CITK Loss Inhibits Growth of Group 3 and Group 4 Medulloblastoma Cells and Sensitizes Them to DNA-Damaging Agents

 ,
,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. CITK Knockdown Impairs Proliferation and Cytokinesis in Group 3 and Group 4 MB
2.2. CITK Knockdown Increases Apoptosis and Cell-Cycle Arrest in D283 and D341 Cell Lines
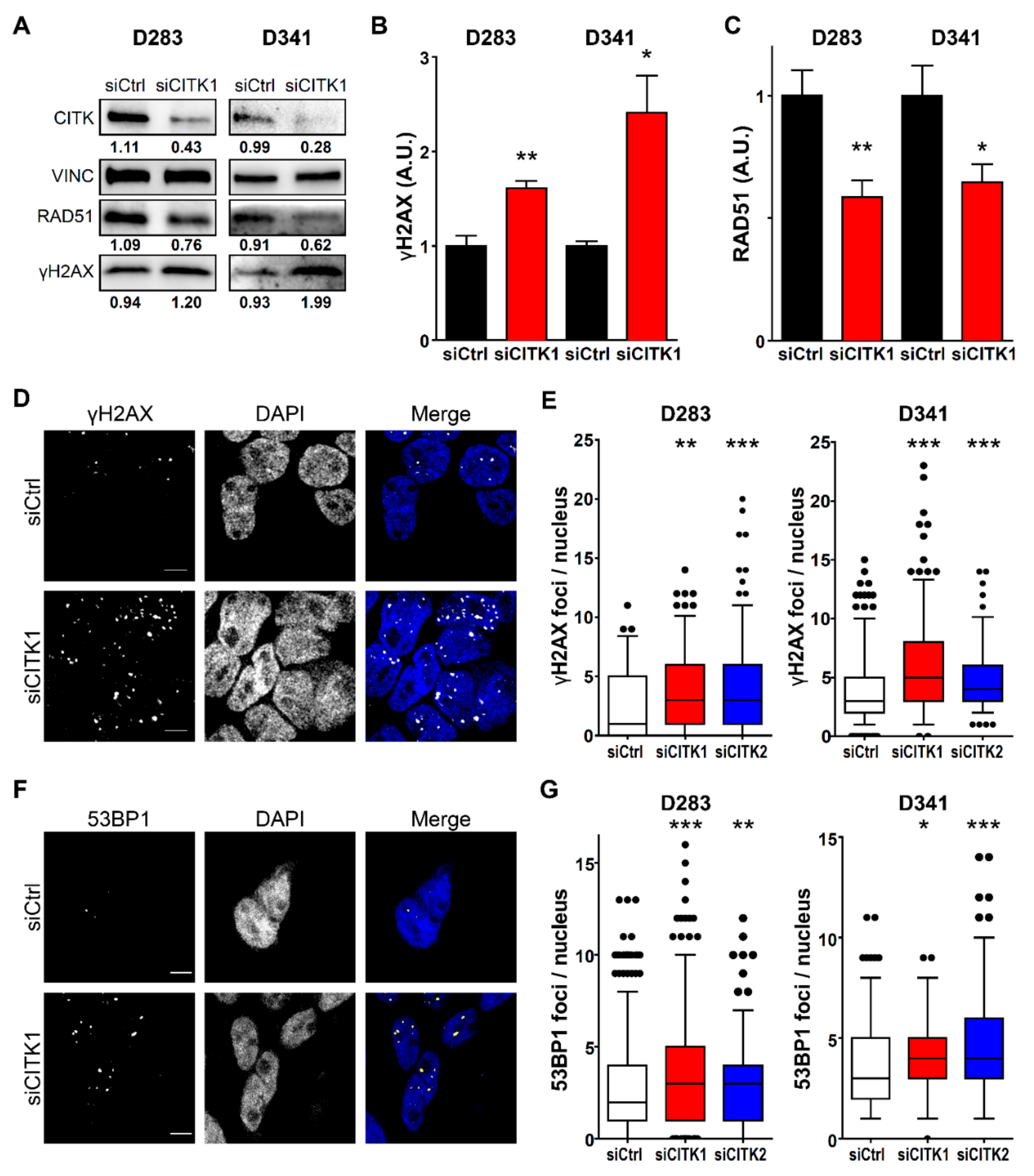
2.3. CITK Loss Induces Double Strand Breaks Accumulation in MB Cells
2.4. CITK Knockdown Strongly Reduces Nuclear RAD51 Levels in MB Cells and Impairs Homologous Recombination
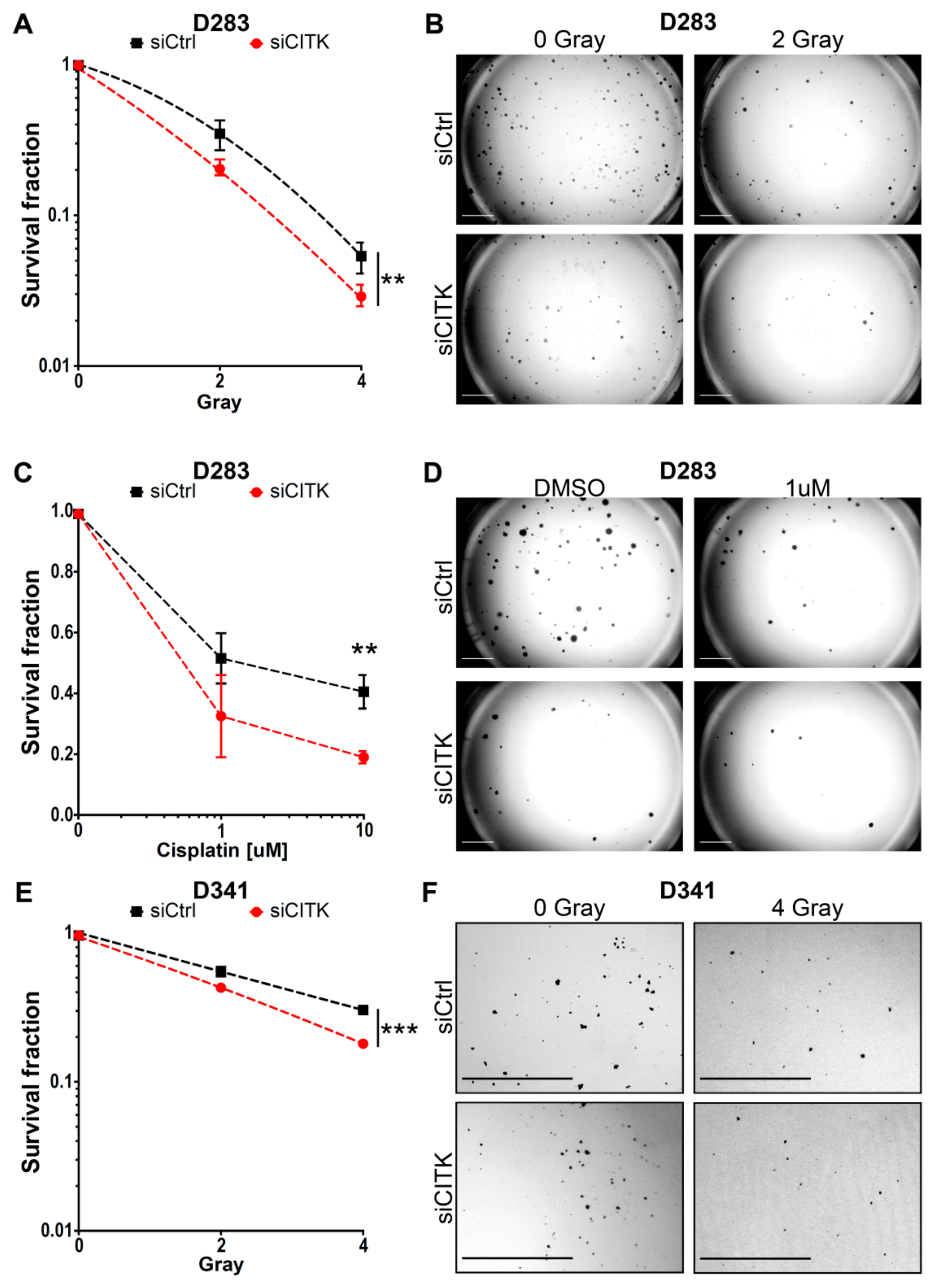
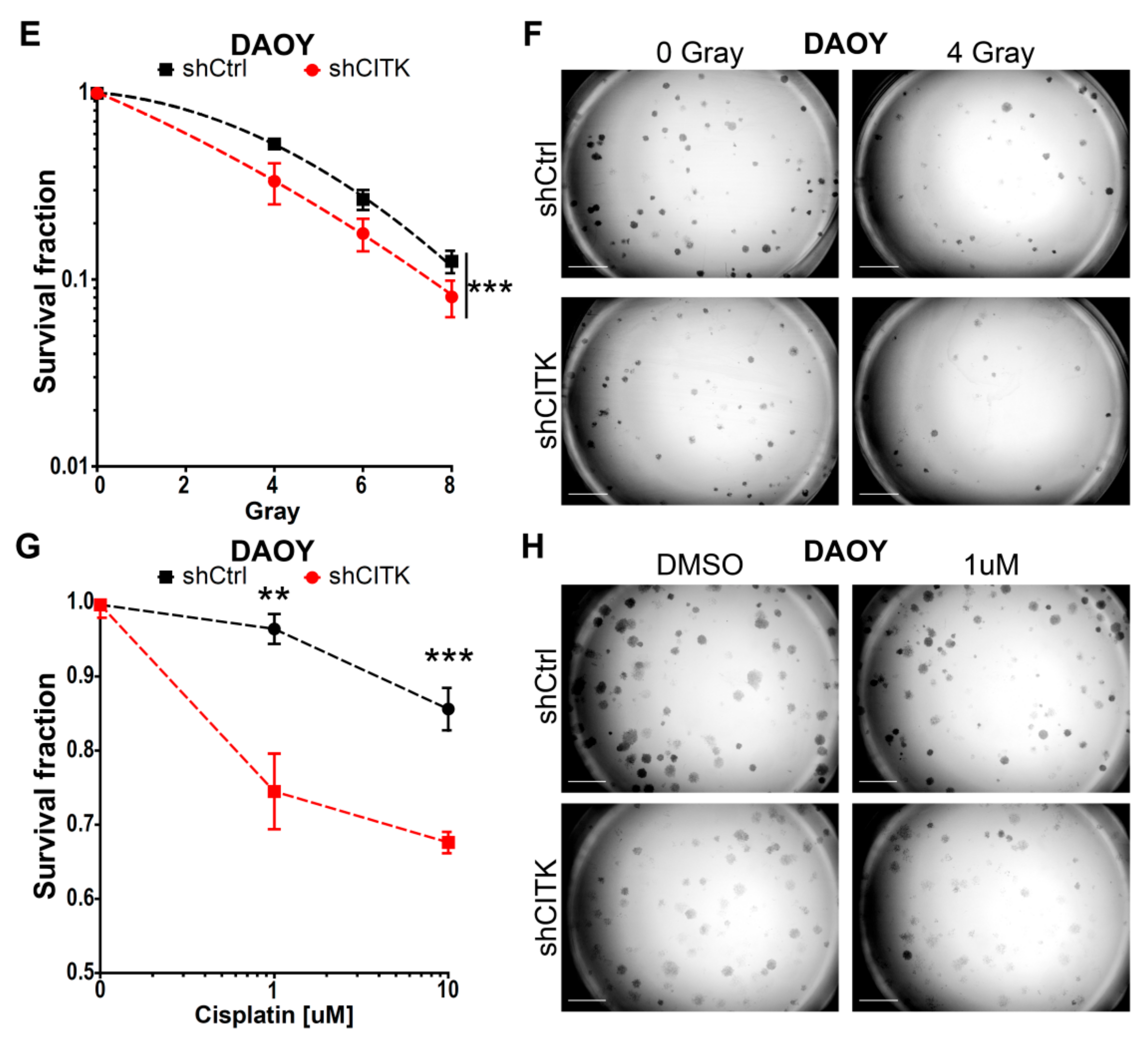
2.5. CITK Downregulation Potentiates the Effects of Ionizing Radiation and Cisplatin in Inhibiting MB Cell Growth
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Transfection and RNAi
4.3. Analysis of Cell Proliferation
4.4. Antibodies
4.5. Western Blotting
4.6. Immunofluorescence
4.7. Cell-Cycle Analysis
4.8. Homologous Recombination Assay
4.9. Radiation Treatment
4.10. Cisplatin Treatment
4.11. Colony-Forming Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Northcott, P.A.; Shih, D.J.H.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stütz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-Specific structural variation across 1000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737.e6–754.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. (Berl.) 2016, 131, 821–831. [Google Scholar] [CrossRef] [Green Version]
- Samkari, A.; White, J.; Packer, R. SHH inhibitors for the treatment of medulloblastoma. Expert Rev. Neurother. 2015, 15, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.; Stewart, C.F.; Ellison, D.W.; Kaste, S.; Kun, L.E.; Packer, R.J.; Goldman, S.; Chintagumpala, M.; Wallace, D.; Takebe, N.; et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: A pediatric brain tumor consortium study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 6305–6312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-Oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Gibson, P.; Tong, Y.; Robinson, G.; Thompson, M.C.; Currle, D.S.; Eden, C.; Kranenburg, T.A.; Hogg, T.; Poppleton, H.; Martin, J.; et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010, 468, 1095–1099. [Google Scholar] [CrossRef]
- Smith, A.W.; Mehta, M.P.; Wernicke, A.G. Neural stem cells, the subventricular zone and radiotherapy: Implications for treating glioblastoma. J. Neurooncol. 2016, 128, 207–216. [Google Scholar] [CrossRef]
- Pallavicini, G.; Berto, G.E.; Di Cunto, F. Precision Revisited: Targeting Microcephaly Kinases in Brain Tumors. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Lang, P.Y.; Gershon, T.R. A New Way to Treat Brain Tumors: Targeting Proteins Coded by Microcephaly Genes?: Brain tumors and microcephaly arise from opposing derangements regulating progenitor growth. Drivers of microcephaly could be attractive brain tumor targets. BioEssays News Rev. Mol. Cell. Dev. Biol. 2018, 40, e1700243. [Google Scholar] [CrossRef] [PubMed]
- Passemard, S.; Kaindl, A.M.; Verloes, A. Microcephaly. Handb. Clin. Neurol. 2013, 111, 129–141. [Google Scholar] [PubMed]
- Morris-Rosendahl, D.J.; Kaindl, A.M. What next-Generation sequencing (NGS) technology has enabled us to learn about primary autosomal recessive microcephaly (MCPH). Mol. Cell. Probes. 2015, 29, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faheem, M.; Naseer, M.I.; Rasool, M.; Chaudhary, A.G.; Kumosani, T.A.; Ilyas, A.M.; Pushparaj, P.; Ahmed, F.; Algahtani, H.A.; Al-Qahtani, M.H.; et al. Molecular genetics of human primary microcephaly: An overview. BMC Med. Genom. 2015, 8 (Suppl. 1). [Google Scholar] [CrossRef] [Green Version]
- O′Neill, R.S.; Schoborg, T.A.; Rusan, N.M. Same but different: Pleiotropy in centrosome-related microcephaly. Mol. Biol. Cell 2018, 29, 241–246. [Google Scholar] [CrossRef]
- Bianchi, F.T.; Tocco, C.; Pallavicini, G.; Liu, Y.; Vernì, F.; Merigliano, C.; Bonaccorsi, S.; El-Assawy, N.; Priano, L.; Gai, M.; et al. Citron Kinase Deficiency Leads to Chromosomal Instability and TP53-Sensitive Microcephaly. Cell Rep. 2017, 18, 1674–1686. [Google Scholar] [CrossRef]
- Williams, S.E.; Garcia, I.; Crowther, A.J.; Li, S.; Stewart, A.; Liu, H.; Lough, K.J.; O’Neill, S.; Veleta, K.; Oyarzabal, E.A.; et al. Aspm sustains postnatal cerebellar neurogenesis and medulloblastoma growth in mice. Dev. Camb. Engl. 2015, 142, 3921–3932. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.-W.; Tapias, A.; Bruhn, C.; Gruber, R.; Sukchev, M.; Wang, Z.-Q. DNA damage response in microcephaly development of MCPH1 mouse model. DNA Repair 2013, 12, 645–655. [Google Scholar] [CrossRef]
- Stouffs, K.; Stergachis, A.B.; Vanderhasselt, T.; Dica, A.; Janssens, S.; Vandervore, L.; Gheldof, A.; Bodamer, O.; Keymolen, K.; Seneca, S.; et al. Expanding the clinical spectrum of biallelic ZNF335 variants. Clin. Genet. 2018, 94, 246–251. [Google Scholar] [CrossRef]
- Yang, Y.J.; Baltus, A.E.; Mathew, R.S.; Murphy, E.A.; Evrony, G.D.; Gonzalez, D.M.; Wang, E.P.; Marshall-Walker, C.A.; Barry, B.J.; Murn, J.; et al. Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 2012, 151, 1097–1112. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, R.; Faqeih, E.; Ansari, S.; Abdel-Salam, G.; Al-Hassnan, Z.N.; Al-Shidi, T.; Alomar, R.; Sogaty, S.; Alkuraya, F.S. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res. 2014, 24, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guemez-Gamboa, A.; Nguyen, L.N.; Yang, H.; Zaki, M.S.; Kara, M.; Ben-Omran, T.; Akizu, N.; Rosti, R.O.; Rosti, B.; Scott, E.; et al. Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome. Nat. Genet. 2015, 47, 809–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzaa, G.M.; Vitre, B.; Carpenter, G.; Abramowicz, I.; Gleeson, J.G.; Paciorkowski, A.R.; Cleveland, D.W.; Dobyns, W.B.; O’Driscoll, M. Mutations in CENPE define a novel kinetochore-Centromeric mechanism for Microcephalic Primordial Dwarfism. Hum. Genet. 2014, 133, 1023–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moawia, A.; Shaheen, R.; Rasool, S.; Waseem, S.S.; Ewida, N.; Budde, B.; Kawalia, A.; Motameny, S.; Khan, K.; Fatima, A.; et al. Mutations of KIF14 cause primary microcephaly by impairing cytokinesis. Ann. Neurol. 2017, 82, 562–577. [Google Scholar] [CrossRef]
- Harding, B.N.; Moccia, A.; Drunat, S.; Soukarieh, O.; Tubeuf, H.; Chitty, L.S.; Verloes, A.; Gressens, P.; El Ghouzzi, V.; Joriot, S.; et al. Mutations in Citron Kinase Cause Recessive Microlissencephaly with Multinucleated Neurons. Am. J. Hum. Genet. 2016, 99, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Bielas, S.L.; Zaki, M.S.; Ismail, S.; Farfara, D.; Um, K.; Rosti, R.O.; Scott, E.C.; Tu, S.; Chi, N.C.; et al. Biallelic Mutations in Citron Kinase Link Mitotic Cytokinesis to Human Primary Microcephaly. Am. J. Hum. Genet. 2016, 99, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, F.T.; Gai, M.; Berto, G.E.; Di Cunto, F. Of rings and spines: The multiple facets of Citron proteins in neural development. Small GTPases 2017, 1–9. [Google Scholar] [CrossRef]
- D’Avino, P.P. Citron kinase-Renaissance of a neglected mitotic kinase. J. Cell Sci. 2017, 130, 1701–1708. [Google Scholar] [CrossRef] [Green Version]
- Sgrò, F.; Bianchi, F.T.; Falcone, M.; Pallavicini, G.; Gai, M.; Chiotto, A.M.A.; Berto, G.E.; Turco, E.; Chang, Y.J.; Huttner, W.B.; et al. Tissue-Specific control of midbody microtubule stability by Citron kinase through modulation of TUBB3 phosphorylation. Cell Death Differ. 2016, 23, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Gai, M.; Bianchi, F.T.; Vagnoni, C.; Vernì, F.; Bonaccorsi, S.; Pasquero, S.; Berto, G.E.; Sgrò, F.; Chiotto, A.M.; Annaratone, L.; et al. ASPM and CITK regulate spindle orientation by affecting the dynamics of astral microtubules. EMBO Rep. 2016, 17, 1396–1409. [Google Scholar] [CrossRef] [Green Version]
- Pallavicini, G.; Sgrò, F.; Garello, F.; Falcone, M.; Bitonto, V.; Berto, G.E.; Bianchi, F.T.; Gai, M.; Chiotto, A.M.A.; Filippi, M.; et al. Inactivation of Citron Kinase Inhibits Medulloblastoma Progression by Inducing Apoptosis and Cell Senescence. Cancer Res. 2018, 78, 4599–4612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, D.P.; Coyle, B.; Walker, D.A.; Grabowska, A.M. In vitro models of medulloblastoma: Choosing the right tool for the job. J. Biotechnol. 2016, 236, 10–25. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, C.; D’Avino, P.P. Investigating cytokinesis failure as a strategy in cancer therapy. Oncotarget 2016, 7, 87323–87341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Capetillo, O.; Chen, H.-T.; Celeste, A.; Ward, I.; Romanienko, P.J.; Morales, J.C.; Naka, K.; Xia, Z.; Camerini-Otero, R.D.; Motoyama, N.; et al. DNA damage-Induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 2002, 4, 993–997. [Google Scholar] [CrossRef]
- DiTullio, R.A.; Mochan, T.A.; Venere, M.; Bartkova, J.; Sehested, M.; Bartek, J.; Halazonetis, T.D. 53BP1 functions in an ATM-Dependent checkpoint pathway that is constitutively activated in human cancer. Nat. Cell Biol. 2002, 4, 998–1002. [Google Scholar] [CrossRef]
- Baumann, P.; Benson, F.E.; West, S.C. Human Rad51 protein promotes ATP-Dependent homologous pairing and strand transfer reactions in vitro. Cell 1996, 87, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ji, W.; Shergalis, A.; Xu, J.; Delaney, A.M.; Calcaterra, A.; Pal, A.; Ljungman, M.; Neamati, N.; Rehemtulla, A. Activation of the Unfolded Protein Response via Inhibition of Protein Disulfide Isomerase Decreases the Capacity for DNA Repair to Sensitize Glioblastoma to Radiotherapy. Cancer Res. 2019, 79, 2923–2932. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.J.; Hwang, I.; Lee, J.Y.; Park, J.N.; Kim, K.C.; Kim, G.-H.; Kang, C.-M.; Kim, I.; Lee, S.-Y.; Kim, H.-S. Hepatocyte Growth Factor Improves the Therapeutic Efficacy of Human Bone Marrow Mesenchymal Stem Cells via RAD51. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 845–859. [Google Scholar] [CrossRef]
- Misic, V.; El-Mogy, M.; Geng, S.; Haj-Ahmad, Y. Effect of endonuclease G depletion on plasmid DNA uptake and levels of homologous recombination in hela cells. Mol. Biol. 2016, 50, 252–261. [Google Scholar] [CrossRef]
- Ohba, S.; Mukherjee, J.; See, W.L.; Pieper, R.O. Mutant IDH1-Driven cellular transformation increases RAD51-Mediated homologous recombination and temozolomide resistance. Cancer Res. 2014, 74, 4836–4844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajjar, A.; Chintagumpala, M.; Ashley, D.; Kellie, S.; Kun, L.E.; Merchant, T.E.; Woo, S.; Wheeler, G.; Ahern, V.; Krasin, M.J.; et al. Risk-Adapted craniospinal radiotherapy followed by high-Dose chemotherapy and stem-Cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): Long-Term results from a prospective, multicentre trial. Lancet Oncol. 2006, 7, 813–820. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-Oncol. 2017, 19, v1–v88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, R.J.; Gajjar, A.; Vezina, G.; Rorke-Adams, L.; Burger, P.C.; Robertson, P.L.; Bayer, L.; LaFond, D.; Donahue, B.R.; Marymont, M.H.; et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-Risk medulloblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 4202–4208. [Google Scholar] [CrossRef]
- Ho, W.S.; Feldman, M.J.; Maric, D.; Amable, L.; Hall, M.D.; Feldman, G.M.; Ray-Chaudhury, A.; Lizak, M.J.; Vera, J.-C.; Robison, R.A.; et al. PP2A inhibition with LB100 enhances cisplatin cytotoxicity and overcomes cisplatin resistance in medulloblastoma cells. Oncotarget 2016, 7, 12447–12463. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; McDonald, D.; Lowe, S.; Bredlau, A.-L.; Vanek, K.; Patel, S.J.; Cheshier, S.; Eskandari, R. Immunological low-Dose radiation modulates the pediatric medulloblastoma antigens and enhances antibody-Dependent cellular cytotoxicity. Childs Nerv. Syst. ChNS Off. J. Int. Soc. Pediatr. Neurosurg. 2017, 33, 429–436. [Google Scholar] [CrossRef]
- Blazek, E.R.; Foutch, J.L.; Maki, G. Daoy medulloblastoma cells that express CD133 are radioresistant relative to CD133-Cells, and the CD133+ sector is enlarged by hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 1–5. [Google Scholar] [CrossRef]
- Di Cunto, F.; Imarisio, S.; Hirsch, E.; Broccoli, V.; Bulfone, A.; Migheli, A.; Atzori, C.; Turco, E.; Triolo, R.; Dotto, G.P.; et al. Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron 2000, 28, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Trojanek, J.; Ho, T.; Del Valle, L.; Nowicki, M.; Wang, J.Y.; Lassak, A.; Peruzzi, F.; Khalili, K.; Skorski, T.; Reiss, K. Role of the insulin-Like growth factor I/insulin receptor substrate 1 axis in Rad51 trafficking and DNA repair by homologous recombination. Mol. Cell. Biol. 2003, 23, 7510–7524. [Google Scholar] [CrossRef] [Green Version]
- Henriksson, S.; Rassoolzadeh, H.; Hedström, E.; Coucoravas, C.; Julner, A.; Goldstein, M.; Imreh, G.; Zhivotovsky, B.; Kastan, M.B.; Helleday, T.; et al. The scaffold protein WRAP53β orchestrates the ubiquitin response critical for DNA double-Strand break repair. Genes Dev. 2014, 28, 2726–2738. [Google Scholar] [CrossRef] [Green Version]
- Poruchynsky, M.S.; Komlodi-Pasztor, E.; Trostel, S.; Wilkerson, J.; Regairaz, M.; Pommier, Y.; Zhang, X.; Maity, T.K.; Robey, R.; Burotto, M.; et al. Microtubule-Targeting agents augment the toxicity of DNA-Damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc. Natl. Acad. Sci. 2015, 112, 1571–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, P.; West, S.C. Role of the human RAD51 protein in homologous recombination and double-Stranded-Break repair. Trends Biochem. Sci. 1998, 23, 247–251. [Google Scholar] [CrossRef]
- Fu, Y.; Huang, J.; Wang, K.-S.; Zhang, X.; Han, Z.-G. RNA interference targeting CITRON can significantly inhibit the proliferation of hepatocellular carcinoma cells. Mol. Biol. Rep. 2011, 38, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Yu, Q.; Feng, L.; Luo, M.; Shao, S.; Huang, S.; Wang, G.; Jing, X.; Tong, Z.; Zhao, X.; et al. Citron kinase (CIT-K) promotes aggressiveness and tumorigenesis of breast cancer cells in vitro and in vivo: Preliminary study of the underlying mechanism. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2019, 21, 910–923. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Zhu, X.; Xu, W.; Zhang, Y.; Chen, L.; Qiu, F.; Zhang, B.; Wu, L.; Peng, Z.; Tang, H. Up-Regulation of CIT promotes the growth of colon cancer cells. Oncotarget 2017, 8, 71954–71964. [Google Scholar] [CrossRef] [Green Version]
- Haiping, C.; Qi, X.; Dawei, L.; Qiang, W. Citron Rho-Interacting serine/threonine kinase knockdown suppresses prostate cancer cell proliferation and metastasis by blocking Hippo-YAP pathway. Nan Fang Yi Ke Da Xue Xue Bao 2019, 39, 257–263. [Google Scholar] [CrossRef]
- Sahin, I.; Kawano, Y.; Sklavenitis-Pistofidis, R.; Moschetta, M.; Mishima, Y.; Manier, S.; Sacco, A.; Carrasco, R.; Fonseca, R.; Roccaro, A.M.; et al. Citron Rho-Interacting kinase silencing causes cytokinesis failure and reduces tumor growth in multiple myeloma. Blood Adv. 2019, 3, 995–1002. [Google Scholar] [CrossRef] [Green Version]
- Dema, A.; Macaluso, F.; Sgrò, F.; Berto, G.E.; Bianchi, F.T.; Chiotto, A.A.; Pallavicini, G.; Di Cunto, F.; Gai, M. Citron kinase-Dependent F-actin maintenance at midbody secondary ingression sites mediates abscission. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pallavicini, G.; Iegiani, G.; Berto, G.E.; Calamia, E.; Trevisiol, E.; Veltri, A.; Allis, S.; Di Cunto, F. CITK Loss Inhibits Growth of Group 3 and Group 4 Medulloblastoma Cells and Sensitizes Them to DNA-Damaging Agents. Cancers 2020, 12, 542. https://doi.org/10.3390/cancers12030542
Pallavicini G, Iegiani G, Berto GE, Calamia E, Trevisiol E, Veltri A, Allis S, Di Cunto F. CITK Loss Inhibits Growth of Group 3 and Group 4 Medulloblastoma Cells and Sensitizes Them to DNA-Damaging Agents. Cancers. 2020; 12(3):542. https://doi.org/10.3390/cancers12030542
Chicago/Turabian StylePallavicini, Gianmarco, Giorgia Iegiani, Gaia Elena Berto, Elisa Calamia, Edoardo Trevisiol, Andrea Veltri, Simona Allis, and Ferdinando Di Cunto. 2020. "CITK Loss Inhibits Growth of Group 3 and Group 4 Medulloblastoma Cells and Sensitizes Them to DNA-Damaging Agents" Cancers 12, no. 3: 542. https://doi.org/10.3390/cancers12030542