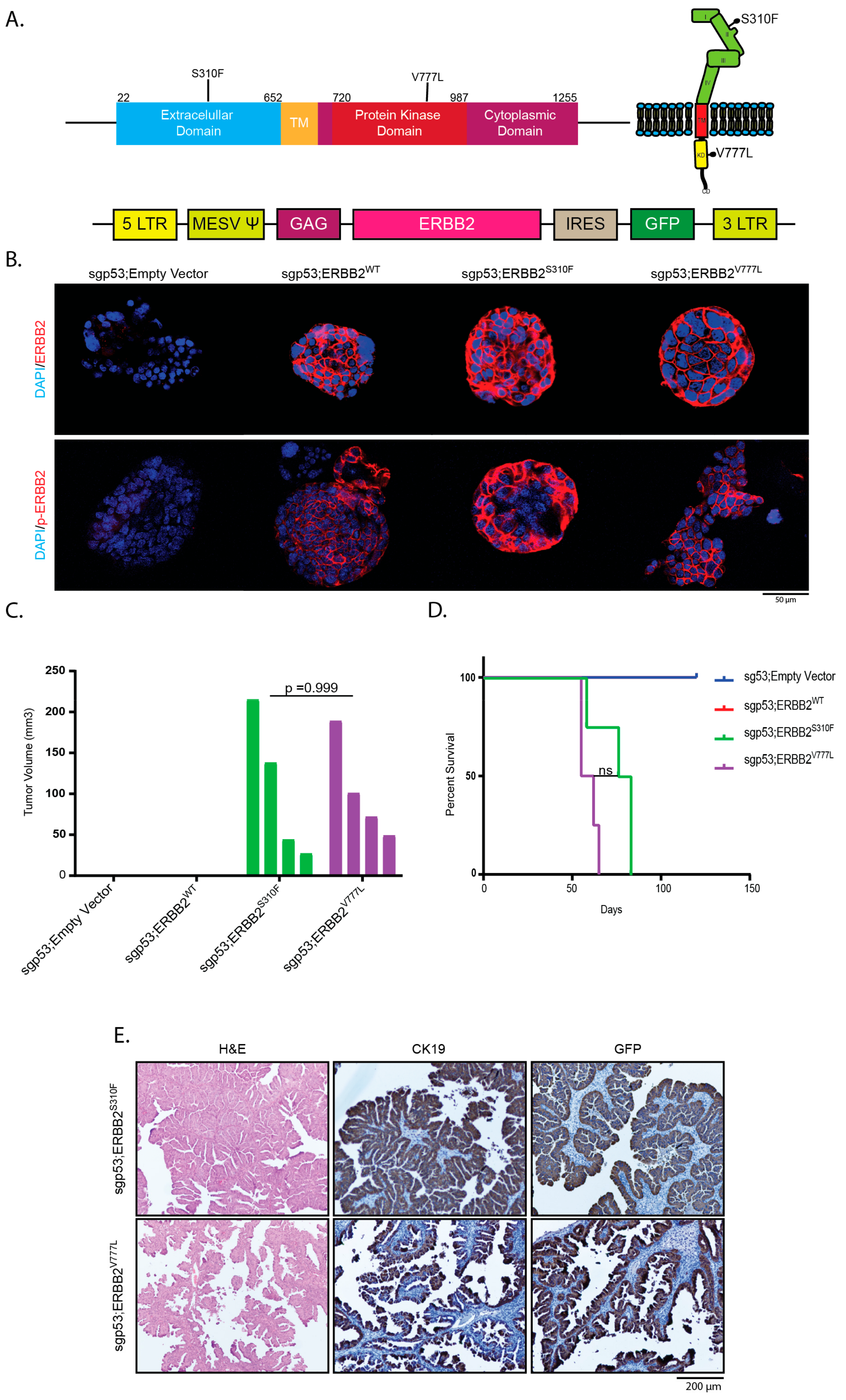
2.3. Overexpression of Activating ERBB2 Mutants Give Rise to GBC
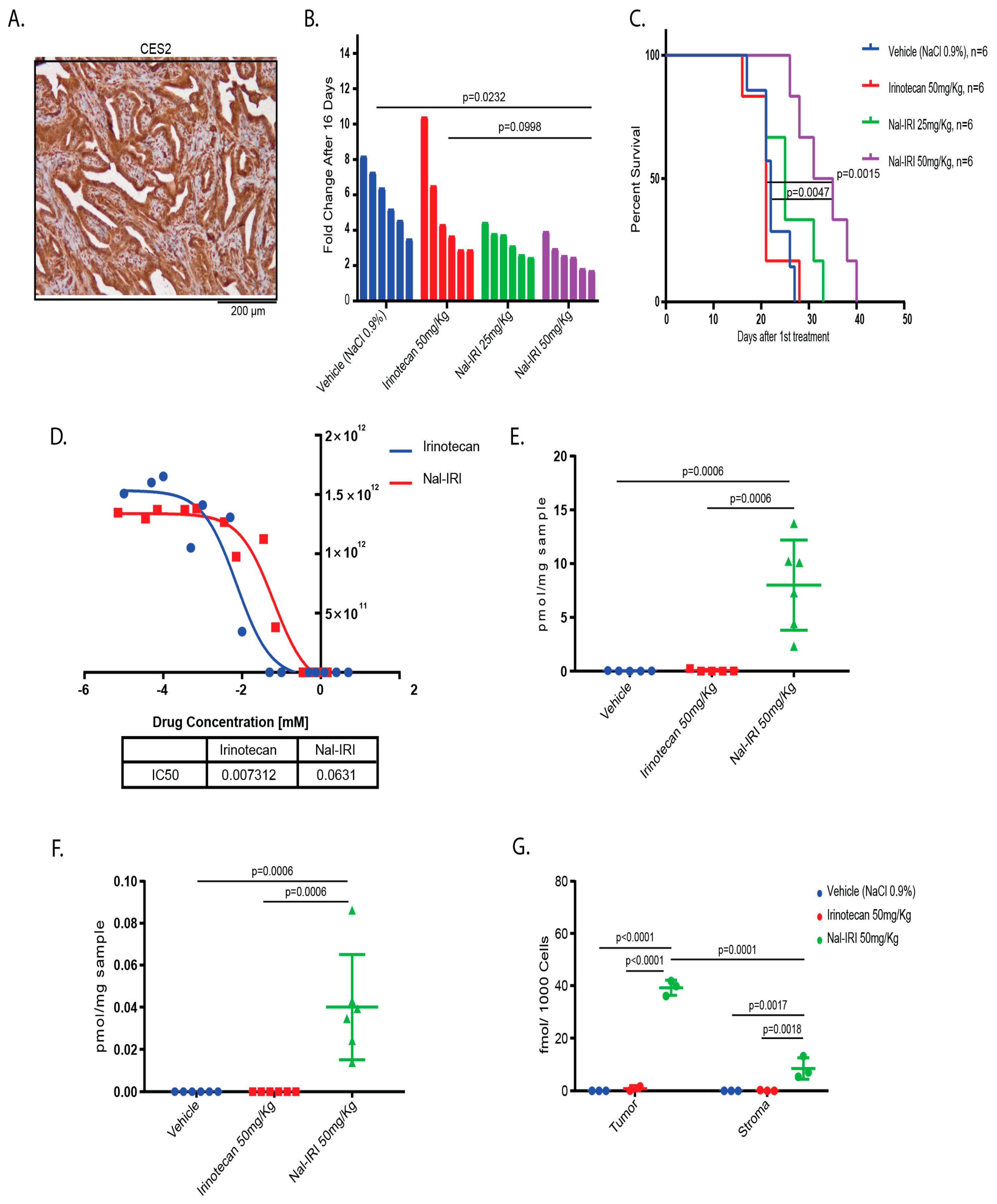
2.4. Antitumor Effects of Nal-IRI Correlate with Increased Intratumoral CPT-11 Concentrations
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Isolation of Murine Gallbladder Organoids
4.3. Tumor Cell Isolation
4.4. IC50 Cell Viability Assay:
4.5. Flow Cytometry and Cell Sorting
4.6. Subcutaneous and Orthotopic Transplantation of Organoids
4.7. Plasmids
4.8. Transfection and Retroviral Transduction of Organoids
4.9. T7-Endonuclease Assays and Quantification of Indel Frequency in Edited orGanoids and Tumor Derived Cell Lines
4.10. Immunohistochemistry, Immunofluorescence and Alcian Blue:
4.11. Immunoblotting
4.12. In Vivo Chemotherapy Treatment
4.13. Determination of CK19-Negative Area
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: Globocan sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Randi, G.; Malvezzi, M.; Levi, F.; Ferlay, J.; Negri, E.; Franceschi, S.; La Vecchia, C. Epidemiology of biliary tract cancers: An update. Ann. Oncol. 2009, 20, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Stinton, L.M.; Shaffer, E.A. Epidemiology of gallbladder disease: Cholelithiasis and cancer. Gut Liver 2012, 6, 172–187. [Google Scholar] [CrossRef]
- Hundal, R.; Shaffer, E.A. Gallbladder cancer: Epidemiology and outcome. Clin. Epidemiol. 2014, 6, 99–109. [Google Scholar]
- Cai, Z.-Q.; Guo, P.; Si, S.-B.; Geng, Z.-M.; Chen, C.; Cong, L.-L. Analysis of prognostic factors for survival after surgery for gallbladder cancer based on a bayesian network. Sci. Rep. 2017, 7, 293. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Chen, C.; Geng, Z.; Shen, H.; Song, H.; Zhao, Y.; Zhang, G.; Li, W.; Ma, L.; Wang, L. Long-term outcomes and prognostic factors in advanced gallbladder cancer: Focus on the advanced t stage. PLoS ONE 2016, 11, e0166361. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. Abc-06 a randomised phase iii, multi-centre, open-label study of active symptom control (asc) alone or asc with oxaliplatin/5-fu chemotherapy (asc+mfolfox) for patients (pts) with locally advanced/metastatic biliary tract cancers (abc) previously-treated with cisplatin/gemcitabine (cisgem) chemotherapy. J. Clin. Oncol. 2019, 37, 4003. [Google Scholar]
- Wang-Gillam, A.; Li, C.P.; Bodoky, G.; Dean, A.; Shan, Y.S.; Jameson, G.; Macarulla, T.; Lee, K.H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (napoli-1): A global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef]
- ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000 Feb 29 -. Identifier NCT03044587, Nal-IRI With 5-fluorouracil (5-FU) and Leucovorin or Gemcitabine Plus Cisplatin in Advanced Biliary-tract Cancer (NIFE); 2017 Feb 07; [about 9 pages]. Available online: https://clinicaltrials.gov/ct2/show/NCT03044587?term=NIFE&draw=2&rank=1 (accessed on 30 August 2019).
- ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000 Feb 29 -. Identifier NCT03043547, Nal-IRI and 5-FU Compared to 5-FU in Patients With Cholangio- and Gallbladder Carcinoma Previously Treated With Gemcitabine-based Therapies (NALIRICC); 2017 Feb 06; [about 9 pages]. Available online: https://clinicaltrials.gov/ct2/show/NCT03043547?term=liposomal+irinotecan&cntry=DE&draw=2&rank=1 (accessed on 30 August 2019).
- Mathijssen, R.H.; van Alphen, R.J.; Verweij, J.; Loos, W.J.; Nooter, K.; Stoter, G.; Sparreboom, A. Clinical pharmacokinetics and metabolism of irinotecan (cpt-11). Clin. Cancer Res. 2001, 7, 2182–2194. [Google Scholar] [PubMed]
- Kalra, A.V.; Kim, J.; Klinz, S.G.; Paz, N.; Cain, J.; Drummond, D.C.; Nielsen, U.B.; Fitzgerald, J.B. Preclinical activity of nanoliposomal irinotecan is governed by tumor deposition and intratumor prodrug conversion. Cancer Res. 2014, 74, 7003–7013. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.C.; Lee, H.; Gaddy, D.F.; Klinz, S.G.; Paz, N.; Kalra, A.V.; Drummond, D.C.; Chan, D.C.; Bunn, P.A.; Fitzgerald, J.B.; et al. Extended topoisomerase 1 inhibition through liposomal irinotecan results in improved efficacy over topotecan and irinotecan in models of small-cell lung cancer. Anticancer Drugs 2017, 28, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Z.; Li, X.; Ye, J.; Wu, X.; Tan, Z.; Liu, C.; Shen, B.; Wang, X.-A.; Wu, W.; et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the erbb pathway. Nat. Genet. 2014, 46, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.R.; Creasy, J.M.; Goldman, D.A.; Gönen, M.; Kandoth, C.; Kundra, R.; Solit, D.B.; Askan, G.; Klimstra, D.S.; Basturk, O.; et al. Regional differences in gallbladder cancer pathogenesis: Insights from a multi-institutional comparison of tumor mutations. Cancer 2019, 125, 575–585. [Google Scholar] [CrossRef]
- Javle, M.M.; Catenacci, D.; Jain, A.; Young, L.; Wang, K.; Chung, J.; Hezel, A.F.; Schrock, A.B.; Goyal, L.; Gay, L.M.; et al. Precision medicine for gallbladder cancer using somatic copy number amplifications (scna) and DNA repair pathway gene alterations. J. Clin. Oncol. 2017, 35, 4076. [Google Scholar] [CrossRef]
- Roa, I.; de Toro, G.; Schalper, K.; de Aretxabala, X.; Churi, C.; Javle, M. Overexpression of the her2/neu gene: A new therapeutic possibility for patients with advanced gallbladder cancer. Gastrointest. Cancer Res. 2014, 7, 42–48. [Google Scholar]
- Kitamura, T.; Srivastava, J.; DiGiovanni, J.; Kiguchi, K. Bile acid accelerates erbb2-induced pro-tumorigenic activities in biliary tract cancer. Mol. Carcinog. 2013, 54, 459–472. [Google Scholar] [CrossRef]
- Han, T.; Schatoff, E.M.; Murphy, C.; Zafra, M.P.; Wilkinson, J.E.; Elemento, O.; Dow, L.E. R-spondin chromosome rearrangements drive wnt-dependent tumour initiation and maintenance in the intestine. Nat. Commun. 2017, 8, 15945. [Google Scholar] [CrossRef]
- Dow, L.E.; Fisher, J.; O’Rourke, K.P.; Muley, A.; Kastenhuber, E.R.; Livshits, G.; Tschaharganeh, D.F.; Socci, N.D.; Lowe, S.W. Inducible in vivo genome editing with crispr-cas9. Nat. Biotechnol. 2015, 33, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, F.; Zhang, F.; Zhou, W.; Jiang, X.; Yang, Y.; Qu, K.; Wang, Y.; Ma, Q.; Wang, T.; et al. Genomic erbb2/erbb3 mutations promote pd-l1-mediated immune escape in gallbladder cancer: A whole-exome sequencing analysis. Gut 2019, 68, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Churi, C.; Kang, H.C.; Shroff, R.; Janku, F.; Surapaneni, R.; Zuo, M.; Barrera, C.; Alshamsi, H.; Krishnan, S.; et al. Her2/neu-directed therapy for biliary tract cancer. J. Hematol. Oncol. 2015, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Iyer, P.; Shrikhande, S.V.; Ranjan, M.; Joshi, A.; Gardi, N.; Prasad, R.; Dharavath, B.; Thorat, R.; Salunkhe, S.; Sahoo, B.; et al. Erbb2 and kras alterations mediate response to egfr inhibitors in early stage gallbladder cancer. Int. J. Cancer 2019, 144, 2008–2019. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.E.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2018, 67, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, K.; Carbajal, S.; Chan, K.; Beltrán, L.; Ruffino, L.; Shen, J.; Matsumoto, T.; Yoshimi, N.; DiGiovanni, J. Constitutive expression of erbb-2 in gallbladder epithelium results in development of adenocarcinoma. Cancer Res. 2001, 61, 6971–6976. [Google Scholar]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Roa, I.; de Toro, G.; Fernández, F.; Game, A.; Muñoz, S.; de Aretxabala, X.; Javle, M. Inactivation of tumor suppressor gene pten in early and advanced gallbladder cancer. Diagn. Pathol. 2015, 10, 148. [Google Scholar] [CrossRef]
- Moasser, M.M. The oncogene her2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef]
- Abrahao-Machado, L.F.; Scapulatempo-Neto, C. Her2 testing in gastric cancer: An update. World J. Gastroenterol. 2016, 22, 4619–4625. [Google Scholar] [CrossRef] [PubMed]
- Sircoulomb, F.; Bekhouche, I.; Finetti, P.; Adélaïde, J.; Ben Hamida, A.; Bonansea, J.; Raynaud, S.; Innocenti, C.; Charafe-Jauffret, E.; Tarpin, C.; et al. Genome profiling of erbb2-amplified breast cancers. BMC Cancer 2010, 10, 539. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Fakih, M.; Ali, S.M.; Elvin, J.A.; Schrock, A.B.; Suh, J.; Vergilio, J.-A.; Ramkissoon, S.; Severson, E.; Daniel, S.; et al. Targeting her2 in colorectal cancer: The landscape of amplification and short variant mutations in erbb2 and erbb3. Cancer 2018, 124, 1358–1373. [Google Scholar] [CrossRef] [PubMed]
- Greulich, H.; Kaplan, B.; Mertins, P.; Chen, T.-H.; Tanaka, K.E.; Yun, C.-H.; Zhang, X.; Lee, S.-H.; Cho, J.; Ambrogio, L.; et al. Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of erbb2. Proc. Natl. Acad. Sci. USA 2012, 109, 14476–14481. [Google Scholar] [CrossRef]
- Bose, R.; Kavuri, S.M.; Searleman, A.C.; Shen, W.; Shen, D.; Koboldt, D.C.; Monsey, J.; Goel, N.; Aronson, A.B.; Li, S.; et al. Activating her2 mutations in her2 gene amplification negative breast cancer. Cancer Discov. 2013, 3, 224–237. [Google Scholar] [CrossRef] [Green Version]
- Marchini, C.; Pietrella, L.; Kalogris, C.; Garulli, C.; Gabrielli, F.; Quaglino, E.; Iezzi, M.; Pupa, S.M.; Tagliabue, E.; Amici, A. Her2-driven carcinogenesis: New mouse models for novel immunotherapies. Oncogene Cancer Bench Clin. 2013. [Google Scholar] [CrossRef] [Green Version]
- Hamada, S.; Masamune, A.; Shimosegawa, T. Novel therapeutic strategies targeting tumor-stromal interactions in pancreatic cancer. Front. Physiol. 2013, 4, 331. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Sirica, A.E.; Gores, G.J. Desmoplastic stroma and cholangiocarcinoma: Clinical implications and therapeutic targeting. Hepatology 2014, 59, 2397–2402. [Google Scholar] [CrossRef] [Green Version]
- Gentilini, A.; Pastore, M.; Marra, F.; Raggi, C. The role of stroma in cholangiocarcinoma: The intriguing interplay between fibroblastic component, immune cell subsets and tumor epithelium. Int. J. Mol. Sci. 2018, 19, 2885. [Google Scholar] [CrossRef] [Green Version]
- Perrin, S. Preclinical research: Make mouse studies work. Nature 2014, 507, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Modeling credentials. Nat. Biotechnol. 2015, 33, 671. [CrossRef] [PubMed] [Green Version]
- Drummond, D.C.; Noble, C.O.; Guo, Z.; Hong, K.; Park, J.W.; Kirpotin, D.B. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006, 66, 3271–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic k-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broutier, L.; Andersson-Rolf, A.; Hindley, C.J.; Boj, S.F.; Clevers, H.; Koo, B.-K.; Huch, M. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3d organoids and their genetic manipulation. Nat. Protoc. 2016, 11, 1724–1743. [Google Scholar] [CrossRef]
- O’Rourke, K.P.; Loizou, E.; Livshits, G.; Schatoff, E.M.; Baslan, T.; Manchado, E.; Simon, J.; Romesser, P.B.; Leach, B.; Han, T.; et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat. Biotechnol. 2017, 35, 577–582. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Ann Ran, F.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the crispr-cas9 system. Nat. Protocols 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Labun, K.; Guo, X.; Chavez, A.; Church, G.; Gagnon, J.A.; Valen, E. Accurate analysis of genuine crispr editing events with amplican. Genome Res. 2019, 29, 843–847. [Google Scholar] [CrossRef] [Green Version]
- Perez, F.; Granger, B.E. Project jupyter: Computational narratives as the engine of collaborative data science. Retrieved Sept. 2015, 11, 108. [Google Scholar]
- Wilkinson, L. Ggplot2: Elegant graphics for data analysis by wickham, H. Biometrics 2011, 67, 678–679. [Google Scholar] [CrossRef]
- Saborowski, M.; Saborowski, A.; Morris, J.P.T.; Bosbach, B.; Dow, L.E.; Pelletier, J.; Klimstra, D.S.; Lowe, S.W. A modular and flexible esc-based mouse model of pancreatic cancer. Genes Dev. 2014, 28, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, A.; Pandey, A. Post-hoc comparison in survival analysis: An easy approach. J. Biosci. Med. 2017, 5, 112. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Guide RNA Sequences | |
|---|---|
| p53 sgRNA | CCTCGAGCTCCCTCTGAGCC |
| Pten sgRNA | GAGATCGTTAGCAGAAACAAA |
| Cr8 sgRNA | GACATTTCTTTCCCCACTGG |
| Primers used in T7 Endonuclease Mutation Detection Assay | |
| T7 Mut PCR p53 fwd | GCCATCTTGGGTCCTGACTT |
| T7 Mut PCR p53 rev | CCCCGCAGGATTTACAGACA |
| T7 Mut PCR Pten fwd | GAGCCATTTCCATCCTGCAG |
| T7 Mut PCR Pten rev | CTAGCCGAACACTCCCTAGG |
| T7 Mut PCR Cr8 fwd | TAAGATGATTATCTGAATTCCTGGG |
| T7 Mut PCR Cr8 rev | TCTTATCCCCTGTGTTGGAA |
| Primers Used in NGS | |
| NGS PCR p53 fwd | CCATAGGGGTTTGTTTGTTTGT |
| NGS PCR p53 rev | CGCAGGATTTACAGACACCC |
| NGS PCR Pten fwd | GAGCCATTTCCATCCTGCAG |
| NGS PCR Pten rev | CACGATCTAGAAATGCGCCC |
| NGS PCR Cr8 fwd | TCTGAATTCCTGGGATGGGG |
| NGS PCR Cr8 rev | TGTGTGGCTACCCTGTTCTT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erlangga, Z.; Wolff, K.; Poth, T.; Peltzer, A.; Nahnsen, S.; Spielberg, S.; Timrott, K.; Woller, N.; Kühnel, F.; Manns, M.P.; et al. Potent Antitumor Activity of Liposomal Irinotecan in an Organoid- and CRISPR-Cas9-Based Murine Model of Gallbladder Cancer. Cancers 2019, 11, 1904. https://doi.org/10.3390/cancers11121904
Erlangga Z, Wolff K, Poth T, Peltzer A, Nahnsen S, Spielberg S, Timrott K, Woller N, Kühnel F, Manns MP, et al. Potent Antitumor Activity of Liposomal Irinotecan in an Organoid- and CRISPR-Cas9-Based Murine Model of Gallbladder Cancer. Cancers. 2019; 11(12):1904. https://doi.org/10.3390/cancers11121904
Chicago/Turabian StyleErlangga, Zulrahman, Katharina Wolff, Tanja Poth, Alexander Peltzer, Sven Nahnsen, Steffi Spielberg, Kai Timrott, Norman Woller, Florian Kühnel, Michael P. Manns, and et al. 2019. "Potent Antitumor Activity of Liposomal Irinotecan in an Organoid- and CRISPR-Cas9-Based Murine Model of Gallbladder Cancer" Cancers 11, no. 12: 1904. https://doi.org/10.3390/cancers11121904