Genetically Engineered Mouse Models of Gliomas: Technological Developments for Translational Discoveries

Abstract
:1. Introduction
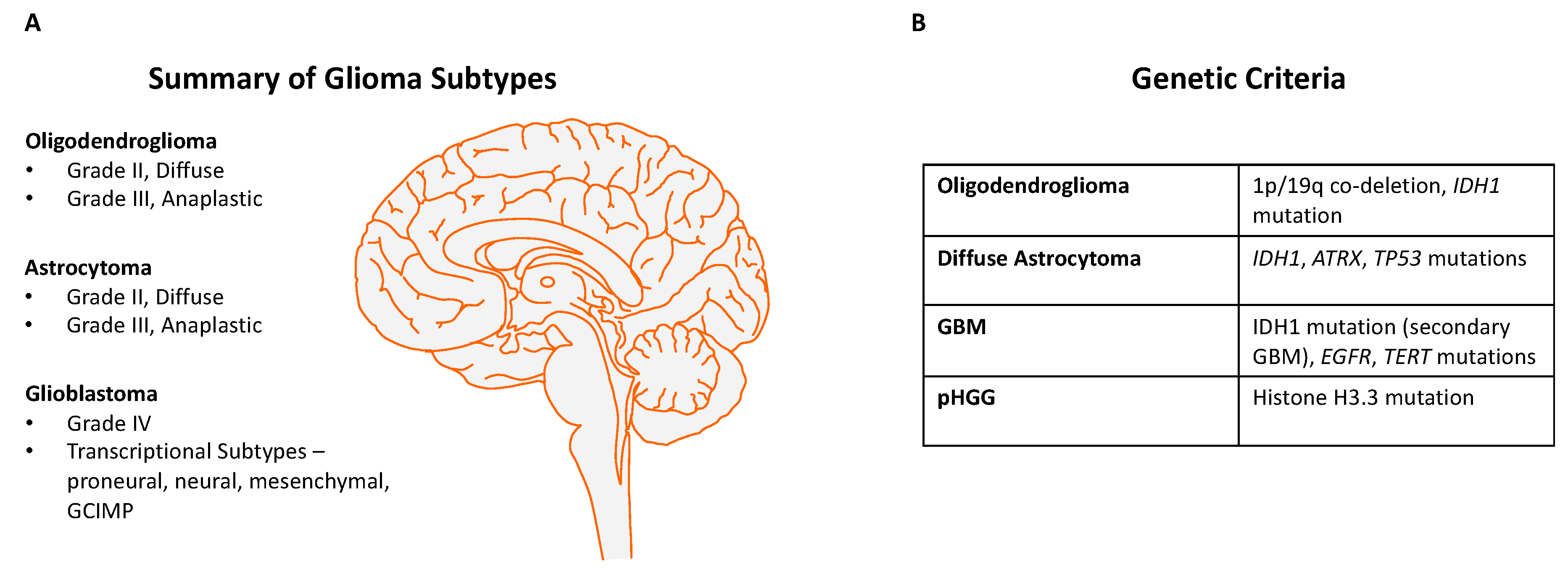
2. Genetic Landscape of Human Gliomas
3. Glioma Mouse Models
4. Cre/LoxP, Flp/FRT, RCAS Technology
5. Cre Lines Used for Glioma GEMMs
6. Glioma Mouse Models and Genetic Drivers
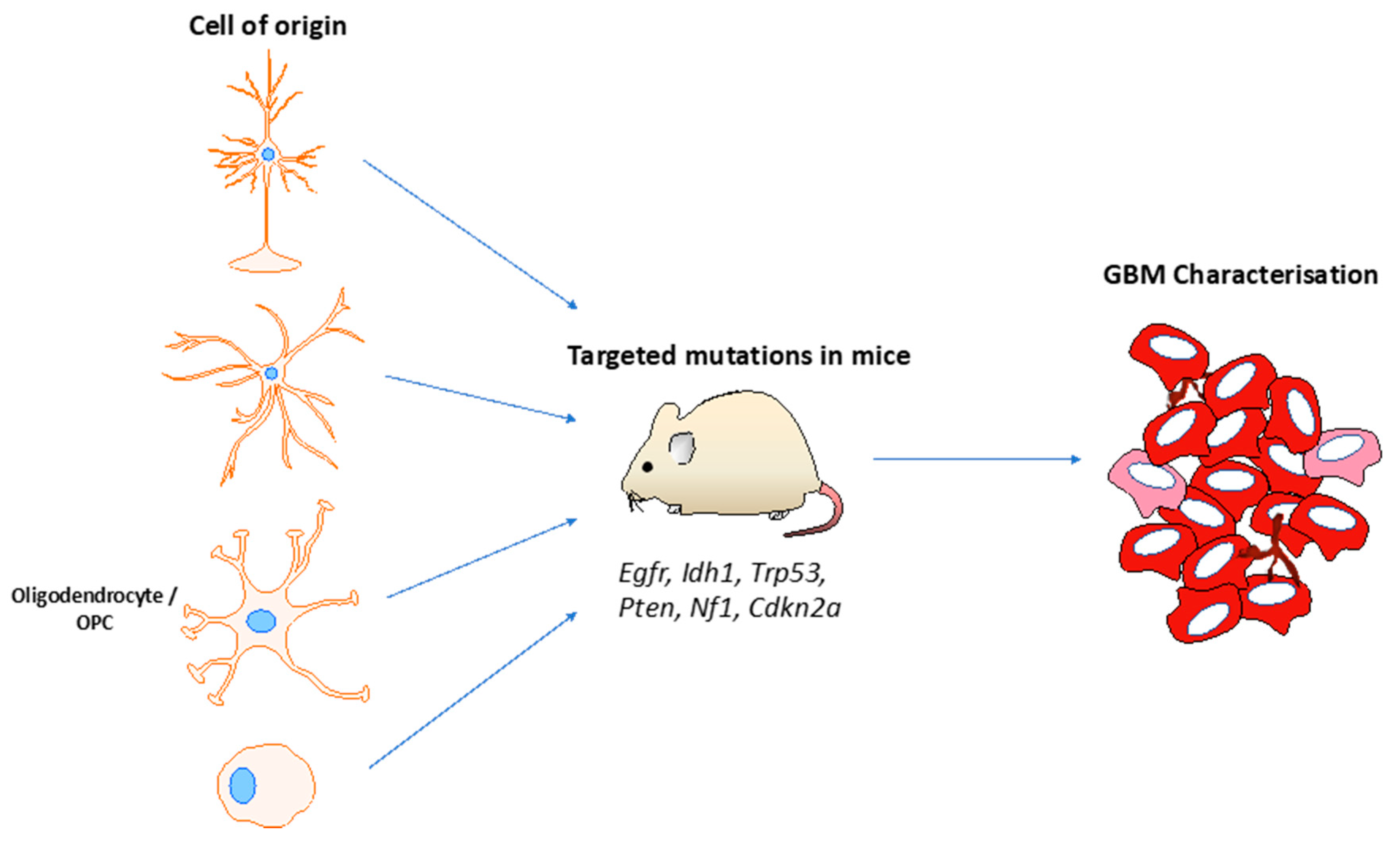
7. Glioma Cell of Origin
8. Sleeping Beauty Transposon-Based GEMMs
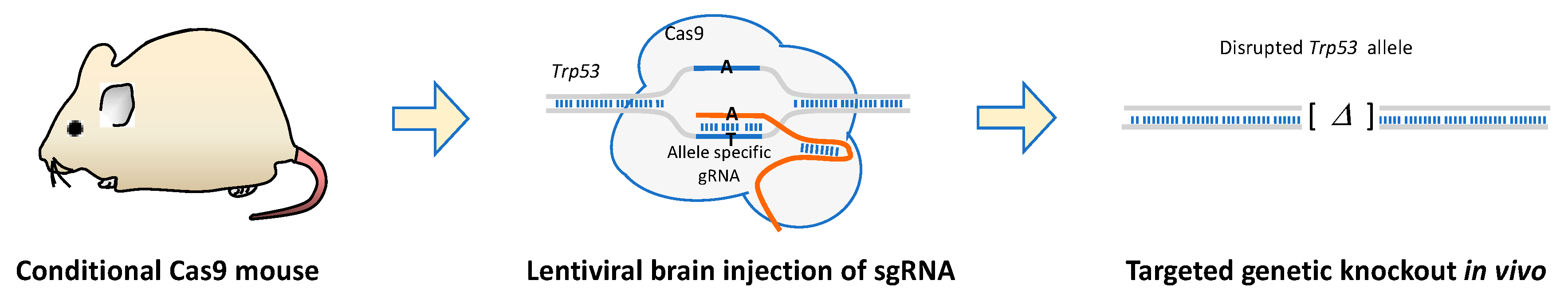
9. CRISPR-Cas9 Genetic Engineering and Screens in Mice
10. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Noorani, I.; Petty, G.; Grundy, P.L.; Sharpe, G.; Willaime-Morawek, S.; Harris, S.; Thomas, G.J.; Nicoll, J.A.; Boche, D. Novel association between microglia and stem cells in human gliomas: A contributor to tumour proliferation? J. Pathol. Clin. Res. 2015, 1, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Parada, L.F.; Holland, E.C.; Charest, A. Genetic modeling of gliomas in mice: New tools to tackle old problems. Glia 2011, 59, 1155–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenting, K.; Verhaak, R.; Ter Laan, M.; Wesseling, P.; Leenders, W. Glioma: Experimental models and reality. Acta Neuropathol. 2017, 133, 263–282. [Google Scholar] [CrossRef] [PubMed]
- Kurzwelly, D.; Herrlinger, U.; Simon, M. Seizures in patients with low-grade gliomas—Incidence, pathogenesis, surgical management, and pharmacotherapy. Adv. Tech. Stand. Neurosurg. 2010, 35, 81–111. [Google Scholar] [PubMed]
- Noorani, I.; Sanai, N. Surgical Management of Incidental Gliomas. Neurosurg. Clin. N. Am. 2017, 28, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Mandonnet, E.; de Witt Hamer, P.; Pallud, J.; Bauchet, L.; Whittle, I.; Duffau, H. Silent diffuse low-grade glioma: Toward screening and preventive treatment? Cancer 2014, 120, 1758–1762. [Google Scholar] [CrossRef]
- Pallud, J.; Capelle, L.; Taillandier, L.; Badoual, M.; Duffau, H.; Mandonnet, E. The silent phase of diffuse low-grade gliomas. Is it when we missed the action? Acta Neurochir. (Wien.) 2013, 155, 2237–2242. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Perry, J.R.; Laperriere, N.; O’Callaghan, C.J.; Brandes, A.A.; Menten, J.; Phillips, C.; Fay, M.; Nishikawa, R.; Cairncross, J.G.; Roa, W.; et al. Short-Course Radiation plus Temozolomide in Elderly Patients with Glioblastoma. N. Engl. J. Med. 2017, 376, 1027–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Hassiotou, F.; Nowak, A. Glioblastoma stem-like cells: At the root of tumor recurrence and a therapeutic target. Carcinogenesis 2015, 36, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Holland, E.C.; Celestino, J.; Dai, C.; Schaefer, L.; Sawaya, R.E.; Fuller, G.N. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat. Genet. 2000, 25, 55–57. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; van Noorden, C.J.F. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef] [Green Version]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Jaunmuktane, Z.; Capper, D.; Jones, D.T.; Schrimpf, D.; Sill, M.; Dutt, M.; Suraweera, N.; Pfister, S.M.; von Deimling, A.; Brandner, S. Methylation array profiling of adult brain tumours: Diagnostic outcomes in a large, single centre. Acta Neuropathol. Commun. 2019, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Jones, D.T.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Laug, D.; Glasgow, S.M.; Deneen, B. A glial blueprint for gliomagenesis. Nat. Rev. Neurosci. 2018, 19, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Golic, K.G.; Lindquist, S. The FLP recombinase of yeast catalyzes site-specific recombination in the Drosophila genome. Cell 1989, 59, 499–509. [Google Scholar] [CrossRef]
- Tsien, J.Z. Cre-Lox Neurogenetics: 20 Years of Versatile Applications in Brain Research and Counting. Front. Genet. 2016, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Friedel, R.H.; Wurst, W.; Wefers, B.; Kuhn, R. Generating conditional knockout mice. Methods Mol. Biol. 2011, 693, 205–231. [Google Scholar] [PubMed]
- Fisher, G.H.; Orsulic, S.; Holland, E.; Hively, W.P.; Li, Y.; Lewis, B.C.; Williams, B.O.; Varmus, H.E. Development of a flexible and specific gene delivery system for production of murine tumor models. Oncogene 1999, 18, 5253–5260. [Google Scholar] [CrossRef] [Green Version]
- Tronche, F.; Kellendonk, C.; Kretz, O.; Gass, P.; Anlag, K.; Orban, P.C.; Bock, R.; Klein, R.; Schütz, G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999, 23, 99–103. [Google Scholar] [CrossRef]
- Dubois, N.C.; Hofmann, D.; Kaloulis, K.; Bishop, J.M.; Trumpp, A. Nestin-Cre transgenic mouse line Nes-Cre1 mediates highly efficient Cre/loxP mediated recombination in the nervous system, kidney, and somite-derived tissues. Genesis 2006, 44, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Hippenmeyer, S.; Ghashghaei, H.T. A Nestin-cre transgenic mouse is insufficient for recombination in early embryonic neural progenitors. Biol. Open 2012, 1, 1200–1203. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, L.; Theis, M.; Alvarez-Maya, I.; Brenner, M.; Willecke, K.; Messing, A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis 2001, 31, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Downward, J.; Yarden, Y.; Mayes, E.; Scrace, G.; Totty, N.; Stockwell, P.; Ullrich, A.; Schlessinger, J.; Waterfield, M.D. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature 1984, 307, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Holland, E.C.; Hively, W.P.; DePinho, R.A.; Varmus, H.E. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998, 12, 3675–3685. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Ying, H.; Yan, H.; Kimmelman, A.C.; Hiller, D.J.; Chen, A.J.; Perry, S.R.; Tonon, G.; Chu, G.C.; Ding, Z.; et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 2008, 455, 1129–1133. [Google Scholar] [CrossRef]
- Llaguno, S.A.; Chen, J.; Kwon, C.H.; Jackson, E.L.; Li, Y.; Burns, D.K.; Alvarez-Buylla, A.; Parada, L.F. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 2009, 15, 45–56. [Google Scholar] [CrossRef]
- Zhu, H.; Acquaviva, J.; Ramachandran, P.; Boskovitz, A.; Woolfenden, S.; Pfannl, R.; Bronson, R.T.; Chen, J.W.; Weissleder, R.; Housman, D.E.; et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 2712–2716. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brüstle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; Knobbe, C.B.; Itsumi, M.; Elia, A.J.; Harris, I.S.; Chio, I.I.; Cairns, R.A.; McCracken, S.; Wakeham, A.; Haight, J.; et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012, 26, 2038–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardella, C.; Al-Dalahmah, O.; Krell, D.; Brazauskas, P.; Al-Qahtani, K.; Tomkova, M.; Adam, J.; Serres, S.; Lockstone, H.; Freeman-Mills, L.; et al. Expression of Idh1(R132H) in the Murine Subventricular Zone Stem Cell Niche Recapitulates Features of Early Gliomagenesis. Cancer Cell 2016, 30, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Kharbanda, S.; Pope, W.B.; Tran, A.; Solis, O.E.; Peale, F.; Forrest, W.F.; Pujara, K.; Carrillo, J.A.; Pandita, A.; et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J. Clin. Oncol. 2011, 29, 4482–4490. [Google Scholar] [CrossRef] [PubMed]
- Jacques, T.S.; Swales, A.; Brzozowski, M.J.; Henriquez, N.V.; Linehan, J.M.; Mirzadeh, Z.; O’Malley, C.; Naumann, H.; Alvarez-Buylla, A.; Brandner, S. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. EMBO J. 2010, 29, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Bachoo, R.M.; Maher, E.A.; Ligon, K.L.; Sharpless, N.E.; Chan, S.S.; You, M.J.; Tang, Y.; DeFrances, J.; Stover, E.; Weissleder, R.; et al. Epidermal growth factor receptor and Ink4a/Arf: Convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 2002, 1, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Friedmann-Morvinski, D.; Bushong, E.A.; Ke, E.; Soda, Y.; Marumoto, T.; Singer, O.; Ellisman, M.H.; Verma, I.M. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 2012, 338, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Doetsch, F.; Petreanu, L.; Caille, I.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron 2002, 36, 1021–1034. [Google Scholar] [CrossRef]
- Zong, H.; Espinosa, J.S.; Su, H.H.; Muzumdar, M.D.; Luo, L. Mosaic analysis with double markers in mice. Cell 2005, 121, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sage, J.C.; Miller, M.R.; Verhaak, R.G.; Hippenmeyer, S.; Vogel, H.; Foreman, O.; Bronson, R.T.; Nishiyama, A.; Luo, L.; et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 2011, 146, 209–221. [Google Scholar] [CrossRef]
- Kim, E.J.; Leung, C.T.; Reed, R.R.; Johnson, J.E. In vivo analysis of Ascl1 defined progenitors reveals distinct developmental dynamics during adult neurogenesis and gliogenesis. J. Neurosci. 2007, 27, 12764–12774. [Google Scholar] [CrossRef]
- Llaguno, S.R.; Wang, Z.; Sun, D.; Chen, J.; Xu, J.; Kim, E.; Hatanpaa, K.J.; Raisanen, J.M.; Burns, D.K.; Johnson, J.E.; et al. Adult Lineage-Restricted CNS Progenitors Specify Distinct Glioblastoma Subtypes. Cancer Cell 2015, 28, 429–440. [Google Scholar] [Green Version]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.Y.; Kim, W.K.; Lee, J.K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvak, Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef]
- Cui, Z.; Geurts, A.M.; Liu, G.; Kaufman, C.D.; Hackett, P.B. Structure-function analysis of the inverted terminal repeats of the sleeping beauty transposon. J. Mol. Biol. 2002, 318, 1221–1235. [Google Scholar] [CrossRef]
- Geurts, A.M.; Yang, Y.; Clark, K.J.; Liu, G.; Cui, Z.; Dupuy, A.J.; Bell, J.B.; Largaespada, D.A.; Hackett, P.B. Gene transfer into genomes of human cells by the sleeping beauty transposon system. Mol. Ther. 2003, 8, 108–117. [Google Scholar] [CrossRef]
- Mátés, L.; Chuah, M.K.; Belay, E.; Jerchow, B.; Manoj, N.; Acosta-Sanchez, A.; Grzela, D.P.; Schmitt, A.; Becker, K.; Matrai, J.; et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 2009, 41, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Wei, Z.; Koso, H.; Rust, A.G.; Yew, C.C.; Mann, M.B.; Ward, J.M.; Adams, D.J.; Copeland, N.G.; Jenkins, N.A. Transposon mutagenesis identifies genes and evolutionary forces driving gastrointestinal tract tumor progression. Nat. Genet. 2015, 47, 142–150. [Google Scholar] [CrossRef]
- Kas, S.M.; de Ruiter, J.R.; Schipper, K.; Annunziato, S.; Schut, E.; Klarenbeek, S.; Drenth, A.P.; van der Burg, E.; Klijn, C.; Ten Hoeve, J.J.; et al. Insertional mutagenesis identifies drivers of a novel oncogenic pathway in invasive lobular breast carcinoma. Nat. Genet. 2017, 49, 1219–1230. [Google Scholar] [CrossRef]
- Bard-Chapeau, E.A.; Nguyen, A.T.; Rust, A.G.; Sayadi, A.; Lee, P.; Chua, B.Q.; New, L.S.; De Jong, J.; Ward, J.M.; Chin, C.K.; et al. Transposon mutagenesis identifies genes driving hepatocellular carcinoma in a chronic hepatitis B mouse model. Nat. Genet. 2014, 46, 24–32. [Google Scholar] [CrossRef]
- Wu, X.; Northcott, P.A.; Dubuc, A.; Dupuy, A.J.; Shih, D.J.; Witt, H.; Croul, S.; Bouffet, E.; Fults, D.W.; Eberhart, C.G.; et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 2012, 482, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Rahrmann, E.P.; Watson, A.L.; Keng, V.W.; Choi, K.; Moriarity, B.S.; Beckmann, D.A.; Wolf, N.K.; Sarver, A.; Collins, M.H.; Moertel, C.L.; et al. Forward genetic screen for malignant peripheral nerve sheath tumor formation identifies new genes and pathways driving tumorigenesis. Nat. Genet. 2013, 45, 756–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriarity, B.S.; Otto, G.M.; Rahrmann, E.P.; Rathe, S.K.; Wolf, N.K.; Weg, M.T.; Manlove, L.A.; LaRue, R.S.; Temiz, N.A.; Molyneux, S.D.; et al. A Sleeping Beauty forward genetic screen identifies new genes and pathways driving osteosarcoma development and metastasis. Nat. Genet. 2015, 47, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Jenjaroenpun, P.; Pillai, A.M.; Ivshina, A.V.; Ow, G.S.; Efthimios, M.; Zhiqun, T.; Tan, T.Z.; Lee, S.C.; Rogers, K.; et al. Transposon insertional mutagenesis in mice identifies human breast cancer susceptibility genes and signatures for stratification. Proc. Natl. Acad. Sci. USA 2017, 114, E2215–E2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, M.B.; Black, M.A.; Jones, D.J.; Ward, J.M.; Yew, C.C.; Newberg, J.Y.; Dupuy, A.J.; Rust, A.G.; Bosenberg, M.W.; McMahon, M.; et al. Transposon mutagenesis identifies genetic drivers of Braf(V600E) melanoma. Nat. Genet. 2015, 47, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, A.J.; Akagi, K.; Largaespada, D.A.; Copeland, N.G.; Jenkins, N.A. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature 2005, 436, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.M.; Ward, J.M.; Yew, C.C.; Kovochich, A.; Dawson, D.W.; Black, M.A.; Brett, B.T.; Sheetz, T.E.; Dupuy, A.J.; Chang, D.K.; et al. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 5934–5941. [Google Scholar] [CrossRef] [PubMed]
- Rahrmann, E.P.; Collier, L.S.; Knutson, T.P.; Doyal, M.E.; Kuslak, S.L.; Green, L.E.; Malinowski, R.L.; Roethe, L.; Akagi, K.; Waknitz, M.; et al. Identification of PDE4D as a proliferation promoting factor in prostate cancer using a Sleeping Beauty transposon-based somatic mutagenesis screen. Cancer Res. 2009, 69, 4388–4397. [Google Scholar] [CrossRef] [PubMed]
- De La Rosa, J.; Weber, J.; Friedrich, M.J.; Li, Y.; Rad, L.; Ponstingl, H.; Liang, Q.; De Quirós, S.B.; Noorani, I.; Metzakopian, E.; et al. A single-copy Sleeping Beauty transposon mutagenesis screen identifies new PTEN-cooperating tumor suppressor genes. Nat. Genet. 2017, 49, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.M.; Collier, L.S.; Rodriguez, F.J.; Tieu, C.; Larson, J.D.; Halder, C.; Mahlum, E.; Kollmeyer, T.M.; Akagi, K.; Sarkar, G.; et al. Sleeping beauty-mediated somatic mutagenesis implicates CSF1 in the formation of high-grade astrocytomas. Cancer Res. 2010, 70, 3557–3565. [Google Scholar] [CrossRef]
- Collier, L.S.; Adams, D.J.; Hackett, C.S.; Bendzick, L.E.; Akagi, K.; Davies, M.N.; Diers, M.D.; Rodriguez, F.J.; Bender, A.M.; Tieu, C.; et al. Whole-body sleeping beauty mutagenesis can cause penetrant leukemia/lymphoma and rare high-grade glioma without associated embryonic lethality. Cancer Res. 2009, 69, 8429–8437. [Google Scholar] [CrossRef]
- Koso, H.; Takeda, H.; Yew, C.C.; Ward, J.M.; Nariai, N.; Ueno, K.; Nagasaki, M.; Watanabe, S.; Rust, A.G.; Adams, D.J.; et al. Transposon mutagenesis identifies genes that transform neural stem cells into glioma-initiating cells. Proc. Natl. Acad. Sci. USA 2012, 109, E2998–E3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesner, S.M.; Decker, S.A.; Larson, J.D.; Ericson, K.; Forster, C.; Gallardo, J.L.; Long, C.; Demorest, Z.L.; Zamora, E.A.; Low, W.C.; et al. De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer Res. 2009, 69, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Koschmann, C.; Calinescu, A.A.; Nunez, F.J.; Mackay, A.; Fazal-Salom, J.; Thomas, D.; Mendez, F.; Kamran, N.; Dzaman, M.; Mulpuri, L.; et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci. Transl. Med. 2016, 8, 328ra28. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lin, C.; Lu, D.; Ning, Z.; Cox, T.; Melvin, D.; Wang, X.; Bradley, A.; Liu, P. Chromosomal transposition of PiggyBac in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9290–9295. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Kong, J.; Stalker, J.; Bradley, A. Chromosomal mobilization and reintegration of Sleeping Beauty and PiggyBac transposons. Genesis 2009, 47, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef]
- Pathania, M.; De Jay, N.; Maestro, N.; Harutyunyan, A.S.; Nitarska, J.; Pahlavan, P.; Henderson, S.; Mikael, L.G.; Richard-Londt, A.; Zhang, Y.; et al. H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 2017, 32, 684–700. [Google Scholar] [CrossRef] [PubMed]
- Rad, R.; Rad, L.; Wang, W.; Cadinanos, J.; Vassiliou, G.; Rice, S.; Campos, L.S.; Yusa, K.; Banerjee, R.; Li, M.A.; et al. PiggyBac transposon mutagenesis: A tool for cancer gene discovery in mice. Science 2010, 330, 1104–1107. [Google Scholar] [CrossRef]
- Rad, R.; Rad, L.; Wang, W.; Strong, A.; Ponstingl, H.; Bronner, I.F.; Mayho, M.; Steiger, K.; Weber, J.; Hieber, M.; et al. A conditional piggyBac transposition system for genetic screening in mice identifies oncogenic networks in pancreatic cancer. Nat. Genet. 2015, 47, 47–56. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [Green Version]
- Mojica, F.J.; Diez-Villasenor, C.; Soria, E.; Juez, G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol. 2000, 36, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.Y.; Dai, J.X.; Zhou, H.H.; Liu, Z.Q.; Jin, W.L. Brain tumor modeling using the CRISPR/Cas9 system: State of the art and view to the future. Oncotarget 2016, 7, 33461–33471. [Google Scholar] [CrossRef] [PubMed]
- Zuckermann, M.; Hovestadt, V.; Knobbe-Thomsen, C.B.; Zapatka, M.; Northcott, P.A.; Schramm, K.; Belic, J.; Jones, D.T.; Tschida, B.; Moriarity, B.; et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat. Commun. 2015, 6, 7391. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef]
- Koike-Yusa, H.; Li, Y.; Tan, E.P.; Velasco-Herrera Mdel, C.; Yusa, K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014, 32, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Chow, R.D.; Guzman, C.D.; Wang, G.; Schmidt, F.; Youngblood, M.W.; Ye, L.; Errami, Y.; Dong, M.B.; Martinez, M.A.; Zhang, S.; et al. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat. Neurosci. 2017, 20, 1329–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldrini, B.; Curiel-García, Á.; Marques, C.; Matia, V.; Uluçkan, Ö.; Graña-Castro, O.; Torres-Ruiz, R.; Rodriguez-Perales, S.; Huse, J.T.; Squatrito, M. Somatic genome editing with the RCAS-TVA-CRISPR-Cas9 system for precision tumor modeling. Nat. Commun. 2018, 9, 1466. [Google Scholar] [CrossRef] [PubMed]
- Squatrito, M.; Brennan, C.W.; Helmy, K.; Huse, J.T.; Petrini, J.H.; Holland, E.C. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell 2010, 18, 619–629. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cancer gene(s) | Technology | Latency | Pathology | Reference |
|---|---|---|---|---|
| Egfr activation (including vIII), Cdkn2a | RCAS, TVA | 2 months for tumors in 13/25 mice | Low grade glioma | [35] |
| KrasG12D, Akt | RCAS | 9 weeks for tumors in 7/27 mice | GBM | [19] |
| Trp53 and Pten loss | hGFAP-cre (diverse glial cell types) | 30 weeks, median latency | Grade III and Grade IV gliomas | [37] |
| EGFRvIII, Ink4a, and Pten | Cre injections in basal ganglia | 7 weeks, median latency | GBM | [39] |
| Idh1R132H | Nestin-creERT2 (neural stem cells and progenitors) | 6 weeks for precursors with 100% incidence | Glioma precursors | [42] |
| Trp53, Pten and Rb loss | Adenoviral-cre | 8 months, mean latency | GBM (Trp53 and Pten); PNET (Trp53, Pten and Rb) | [44] |
| Nf1, Pten and Trp53 loss | hGFAP-cre (diverse glial cell types) | 35 weeks, median latency | Grade III and Grade IV gliomas | [38] |
| Trp53/Nf1 loss; or H-ras overexpression with Trp53 loss | SynI-cre and CamK2a-cre (neurons) | 6-10 weeks for SynI-cre, 9-12 months for CamK2a-cre (full penetrance) | GBM | [46] |
| Nf1, Trp53 and Pten loss | Ascl1-creERTM (neural progenitors and OPCs). | 40 weeks, median latency | GBM | [51] |
| Atrx loss, Trp53 loss and Nras overexpression | Sleeping beauty transposon | 50 days, median latency | GBM | [73] |
| H3.3K27M, and Trp53 loss | PiggyBac transposons, CRISPR-cas9 | 9 months, full penetrance | GBM | [77] |
| Nf1, Pten and Trp53 loss | CRISPR-cas9 | 14 weeks, complete penetrance | GBM | [87] |
| PDGFB expression; Chk2, ATM or Trp53 loss | RCAS, TVA | 60 days, median latency | GBM and low grade gliomas | [96] |
| PDGFB expression; Trp53, Pten or Cdkn2a loss | CRISPR-cas9 | 46-57 days, median latency | GBMs | [95] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noorani, I. Genetically Engineered Mouse Models of Gliomas: Technological Developments for Translational Discoveries. Cancers 2019, 11, 1335. https://doi.org/10.3390/cancers11091335
Noorani I. Genetically Engineered Mouse Models of Gliomas: Technological Developments for Translational Discoveries. Cancers. 2019; 11(9):1335. https://doi.org/10.3390/cancers11091335
Chicago/Turabian StyleNoorani, Imran. 2019. "Genetically Engineered Mouse Models of Gliomas: Technological Developments for Translational Discoveries" Cancers 11, no. 9: 1335. https://doi.org/10.3390/cancers11091335
APA StyleNoorani, I. (2019). Genetically Engineered Mouse Models of Gliomas: Technological Developments for Translational Discoveries. Cancers, 11(9), 1335. https://doi.org/10.3390/cancers11091335