XIAP as a Target of New Small Organic Natural Molecules Inducing Human Cancer Cell Death

 , , , ,
, , , ,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
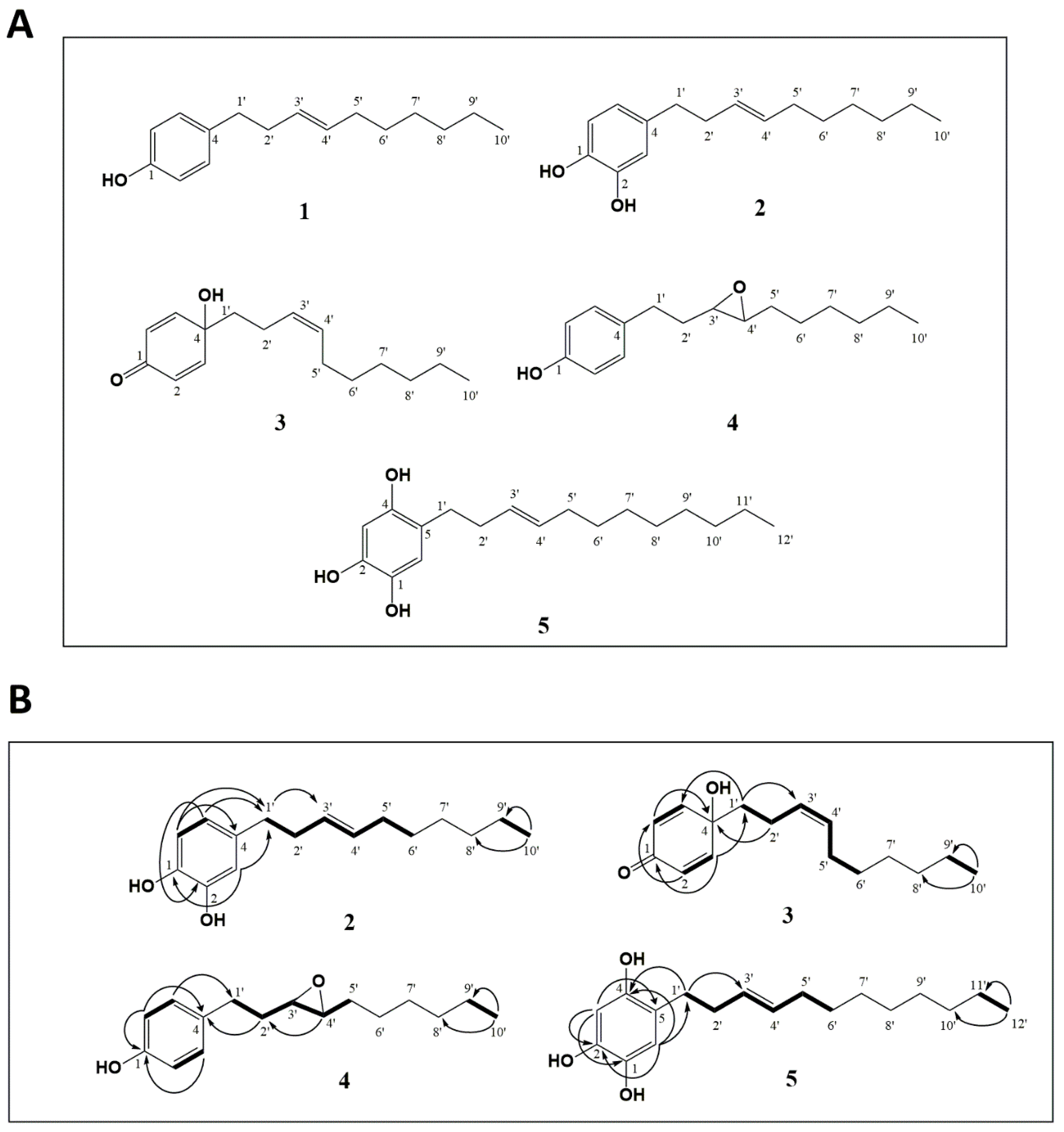
2.1. Structural Identification of New Piper Genus-Derived Compounds
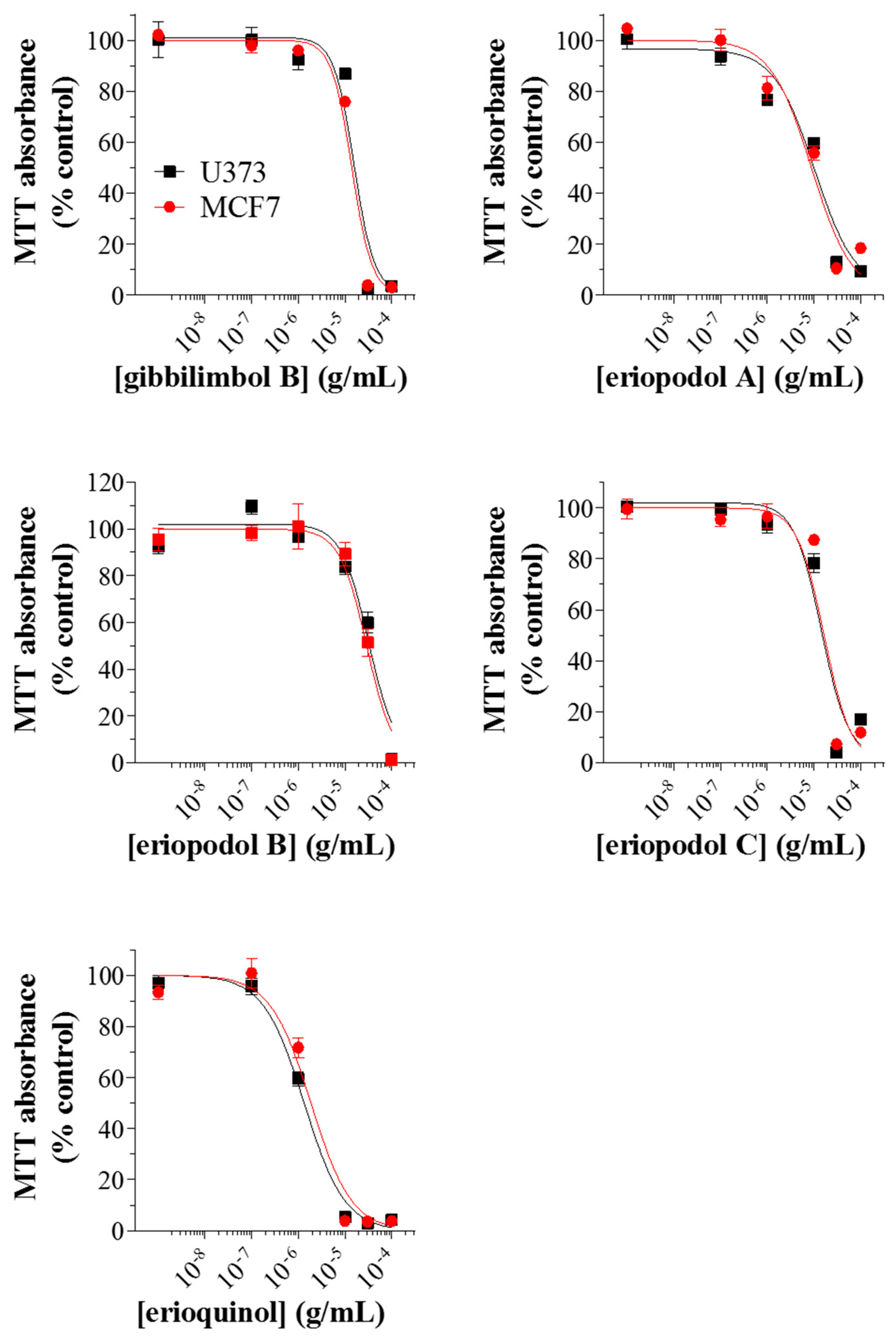
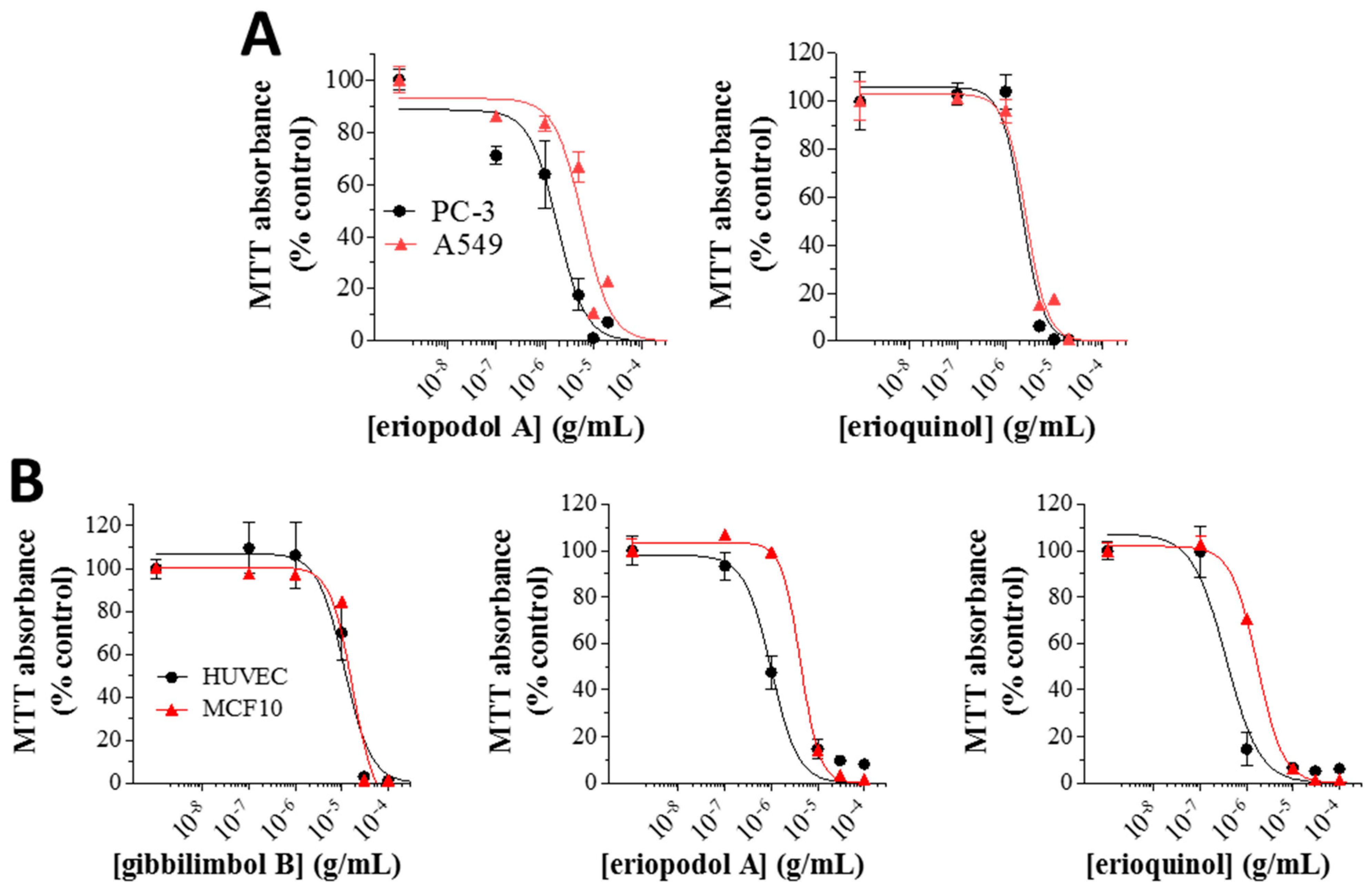
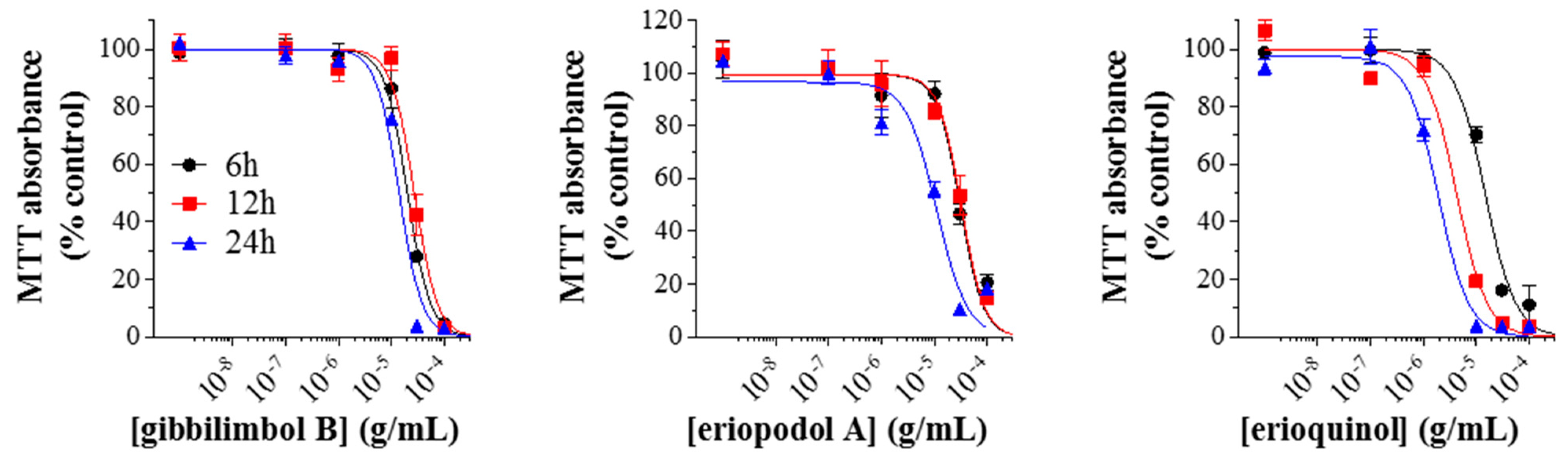
2.2. Piper Genus-Derived Compounds Exhibit Cytotoxic Effects
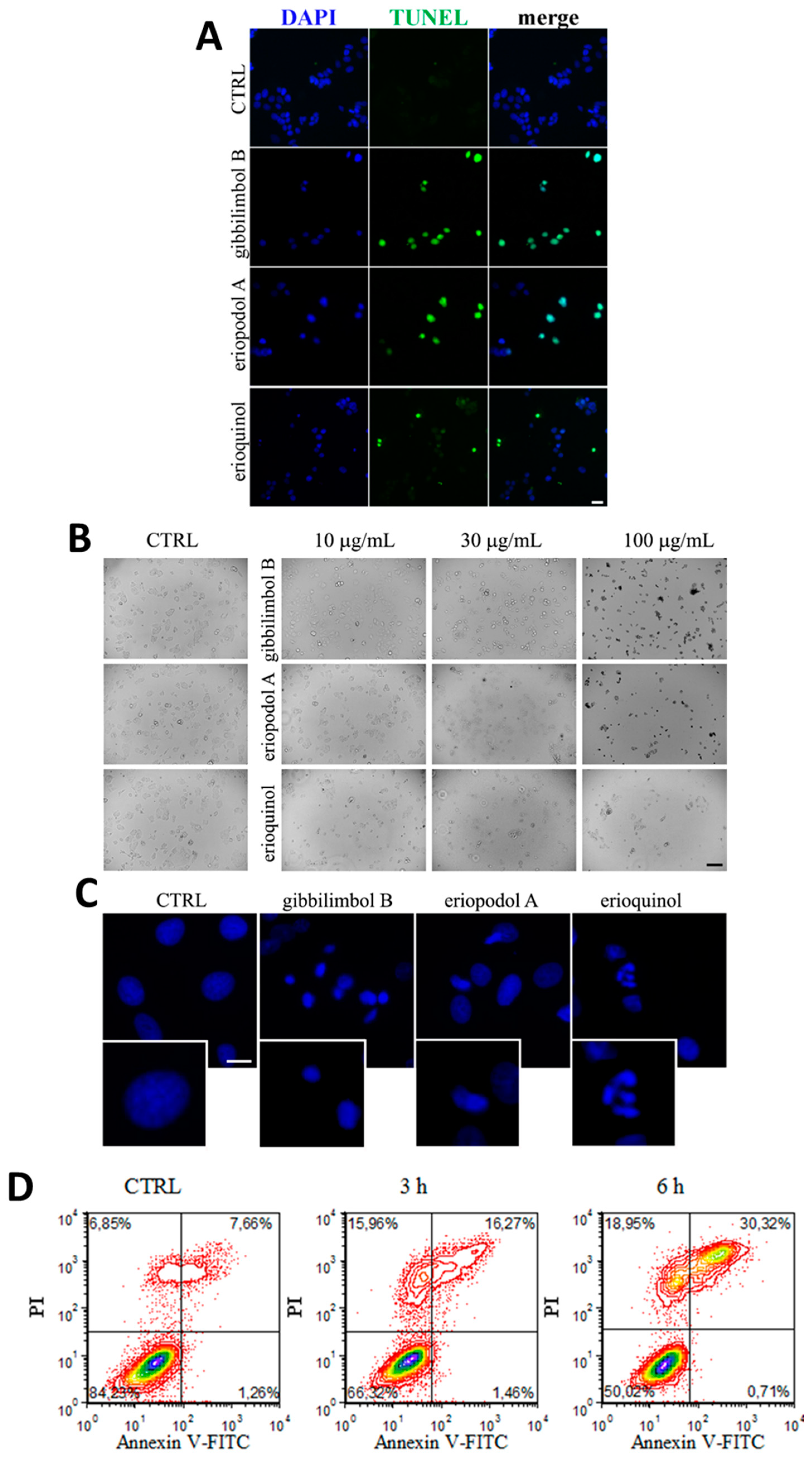
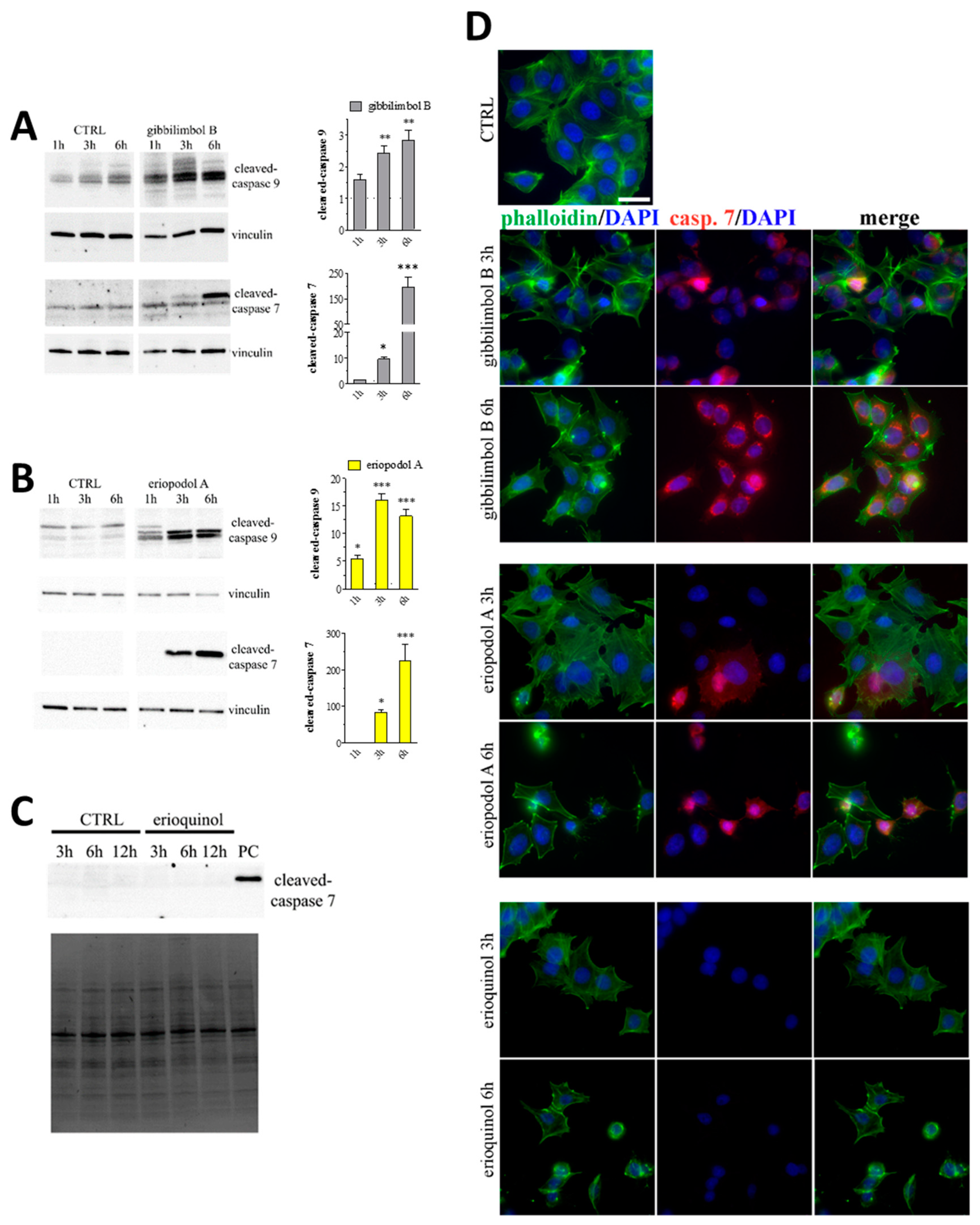
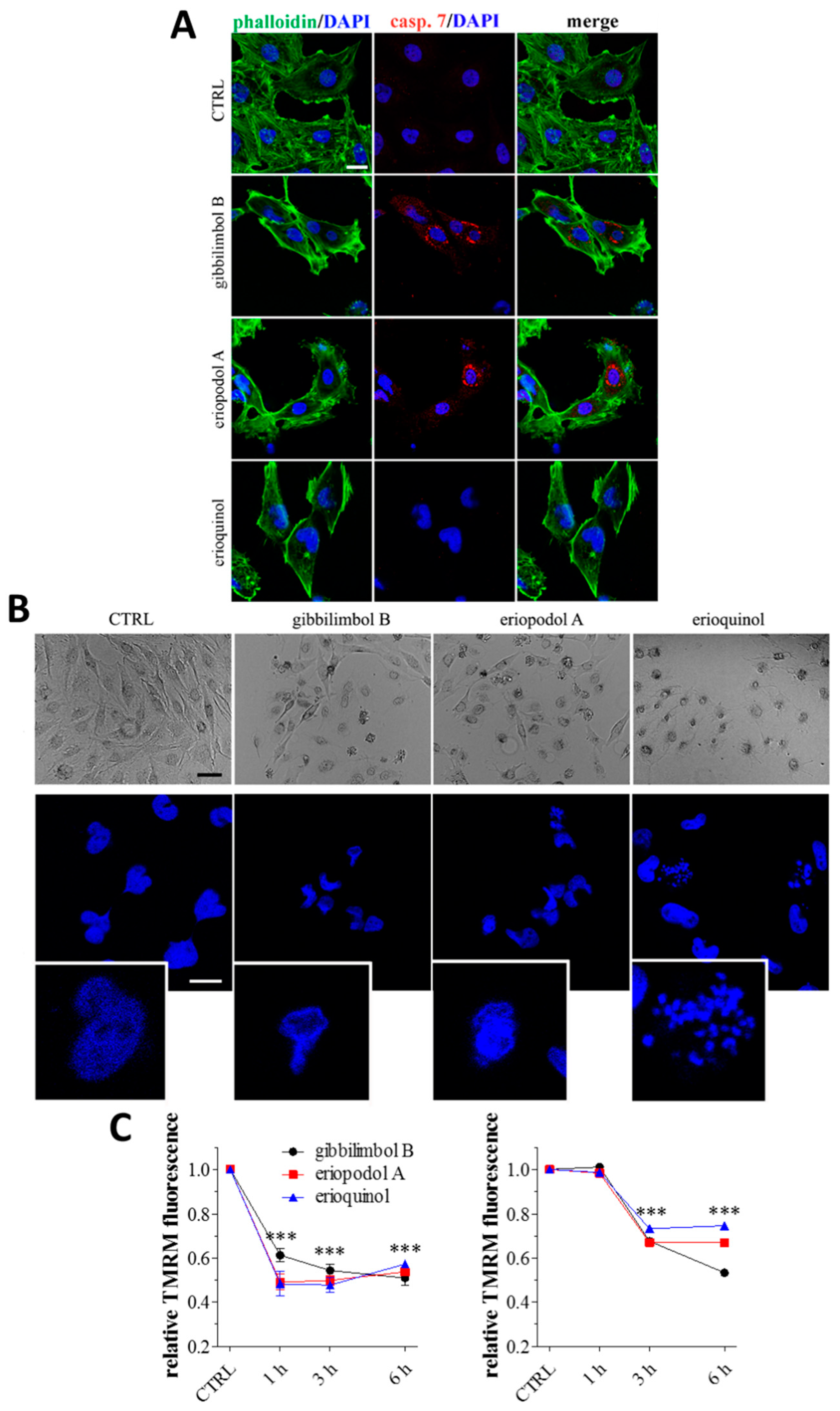
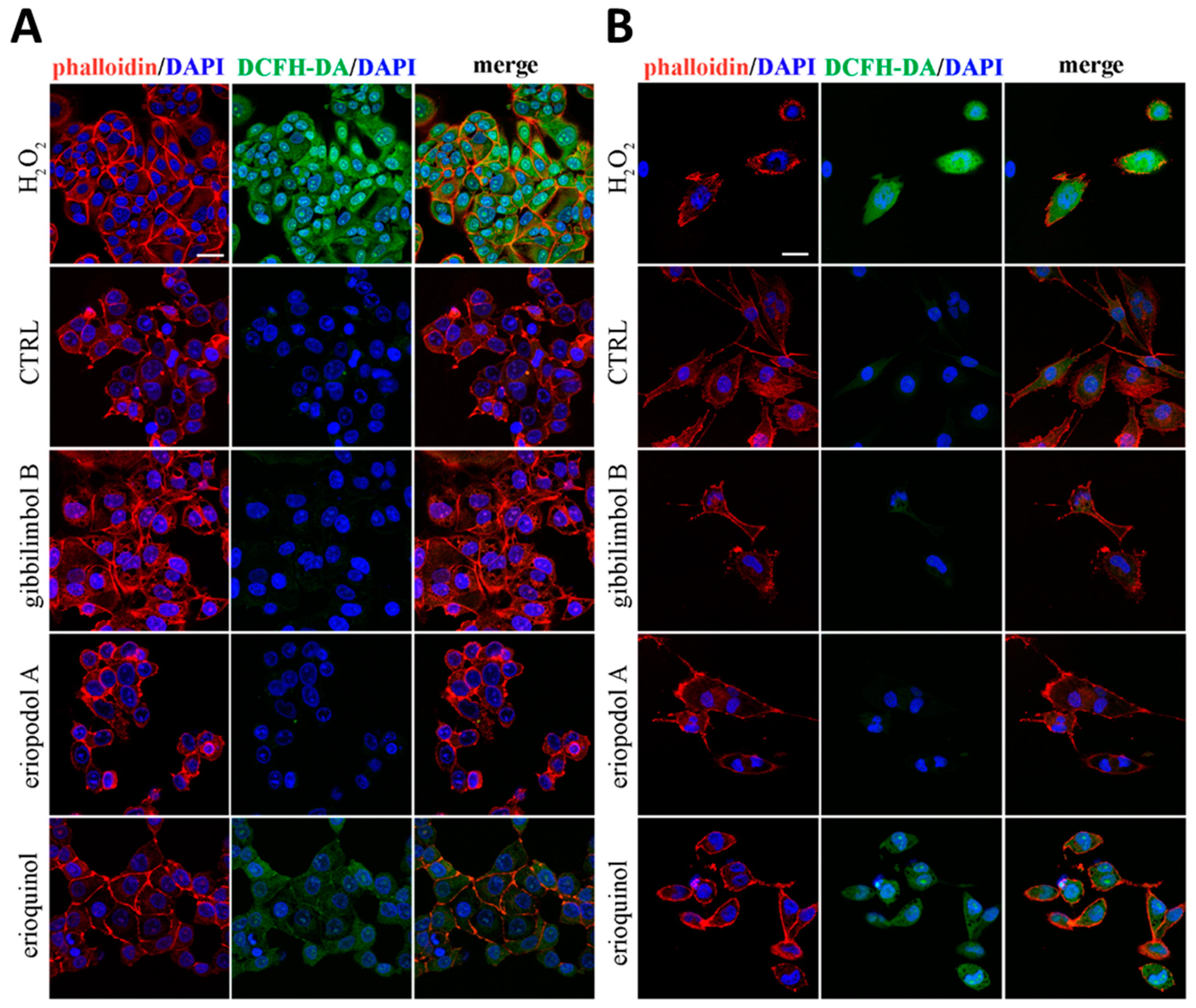
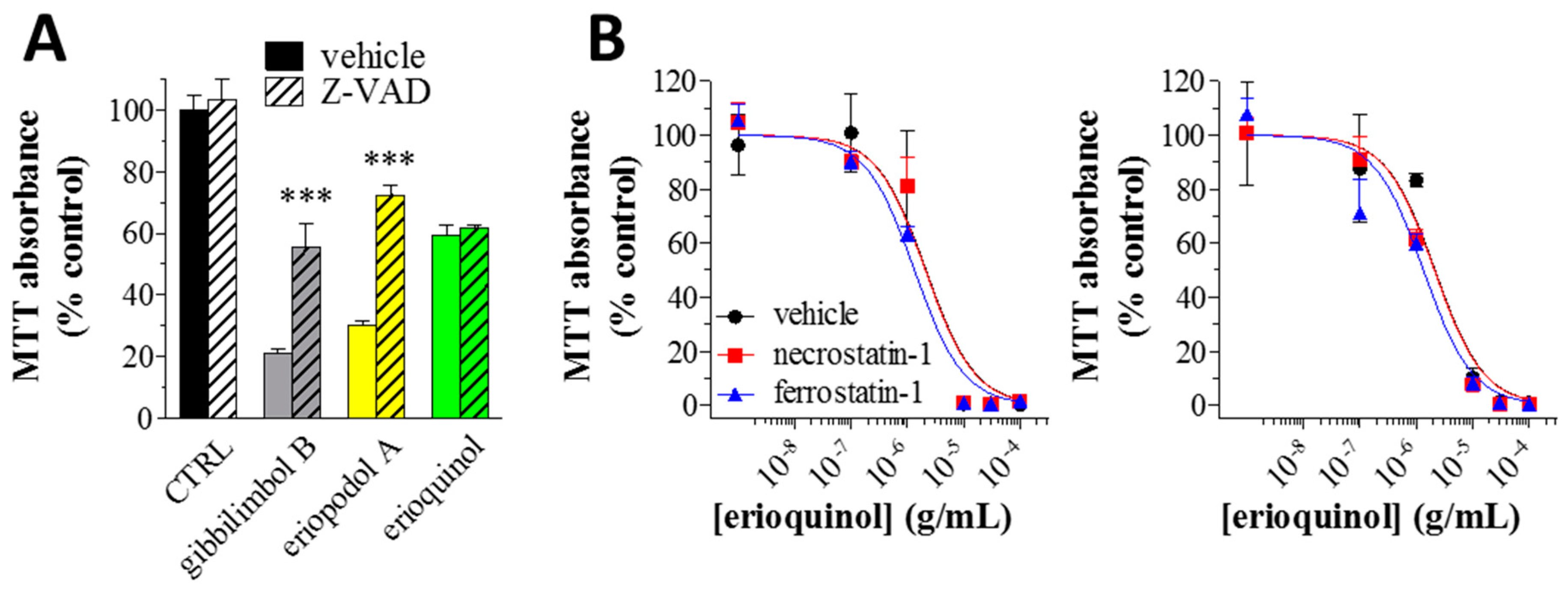
2.3. Piper Genus-Derived Compounds Induce Cell Death
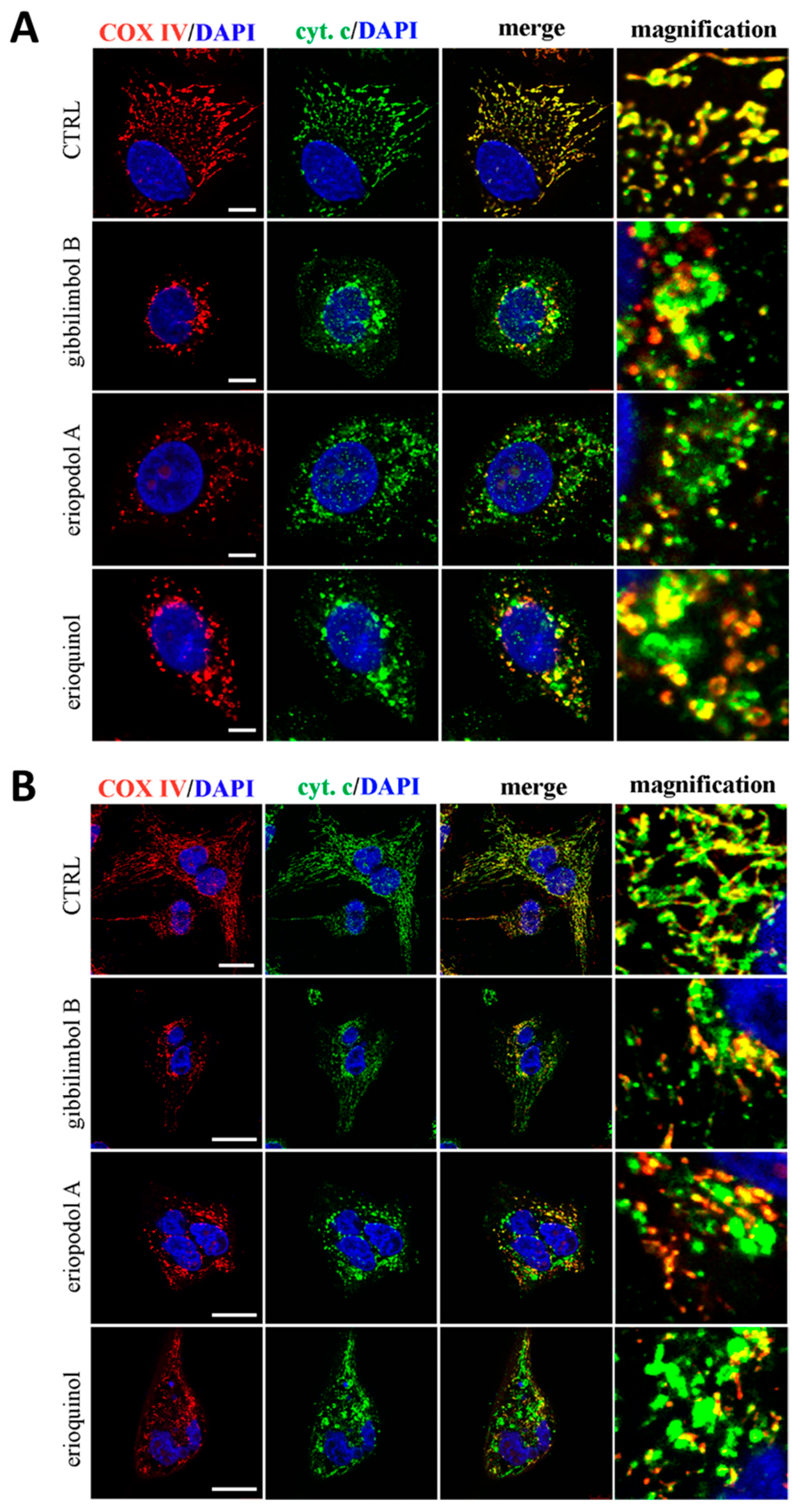
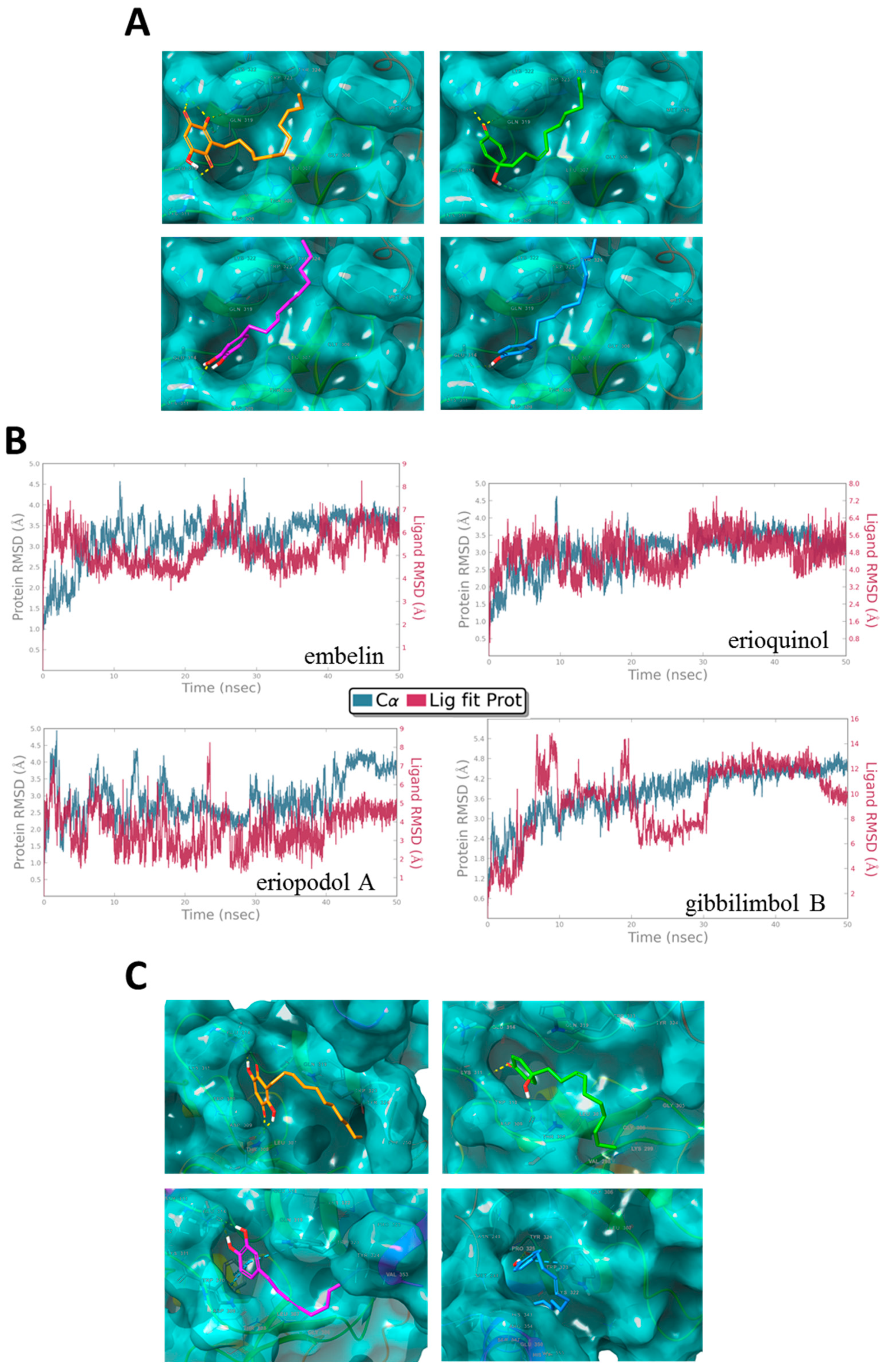
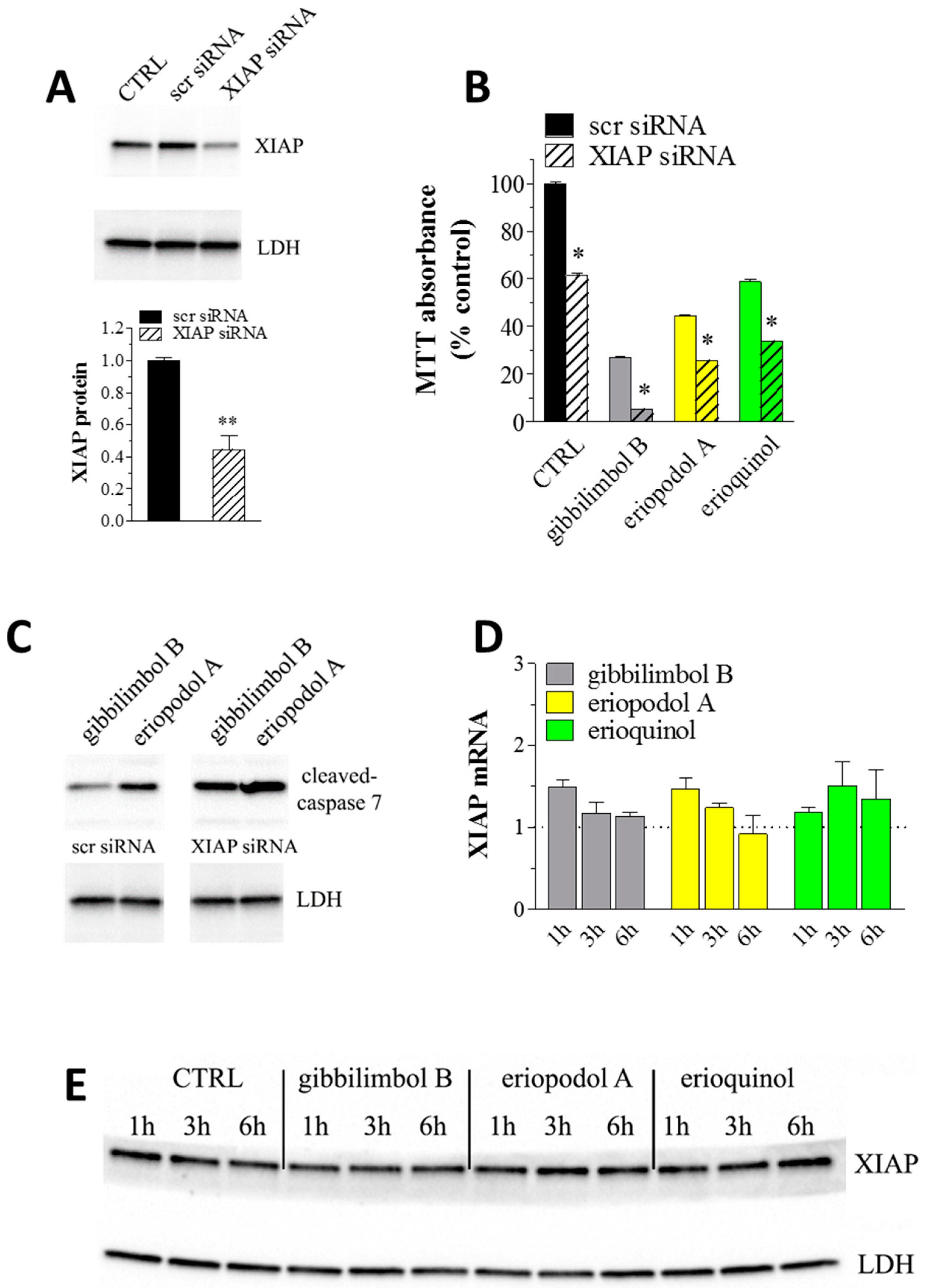
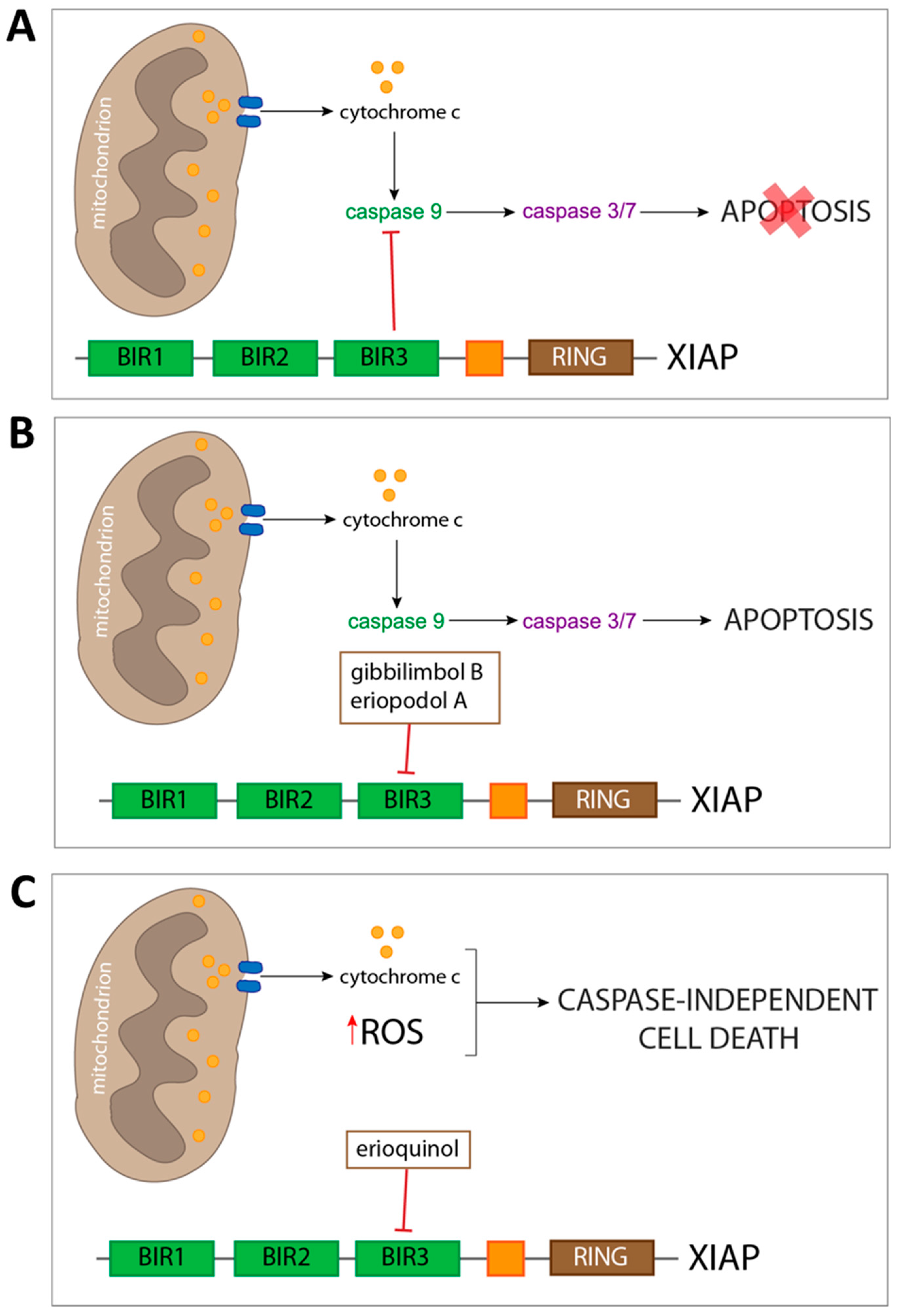
2.4. XIAP as a Molecular Target of Piper Genus-Derived Compounds
3. Materials and Methods
3.1. Extraction and Isolation of Natural Compounds
3.2. General Chemical Methods
3.3. Cell Culture and Chemicals
3.4. MTT Assay
3.5. TUNEL Assay
3.6. Immunofluorescence Microscopy Analysis
3.7. Annexin V Staining
3.8. Western Blotting
3.9. Mitochondrial Membrane Potential Analysis
3.10. Measurement of ROS
3.11. Molecular Modeling
3.12. RNA Interference
3.13. Real-Time PCR
3.14. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gurevich, E.V.; Gurevich, V.V. Therapeutic potential of small molecules and engineered proteins. Handb. Exp. Pharmacol. 2014, 219, 1–12. [Google Scholar]
- Cheng, B.; Yuan, W.E.; Su, J.; Liu, Y.; Chen, J. Recent advances in small molecule based cancer immunotherapy. Eur. J. Med. Chem. 2018, 157, 582–598. [Google Scholar] [CrossRef]
- Huck, B.R.; Kotzner, L.; Urbahns, K. Small Molecules Drive Big Improvements in Immuno-Oncology Therapies. Angew. Chem. Int. Ed. Engl. 2018, 57, 4412–4428. [Google Scholar] [CrossRef] [Green Version]
- Schiavone, S.; Trabace, L. Small Molecules: Therapeutic Application in Neuropsychiatric and Neurodegenerative Disorders. Molecules 2018, 23, 411. [Google Scholar] [CrossRef]
- Dhanak, D.; Edwards, J.P.; Nguyen, A.; Tummino, P.J. Small-Molecule Targets in Immuno-Oncology. Cell Chem. Biol. 2017, 24, 1148–1160. [Google Scholar] [CrossRef] [Green Version]
- Shen, B. A New Golden Age of Natural Products Drug Discovery. Cell 2015, 163, 1297–1300. [Google Scholar] [CrossRef] [Green Version]
- Nobili, S.; Lippi, D.; Witort, E.; Donnini, M.; Bausi, L.; Mini, E.; Capaccioli, S. Natural compounds for cancer treatment and prevention. Pharmacol. Res. 2009, 59, 365–378. [Google Scholar] [CrossRef]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Catalani, E.; Proietti Serafini, F.; Zecchini, S.; Picchietti, S.; Fausto, A.M.; Marcantoni, E.; Buonanno, F.; Ortenzi, C.; Perrotta, C.; Cervia, D. Natural products from aquatic eukaryotic microorganisms for cancer therapy: Perspectives on anti-tumour properties of ciliate bioactive molecules. Pharmacol. Res. 2016, 113, 409–420. [Google Scholar] [CrossRef]
- Carocho, M.; Ferreira, I.C. The role of phenolic compounds in the fight against cancer—A review. Anticancer Agents Med. Chem. 2013, 13, 1236–1258. [Google Scholar] [CrossRef]
- Guerra, A.R.; Duarte, M.F.; Duarte, I.F. Targeting Tumor Metabolism with Plant-Derived Natural Products: Emerging Trends in Cancer Therapy. J. Agric. Food Chem. 2018, 66, 10663–10685. [Google Scholar] [CrossRef]
- Jafari, S.; Saeidnia, S.; Abdollahi, M. Role of natural phenolic compounds in cancer chemoprevention via regulation of the cell cycle. Curr. Pharm. Biotechnol. 2014, 15, 409–421. [Google Scholar] [CrossRef]
- Tungmunnithum, D.; Thongboonyou, A.; Pholboon, A.; Yangsabai, A. Flavonoids and Other Phenolic Compounds from Medicinal Plants for Pharmaceutical and Medical Aspects: An Overview. Medicines 2018, 5, 93. [Google Scholar] [CrossRef]
- Parmar, V.S.; Jain, S.C.; Gupta, S.; Talwar, S.; Rajwanshi, V.K.; Kumar, R.; Azim, A.; Malhotra, S.; Kumar, N.; Jain, R.; et al. Polyphenols and alkaloids from Piper species. Phytochemistry 1998, 49, 1069–1078. [Google Scholar] [CrossRef]
- Xiang, C.P.; Shi, Y.N.; Liu, F.F.; Li, H.Z.; Zhang, Y.J.; Yang, C.R.; Xu, M. A Survey of the Chemical Compounds of Piper spp. (Piperaceae) and Their Biological Activities. Nat. Prod. Commun. 2016, 11, 1403–1408. [Google Scholar] [CrossRef] [Green Version]
- Valdivia, C.; Marquez, N.; Eriksson, J.; Vilaseca, A.; Munoz, E.; Sterner, O. Bioactive alkenylphenols from Piper obliquum. Bioorg. Med. Chem. 2008, 16, 4120–4126. [Google Scholar] [CrossRef]
- Yang, S.X.; Sun, Q.Y.; Yang, F.M.; Hu, G.W.; Luo, J.F.; Wang, Y.H.; Long, C.L. Sarmentosumols A to F, new mono- and dimeric alkenylphenols from Piper sarmentosum. Planta Med. 2013, 79, 693–696. [Google Scholar] [CrossRef]
- Orjala, J.; Mian, P.; Rali, T.; Sticher, O. Gibbilimbols A-D, cytotoxic and antibacterial alkenylphenols from Piper gibbilimbum. J. Nat. Prod. 1998, 61, 939–941. [Google Scholar] [CrossRef]
- Yoshida, N.C.; Benedetti, A.M.; dos Santos, R.A.; Ramos, C.S.; Batista, R.; Yamaguchi, L.F.; Kato, M.J. Alkenylphenols from Piper dilatatum and P. diospyrifolium. Phytochem. Lett. 2018, 25, 136–140. [Google Scholar] [CrossRef]
- De Oliveira, A.; Mesquita, J.T.; Tempone, A.G.; Lago, J.H.G.; Guimaraes, E.F.; Kato, M.J. Leishmanicidal activity of an alkenylphenol from Piper malacophyllum is related to plasma membrane disruption. Exp. Parasitol. 2012, 132, 383–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezerra, D.P.; Pessoa, C.; de Moraes, M.O.; Saker-Neto, N.; Silveira, E.R.; Costa-Lotufo, L.V. Overview of the therapeutic potential of piplartine (piperlongumine). Eur. J. Pharm. Sci. 2013, 48, 453–463. [Google Scholar] [CrossRef] [PubMed]
- D’Sousa Costa, C.O.; Araujo Neto, J.H.; Baliza, I.R.S.; Dias, R.B.; Valverde, L.F.; Vidal, M.T.A.; Sales, C.B.S.; Rocha, C.A.G.; Moreira, D.R.M.; Soares, M.B.P.; et al. Novel piplartine-containing ruthenium complexes: Synthesis, cell growth inhibition, apoptosis induction and ROS production on HCT116 cells. Oncotarget 2017, 8, 104367–104392. [Google Scholar] [CrossRef] [PubMed]
- Piska, K.; Gunia-Krzyzak, A.; Koczurkiewicz, P.; Wojcik-Pszczola, K.; Pekala, E. Piperlongumine (piplartine) as a lead compound for anticancer agents—Synthesis and properties of analogues: A mini-review. Eur. J. Med. Chem. 2018, 156, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Benfica, P.L.; Avila, R.I.; Rodrigues, B.D.S.; Cortez, A.P.; Batista, A.C.; Gaeti, M.P.N.; Lima, E.M.; Rezende, K.R.; Valadares, M.C. 4-Nerolidylcatechol: Apoptosis by mitochondrial mechanisms with reduction in cyclin D1 at G0/G1 stage of the chronic myelogenous K562 cell line. Pharm. Biol. 2017, 55, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Cortez, A.P.; de Avila, R.I.; da Cunha, C.R.; Santos, A.P.; Menegatti, R.; Rezende, K.R.; Valadares, M.C. 4-Nerolidylcatechol analogues as promising anticancer agents. Eur. J. Pharmacol. 2015, 765, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gundala, S.R.; Yang, C.; Mukkavilli, R.; Paranjpe, R.; Brahmbhatt, M.; Pannu, V.; Cheng, A.; Reid, M.D.; Aneja, R. Hydroxychavicol, a betel leaf component, inhibits prostate cancer through ROS-driven DNA damage and apoptosis. Toxicol. Appl. Pharmacol. 2014, 280, 86–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemamalini, V.; Velayutham, D.P.M.; Lakshmanan, L.; Muthusamy, K.; Sivaramakrishnan, S.; Premkumar, K. Inhibitory potential of Hydroxychavicol on Ehrlich ascites carcinoma model and in silico interaction on cancer targets. Nat. Prod. Res. 2018, 1–6. [Google Scholar] [CrossRef]
- Munoz, D.R.; Sandoval-Hernandez, A.G.; Delgado, W.A.; Arboleda, G.H.; Cuca, L.E. In vitro anticancer screening of Colombian plants from Piper genus (Piperaceae). J. Pharmacogn. Phytother. 2018, 10, 174–181. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- Lalaoui, N.; Vaux, D.L. Recent advances in understanding inhibitor of apoptosis proteins. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Rathore, R.; McCallum, J.E.; Varghese, E.; Florea, A.M.; Busselberg, D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 2017, 22, 898–919. [Google Scholar] [CrossRef] [PubMed]
- Schimmer, A.D.; Dalili, S.; Batey, R.A.; Riedl, S.J. Targeting XIAP for the treatment of malignancy. Cell Death Differ. 2006, 13, 179–188. [Google Scholar] [CrossRef]
- Tamanini, E.; Buck, I.M.; Chessari, G.; Chiarparin, E.; Day, J.E.H.; Frederickson, M.; Griffiths-Jones, C.M.; Hearn, K.; Heightman, T.D.; Iqbal, A.; et al. Discovery of a Potent Nonpeptidomimetic, Small-Molecule Antagonist of Cellular Inhibitor of Apoptosis Protein 1 (cIAP1) and X-Linked Inhibitor of Apoptosis Protein (XIAP). J. Med. Chem. 2017, 60, 4611–4625. [Google Scholar] [CrossRef]
- Sun, H.; Stuckey, J.A.; Nikolovska-Coleska, Z.; Qin, D.; Meagher, J.L.; Qiu, S.; Lu, J.; Yang, C.Y.; Saito, N.G.; Wang, S. Structure-based design, synthesis, evaluation, and crystallographic studies of conformationally constrained Smac mimetics as inhibitors of the X-linked inhibitor of apoptosis protein (XIAP). J. Med. Chem. 2008, 51, 7169–7180. [Google Scholar] [CrossRef]
- Fakler, M.; Loeder, S.; Vogler, M.; Schneider, K.; Jeremias, I.; Debatin, K.M.; Fulda, S. Small molecule XIAP inhibitors cooperate with TRAIL to induce apoptosis in childhood acute leukemia cells and overcome Bcl-2-mediated resistance. Blood 2009, 113, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M.; Walczak, H.; Stadel, D.; Haas, T.L.; Genze, F.; Jovanovic, M.; Bhanot, U.; Hasel, C.; Moller, P.; Gschwend, J.E.; et al. Small molecule XIAP inhibitors enhance TRAIL-induced apoptosis and antitumor activity in preclinical models of pancreatic carcinoma. Cancer Res. 2009, 69, 2425–2434. [Google Scholar] [CrossRef]
- Dean, E.J.; Ward, T.; Pinilla, C.; Houghten, R.; Welsh, K.; Makin, G.; Ranson, M.; Dive, C. A small molecule inhibitor of XIAP induces apoptosis and synergises with vinorelbine and cisplatin in NSCLC. Br. J. Cancer 2010, 102, 97–103. [Google Scholar] [CrossRef]
- Obexer, P.; Ausserlechner, M.J. X-linked inhibitor of apoptosis protein—A critical death resistance regulator and therapeutic target for personalized cancer therapy. Front. Oncol. 2014, 4, 197. [Google Scholar] [CrossRef]
- Cong, H.; Xu, L.; Wu, Y.; Qu, Z.; Bian, T.; Zhang, W.; Xing, C.; Zhuang, C. Inhibitor of Apoptosis Protein (IAP) Antagonists in Anticancer Agent Discovery: Current Status and Perspectives. J. Med. Chem. 2019. [Google Scholar] [CrossRef]
- Fulda, S. Promises and Challenges of Smac Mimetics as Cancer Therapeutics. Clin. Cancer Res. 2015, 21, 5030–5036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimmer, A.D.; Welsh, K.; Pinilla, C.; Wang, Z.; Krajewska, M.; Bonneau, M.J.; Pedersen, I.M.; Kitada, S.; Scott, F.L.; Bailly-Maitre, B.; et al. Small-molecule antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer Cell 2004, 5, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Masaki, M.E.; Harumoto, T.; Terazima, M.N.; Miyake, A.; Usuki, Y.; Iio, H. Climacostol, a defense toxin of the heterotrich ciliate Climacostomum virens against predators. Tetrahedron Lett. 1999, 40, 8227–8229. [Google Scholar] [CrossRef]
- Varela, M.T.; Dias, R.Z.; Martins, L.F.; Ferreira, D.D.; Tempone, A.G.; Ueno, A.K.; Lago, J.H.G.; Fernandes, J.P.S. Gibbilimbol analogues as antiparasitic agents-Synthesis and biological activity against Trypanosoma cruzi and Leishmania (L.) infantum. Bioorg. Med. Chem. Lett. 2016, 26, 1180–1183. [Google Scholar] [CrossRef] [PubMed]
- Carreno, M.C.; Gonzalez-Lopez, M.; Urbano, A. Oxidative de-aromatization of para-alkyl phenols into para-peroxyquinols and para-quinols mediated by oxone as a source of singlet oxygen. Angew. Chem. Int. Ed. 2006, 45, 2737–2741. [Google Scholar] [CrossRef]
- Freitas, G.C.; Batista, J.M.; Franchi, G.C.; Nowill, A.E.; Yamaguchi, L.F.; Vilcachagua, J.D.; Favaro, D.C.; Furlan, M.; Guimaraes, E.F.; Jeffrey, C.S.; et al. Cytotoxic non-aromatic B-ring flavanones from Piper carniconnectivum C. DC. Phytochemistry 2014, 97, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Nishino, C.; Kobayashi, K.; Fukushima, M. Halleridone, a cytotoxic constituent from Cornus controversa. J. Nat. Prod. 1988, 51, 1281–1282. [Google Scholar] [CrossRef]
- Bradshaw, T.D.; Matthews, C.S.; Cookson, J.; Chew, E.H.; Shah, M.; Bailey, K.; Monks, A.; Harris, E.; Westwell, A.D.; Wells, G.; et al. Elucidation of thioredoxin as a molecular target for antitumor quinols. Cancer Res. 2005, 65, 3911–3919. [Google Scholar] [CrossRef]
- Berry, J.M.; Bradshaw, T.D.; Fichtner, I.; Ren, R.; Schwalbe, C.H.; Wells, G.; Chew, E.H.; Stevens, M.F.; Westwell, A.D. Quinols as novel therapeutic agents. 2.(1) 4-(1-Arylsulfonylindol-2-yl)-4-hydroxycyclohexa-2,5-dien-1-ones and related agents as potent and selective antitumor agents. J. Med. Chem. 2005, 48, 639–644. [Google Scholar] [CrossRef]
- McCarroll, A.J.; Bradshaw, T.D.; Westwell, A.D.; Matthews, C.S.; Stevens, M.F. Quinols as novel therapeutic agents. 7.1 Synthesis of antitumor 4-[1-(arylsulfonyl-1H-indol-2-yl)]-4-hydroxycyclohexa-2,5-dien-1-ones by Sonogashira reactions. J. Med. Chem. 2007, 50, 1707–1710. [Google Scholar] [CrossRef] [PubMed]
- Abu Bakar, A.; Akhtar, M.N.; Mohd Ali, N.; Yeap, S.K.; Quah, C.K.; Loh, W.S.; Alitheen, N.B.; Zareen, S.; Ul-Haq, Z.; Shah, S.A.A. Design, Synthesis and Docking Studies of Flavokawain B Type Chalcones and Their Cytotoxic Effects on MCF-7 and MDA-MB-231 Cell Lines. Molecules 2018, 23, 616. [Google Scholar] [CrossRef] [PubMed]
- Sriwiriyajan, S.; Sukpondma, Y.; Srisawat, T.; Madla, S.; Graidist, P. (-)-Kusunokinin and piperloguminine from Piper nigrum: An alternative option to treat breast cancer. Biomed. Pharm. 2017, 92, 732–743. [Google Scholar] [CrossRef] [PubMed]
- Sriwiriyajan, S.; Ninpesh, T.; Sukpondma, Y.; Nasomyon, T.; Graidist, P. Cytotoxicity Screening of Plants of Genus Piper in Breast Cancer Cell Lines. Trop. J. Pharm. Res. 2014, 13, 921–928. [Google Scholar] [CrossRef]
- Fan, L.; Cao, X.; Yan, H.; Wang, Q.; Tian, X.; Zhang, L.; He, X.; Borjihan, G.; Morigen. The synthetic antihyperlipidemic drug potassium piperate selectively kills breast cancer cells through inhibiting G1-S-phase transition and inducing apoptosis. Oncotarget 2017, 8, 47250–47268. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Lee, D.E.; Shim, M.K.; Jang, E.H.; Lee, J.K.; Jeong, S.Y.; Kim, J.H. Piperlongumine inhibits TGF-beta-induced epithelial-to-mesenchymal transition by modulating the expression of E-cadherin, Snail1, and Twist1. Eur. J. Pharmacol. 2017, 812, 243–249. [Google Scholar] [CrossRef] [PubMed]
- De Souza Grinevicius, V.M.; Kviecinski, M.R.; Santos Mota, N.S.; Ourique, F.; Porfirio Will Castro, L.S.; Andreguetti, R.R.; Gomes Correia, J.F.; Filho, D.W.; Pich, C.T.; Pedrosa, R.C. Piper nigrum ethanolic extract rich in piperamides causes ROS overproduction, oxidative damage in DNA leading to cell cycle arrest and apoptosis in cancer cells. J. Ethnopharmacol. 2016, 189, 139–147. [Google Scholar] [CrossRef]
- Deng, Y.; Sriwiriyajan, S.; Tedasen, A.; Hiransai, P.; Graidist, P. Anti-cancer effects of Piper nigrum via inducing multiple molecular signaling in vivo and in vitro. J. Ethnopharmacol. 2016, 188, 87–95. [Google Scholar] [CrossRef]
- Da Nobrega, F.R.; Ozdemir, O.; Nascimento Sousa, S.C.S.; Barboza, J.N.; Turkez, H.; de Sousa, D.P. Piplartine Analogues and Cytotoxic Evaluation against Glioblastoma. Molecules 2018, 23, 1382. [Google Scholar] [CrossRef]
- Abdul Rahman, A.; Jamal, A.R.; Harun, R.; Mohd Mokhtar, N.; Wan Ngah, W.Z. Gamma-tocotrienol and hydroxy-chavicol synergistically inhibits growth and induces apoptosis of human glioma cells. BMC Complement. Altern. Med. 2014, 14, 213. [Google Scholar] [CrossRef]
- Subramanian, U.; Poongavanam, S.; Vanisree, A.J. Studies on the neuroprotective role of Piper longum in C6 glioma induced rats. Investing. New Drugs 2010, 28, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Buonanno, F.; Catalani, E.; Cervia, D.; Proietti Serafini, F.; Picchietti, S.; Fausto, A.M.; Giorgi, S.; Lupidi, G.; Rossi, F.V.; Marcantoni, E.; et al. Bioactivity and Structural Properties of Novel Synthetic Analogues of the Protozoan Toxin Climacostol. Toxins 2019, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Caspase-independent cell death: Leaving the set without the final cut. Oncogene 2008, 27, 6452–6461. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Chan, F.K.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. The Coming Decade of Cell Death Research: Five Riddles. Cell 2019, 177, 1094–1107. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhang, R.; Wu, F.; Zhai, L.; Wang, K.; Xiao, M.; Xie, T.; Sui, X. Non-apoptotic cell death in malignant tumor cells and natural compounds. Cancer Lett. 2018, 420, 210–227. [Google Scholar] [CrossRef]
- Holze, C.; Michaudel, C.; Mackowiak, C.; Haas, D.A.; Benda, C.; Hubel, P.; Pennemann, F.L.; Schnepf, D.; Wettmarshausen, J.; Braun, M.; et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat. Immunol. 2018, 19, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Shiozaki, E.; Srinivasula, S.M.; Wu, Q.; Datta, P.; Alnemri, E.S.; Shi, Y. Structural basis of caspase-7 inhibition by XIAP. Cell 2001, 104, 769–780. [Google Scholar] [CrossRef]
- Shiozaki, E.N.; Chai, J.; Rigotti, D.J.; Riedl, S.J.; Li, P.; Srinivasula, S.M.; Alnemri, E.S.; Fairman, R.; Shi, Y. Mechanism of XIAP-mediated inhibition of caspase-9. Mol. Cell 2003, 11, 519–527. [Google Scholar] [CrossRef]
- Suzuki, Y.; Nakabayashi, Y.; Nakata, K.; Reed, J.C.; Takahashi, R. X-linked inhibitor of apoptosis protein (XIAP) inhibits caspase-3 and -7 in distinct modes. J. Biol. Chem. 2001, 276, 27058–27063. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Li, C.; Duan, J.; Lee, S.; Kim, K.; Park, Y.; Yang, Y.; Kim, K.I.; Lim, J.S.; Cheon, C.I.; et al. TRIP-Br1 oncoprotein inhibits autophagy, apoptosis, and necroptosis under nutrient/serum-deprived condition. Oncotarget 2015, 6, 29060–29075. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Burstein, E.; Reffey, S.B.; Bratton, S.B.; Roberts, A.B.; Duckett, C.S. Uncoupling of the signaling and caspase-inhibitory properties of X-linked inhibitor of apoptosis. J. Biol. Chem. 2004, 279, 9023–9029. [Google Scholar] [CrossRef] [PubMed]
- Burstein, E.; Ganesh, L.; Dick, R.D.; van De Sluis, B.; Wilkinson, J.C.; Klomp, L.W.; Wijmenga, C.; Brewer, G.J.; Nabel, G.J.; Duckett, C.S. A novel role for XIAP in copper homeostasis through regulation of MURR1. EMBO J. 2004, 23, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Sanna, M.G.; da Silva Correia, J.; Ducrey, O.; Lee, J.; Nomoto, K.; Schrantz, N.; Deveraux, Q.L.; Ulevitch, R.J. IAP suppression of apoptosis involves distinct mechanisms: The TAK1/JNK1 signaling cascade and caspase inhibition. Mol. Cell Biol. 2002, 22, 1754–1766. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.M.; Wilkinson, A.S.; Davis, N.Y.; Horita, D.A.; Wilkinson, J.C. Nondegradative ubiquitination of apoptosis inducing factor (AIF) by X-linked inhibitor of apoptosis at a residue critical for AIF-mediated chromatin degradation. Biochemistry 2011, 50, 11084–11096. [Google Scholar] [CrossRef] [PubMed]
- Levkau, B.; Garton, K.J.; Ferri, N.; Kloke, K.; Nofer, J.R.; Baba, H.A.; Raines, E.W.; Breithardt, G. xIAP induces cell-cycle arrest and activates nuclear factor-kappaB: New survival pathways disabled by caspase-mediated cleavage during apoptosis of human endothelial cells. Circ. Res. 2001, 88, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Mufti, A.R.; Burstein, E.; Csomos, R.A.; Graf, P.C.; Wilkinson, J.C.; Dick, R.D.; Challa, M.; Son, J.K.; Bratton, S.B.; Su, G.L.; et al. XIAP Is a copper binding protein deregulated in Wilson’s disease and other copper toxicosis disorders. Mol. Cell 2006, 21, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Wicki, S.; Gurzeler, U.; Wei-Lynn Wong, W.; Jost, P.J.; Bachmann, D.; Kaufmann, T. Loss of XIAP facilitates switch to TNFalpha-induced necroptosis in mouse neutrophils. Cell Death Dis. 2016, 7, e2422. [Google Scholar] [CrossRef] [PubMed]
- Yabal, M.; Jost, P.J. XIAP as a regulator of inflammatory cell death: The TNF and RIP3 angle. Mol. Cell. Oncol. 2015, 2, e964622. [Google Scholar] [CrossRef] [PubMed]
- Yabal, M.; Muller, N.; Adler, H.; Knies, N.; Gross, C.J.; Damgaard, R.B.; Kanegane, H.; Ringelhan, M.; Kaufmann, T.; Heikenwalder, M.; et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 2014, 7, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, K.E.; Feltham, R.; Yabal, M.; Conos, S.A.; Chen, K.W.; Ziehe, S.; Grass, C.; Zhan, Y.; Nguyen, T.A.; Hall, C.; et al. XIAP Loss Triggers RIPK3- and Caspase-8-Driven IL-1beta Activation and Cell Death as a Consequence of TLR-MyD88-Induced cIAP1-TRAF2 Degradation. Cell Rep. 2017, 20, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Poojari, R. Embelin—A drug of antiquity: Shifting the paradigm towards modern medicine. Expert Opin. Investig. Drugs 2014, 23, 427–444. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Lee, S.G.; Yang, W.M.; Um, J.Y.; Sethi, G.; Mishra, S.; Shanmugam, M.K.; Ahn, K.S. The Application of Embelin for Cancer Prevention and Therapy. Molecules 2018, 23, 621. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, K.S.; Achkar, I.W.; Kuttikrishnan, S.; Akhtar, S.; Khan, A.Q.; Siveen, K.S.; Uddin, S. Embelin: A benzoquinone possesses therapeutic potential for the treatment of human cancer. Future Med. Chem. 2018, 10, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Nikolovska-Coleska, Z.; Xu, L.; Hu, Z.; Tomita, Y.; Li, P.; Roller, P.P.; Wang, R.; Fang, X.; Guo, R.; Zhang, M.; et al. Discovery of embelin as a cell-permeable, small-molecular weight inhibitor of XIAP through structure-based computational screening of a traditional herbal medicine three-dimensional structure database. J. Med. Chem. 2004, 47, 2430–2440. [Google Scholar] [CrossRef]
- Chessari, G.; Buck, I.M.; Day, J.E.; Day, P.J.; Iqbal, A.; Johnson, C.N.; Lewis, E.J.; Martins, V.; Miller, D.; Reader, M.; et al. Fragment-Based Drug Discovery Targeting Inhibitor of Apoptosis Proteins: Discovery of a Non-Alanine Lead Series with Dual Activity Against cIAP1 and XIAP. J. Med. Chem. 2015, 58, 6574–6588. [Google Scholar] [CrossRef]
- Jin, X.; Lee, K.; Kim, N.H.; Kim, H.S.; Yook, J.I.; Choi, J.; No, K.T. Natural products used as a chemical library for protein-protein interaction targeted drug discovery. J. Mol. Graph. Model. 2018, 79, 46–58. [Google Scholar] [CrossRef]
- Johnson, C.N.; Ahn, J.S.; Buck, I.M.; Chiarparin, E.; Day, J.E.H.; Hopkins, A.; Howard, S.; Lewis, E.J.; Martins, V.; Millemaggi, A.; et al. A Fragment-Derived Clinical Candidate for Antagonism of X-Linked and Cellular Inhibitor of Apoptosis Proteins: 1-(6-[(4-Fluorophenyl)methyl]-5-(hydroxymethyl)-3,3-dimethyl-1H,2H,3H-pyrrolo[3,2-b]pyridin-1-yl)-2-[(2R,5R)-5-methyl-2-([(3R)-3-methylmorpholin-4-yl]methyl)piperazin-1-yl]ethan-1-one (ASTX660). J. Med. Chem. 2018, 61, 7314–7329. [Google Scholar]
- Kashkar, H. X-linked inhibitor of apoptosis: A chemoresistance factor or a hollow promise. Clin. Cancer Res. 2010, 16, 4496–4502. [Google Scholar] [CrossRef]
- Shah, P.; Djisam, R.; Damulira, H.; Aganze, A.; Danquah, M. Embelin inhibits proliferation, induces apoptosis and alters gene expression profiles in breast cancer cells. Pharmacol. Rep. 2016, 68, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Hahm, E.R.; Singh, S.V. Withaferin A-induced apoptosis in human breast cancer cells is associated with suppression of inhibitor of apoptosis family protein expression. Cancer Lett. 2013, 334, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhang, X.; Liu, A.; Liu, S.; Zhang, L.; Wu, B.; Hu, Q. Berberine induces apoptosis in p53-null leukemia cells by down-regulating XIAP at the post-transcriptional level. Cell. Physiol. Biochem. 2013, 32, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.T.; Martins, L.M.; Guimaraes, J.E.; Sambade, C.; Vasconcelos, M.H. Specific downregulation of bcl-2 and xIAP by RNAi enhances the effects of chemotherapeutic agents in MCF-7 human breast cancer cells. Cancer Gene Ther. 2004, 11, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sensintaffar, J.; Scott, F.L.; Peach, R.; Hager, J.H. XIAP is not required for human tumor cell survival in the absence of an exogenous death signal. BMC Cancer 2010, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Gao, W.; Zhang, R.; Han, X.; Jia, M.; Guan, W. Transfer of siRNA against XIAP induces apoptosis and reduces tumor cells growth potential in human breast cancer in vitro and in vivo. Breast Cancer Res. Treat 2006, 96, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Foster, F.M.; Owens, T.W.; Tanianis-Hughes, J.; Clarke, R.B.; Brennan, K.; Bundred, N.J.; Streuli, C.H. Targeting inhibitor of apoptosis proteins in combination with ErbB antagonists in breast cancer. Breast Cancer Res. BCR 2009, 11, R41. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Letai, A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018, 25, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Giampazolias, E.; Zunino, B.; Dhayade, S.; Bock, F.; Cloix, C.; Cao, K.; Roca, A.; Lopez, J.; Ichim, G.; Proics, E.; et al. Mitochondrial permeabilization engages NF-kappaB-dependent anti-tumour activity under caspase deficiency. Nat. Cell Biol. 2017, 19, 1116–1129. [Google Scholar] [CrossRef] [PubMed]
- Armani, C.; Catalani, E.; Balbarini, A.; Bagnoli, P.; Cervia, D. Expression, pharmacology, and functional role of somatostatin receptor subtypes 1 and 2 in human macrophages. J. Leukoc. Biol. 2007, 81, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Cervia, D.; Martini, D.; Garcia-Gil, M.; Di Giuseppe, G.; Guella, G.; Dini, F.; Bagnoli, P. Cytotoxic effects and apoptotic signalling mechanisms of the sesquiterpenoid euplotin C, a secondary metabolite of the marine ciliate Euplotes crassus, in tumour cells. Apoptosis 2006, 11, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Cervia, D.; Garcia-Gil, M.; Simonetti, E.; Di Giuseppe, G.; Guella, G.; Bagnoli, P.; Dini, F. Molecular mechanisms of euplotin C-induced apoptosis: Involvement of mitochondrial dysfunction, oxidative stress and proteases. Apoptosis 2007, 12, 1349–1363. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, C.; Buldorini, M.; Assi, E.; Cazzato, D.; De Palma, C.; Clementi, E.; Cervia, D. The thyroid hormone triiodothyronine controls macrophage maturation and functions: Protective role during inflammation. Am. J. Pathol. 2014, 184, 230–247. [Google Scholar] [CrossRef]
- Di Giuseppe, G.; Cervia, D.; Vallesi, A. Divergences in the Response to Ultraviolet Radiation Between Polar and Non-Polar Ciliated Protozoa: UV Radiation Effects in Euplotes. Microb. Ecol. 2011, 63, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, C.; Buonanno, F.; Zecchini, S.; Giavazzi, A.; Proietti Serafini, F.; Catalani, E.; Guerra, L.; Belardinelli, M.C.; Picchietti, S.; Fausto, A.M.; et al. Climacostol reduces tumour progression in a mouse model of melanoma via the p53-dependent intrinsic apoptotic programme. Sci. Rep. 2016, 6, 27281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bizzozero, L.; Cazzato, D.; Cervia, D.; Assi, E.; Simbari, F.; Pagni, F.; De Palma, C.; Monno, A.; Verdelli, C.; Querini, P.R.; et al. Acid sphingomyelinase determines melanoma progression and metastatic behaviour via the microphtalmia-associated transcription factor signalling pathway. Cell Death Differ. 2014, 21, 507–520. [Google Scholar] [CrossRef]
- Zecchini, S.; Proietti Serafini, F.; Catalani, E.; Giovarelli, M.; Coazzoli, M.; Di Renzo, I.; De Palma, C.; Perrotta, C.; Clementi, E.; Buonanno, F.; et al. Dysfunctional autophagy induced by the pro-apoptotic natural compound climacostol in tumour cells. Cell Death Dis. 2019, 10, 10. [Google Scholar] [CrossRef]
- Assi, E.; Cervia, D.; Bizzozero, L.; Capobianco, A.; Pambianco, S.; Morisi, F.; De Palma, C.; Moscheni, C.; Pellegrino, P.; Clementi, E.; et al. Modulation of Acid Sphingomyelinase in Melanoma Reprogrammes the Tumour Immune Microenvironment. Mediat. Inflamm 2015, 2015, 370482. [Google Scholar] [CrossRef]
- Cervia, D.; Assi, E.; De Palma, C.; Giovarelli, M.; Bizzozero, L.; Pambianco, S.; Di Renzo, I.; Zecchini, S.; Moscheni, C.; Vantaggiato, C.; et al. Essential role for acid sphingomyelinase-inhibited autophagy in melanoma response to cisplatin. Oncotarget 2016, 7, 24995–25009. [Google Scholar] [CrossRef]
- Perrotta, C.; De Palma, C.; Clementi, E.; Cervia, D. Hormones and immunity in cancer: Are thyroid hormones endocrine players in the microglia/glioma cross-talk? Front. Cell. Neurosci. 2015, 9, 236. [Google Scholar] [CrossRef] [PubMed]
- Vantaggiato, C.; Castelli, M.; Giovarelli, M.; Orso, G.; Bassi, M.T.; Clementi, E.; De Palma, C. The Fine Tuning of Drp1-Dependent Mitochondrial Remodeling and Autophagy Controls Neuronal Differentiation. Front. Cell. Neurosci. 2019, 13, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfisterer, P.H.; Wolber, G.; Efferth, T.; Rollinger, J.M.; Stuppner, H. Natural products in structure-assisted design of molecular cancer therapeutics. Curr. Pharm. Des. 2010, 16, 1718–1741. [Google Scholar] [CrossRef] [PubMed]
- Cazzato, D.; Assi, E.; Moscheni, C.; Brunelli, S.; De Palma, C.; Cervia, D.; Perrotta, C.; Clementi, E. Nitric oxide drives embryonic myogenesis in chicken through the upregulation of myogenic differentiation factors. Exp. Cell Res. 2014, 320, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Cervia, D.; Catalani, E.; Belardinelli, M.C.; Perrotta, C.; Picchietti, S.; Alimenti, C.; Casini, G.; Fausto, A.M.; Vallesi, A. The protein pheromone Er-1 of the ciliate Euplotes raikovi stimulates human T-cell activity: Involvement of interleukin-2 system. Exp. Cell Res. 2013, 319, 56–67. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μg/mL) | |
|---|---|---|
| U373 Cells | MCF7 Cells | |
| Gibbilimbol B | 16.79 | 16.44 |
| Eriopodol A | 11.12 | 10.12 |
| Eriopodol B | 31.91 | 29.36 |
| Eriopodol C | 14.30 | 16.30 |
| Erioquinol | 1.78 | 2.63 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz, D.; Brucoli, M.; Zecchini, S.; Sandoval-Hernandez, A.; Arboleda, G.; Lopez-Vallejo, F.; Delgado, W.; Giovarelli, M.; Coazzoli, M.; Catalani, E.; et al. XIAP as a Target of New Small Organic Natural Molecules Inducing Human Cancer Cell Death. Cancers 2019, 11, 1336. https://doi.org/10.3390/cancers11091336
Muñoz D, Brucoli M, Zecchini S, Sandoval-Hernandez A, Arboleda G, Lopez-Vallejo F, Delgado W, Giovarelli M, Coazzoli M, Catalani E, et al. XIAP as a Target of New Small Organic Natural Molecules Inducing Human Cancer Cell Death. Cancers. 2019; 11(9):1336. https://doi.org/10.3390/cancers11091336
Chicago/Turabian StyleMuñoz, Diego, Martina Brucoli, Silvia Zecchini, Adrian Sandoval-Hernandez, Gonzalo Arboleda, Fabian Lopez-Vallejo, Wilman Delgado, Matteo Giovarelli, Marco Coazzoli, Elisabetta Catalani, and et al. 2019. "XIAP as a Target of New Small Organic Natural Molecules Inducing Human Cancer Cell Death" Cancers 11, no. 9: 1336. https://doi.org/10.3390/cancers11091336
APA StyleMuñoz, D., Brucoli, M., Zecchini, S., Sandoval-Hernandez, A., Arboleda, G., Lopez-Vallejo, F., Delgado, W., Giovarelli, M., Coazzoli, M., Catalani, E., Palma, C. D., Perrotta, C., Cuca, L., Clementi, E., & Cervia, D. (2019). XIAP as a Target of New Small Organic Natural Molecules Inducing Human Cancer Cell Death. Cancers, 11(9), 1336. https://doi.org/10.3390/cancers11091336