Nucleolin-Sle A Glycoforms as E-Selectin Ligands and Potentially Targetable Biomarkers at the Cell Surface of Gastric Cancer Cells

 , , ,
, , ,  ,
,
Abstract
:
1. Introduction
2. Material and Methods
2.1. Cell Lines and Culture Conditions
2.2. Population and Ethics Statement
2.3. Antibodies and Lectins
2.4. Flow Cytometry
2.5. O-Glycomics
2.6. Subcellular Fractionation And Protein Extraction From Gastric Tissues
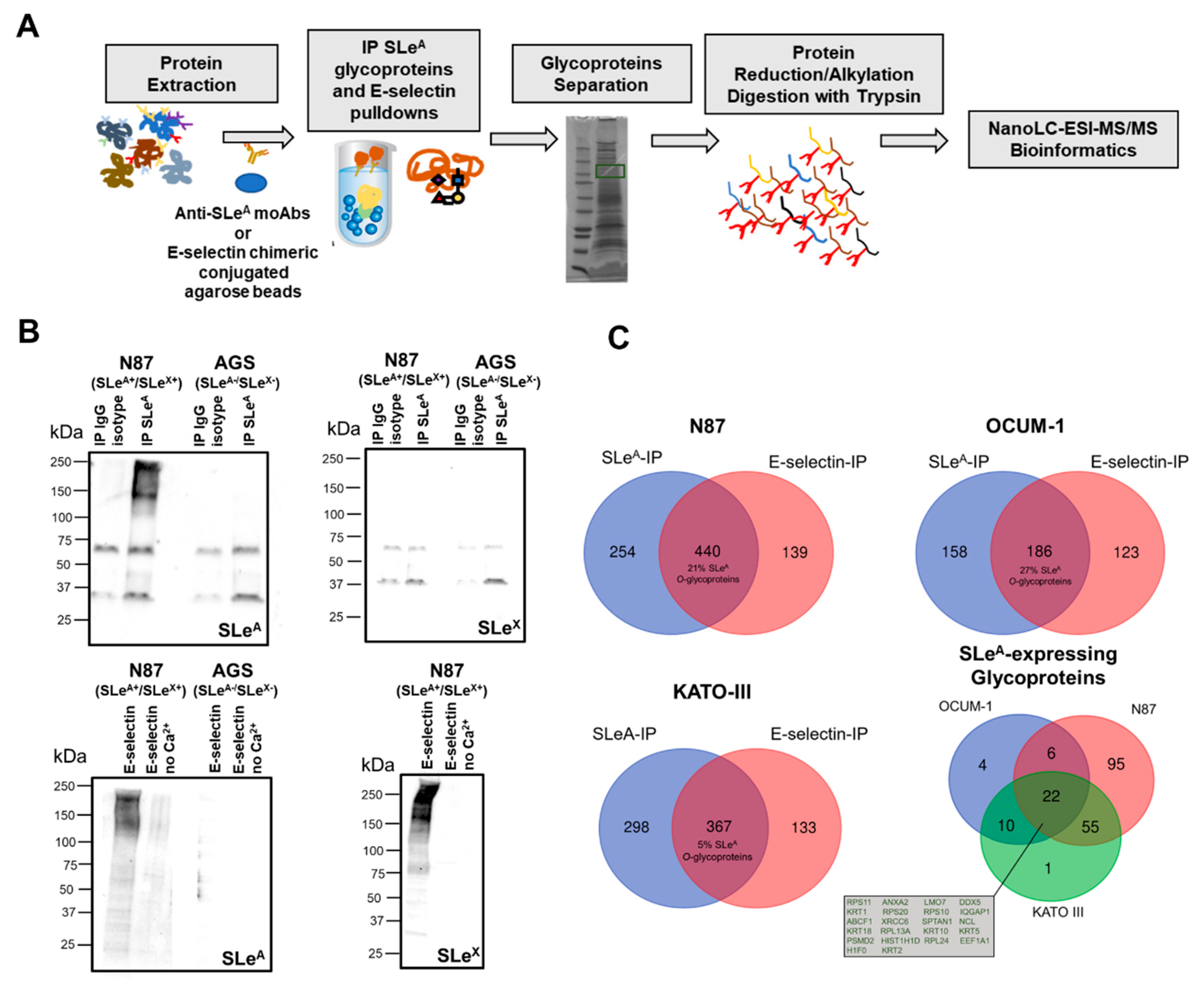
2.7. O-Linked Glycoproteomics
2.8. Bioinformatics for Biomarker Discovery
2.9. Western Blot
2.10. Immunofluorescence Microscopy In Situ Proximity Ligation Assays (PLA)
2.11. Immunohistochemistry
2.12. Statistical Analysis
3. Results
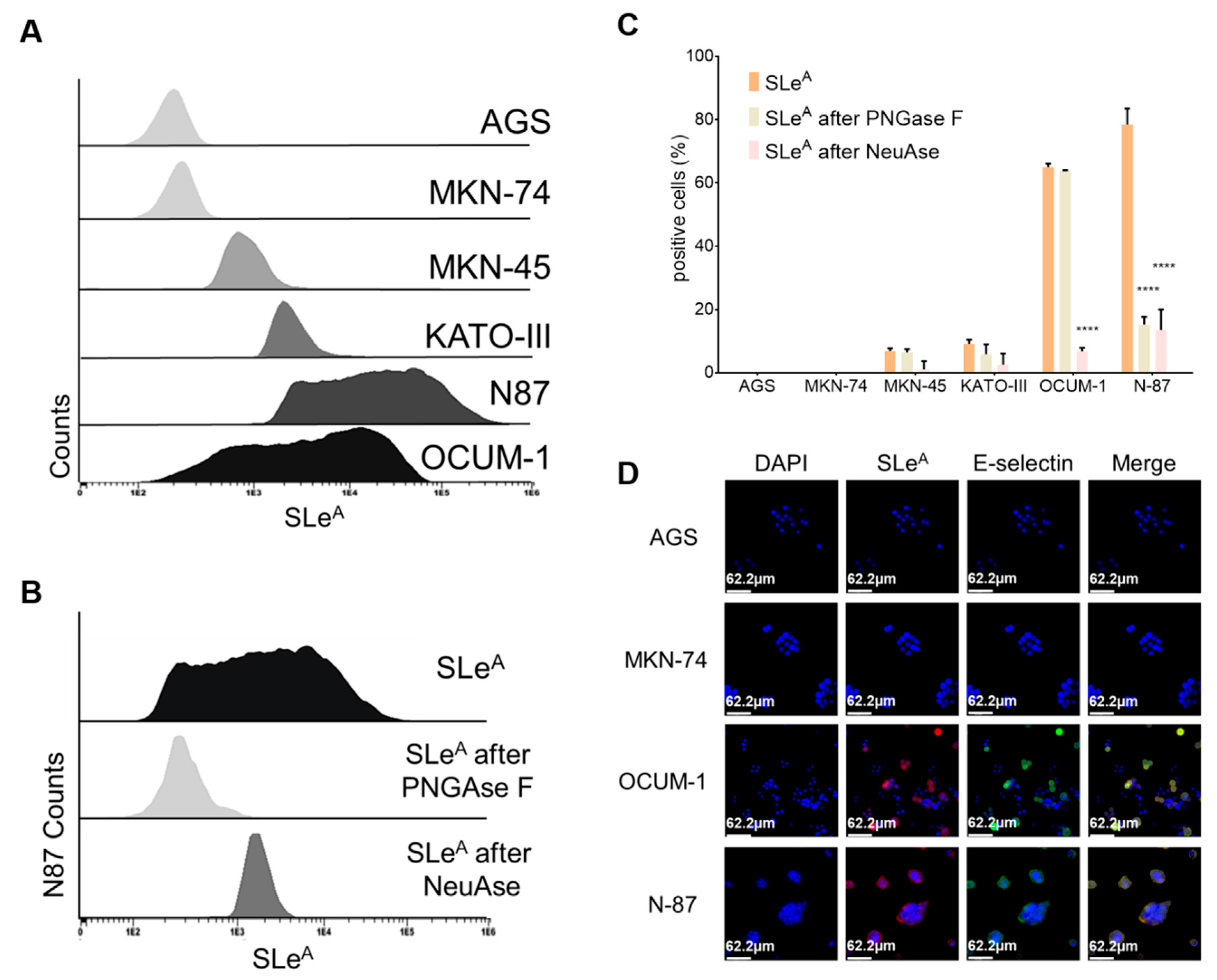
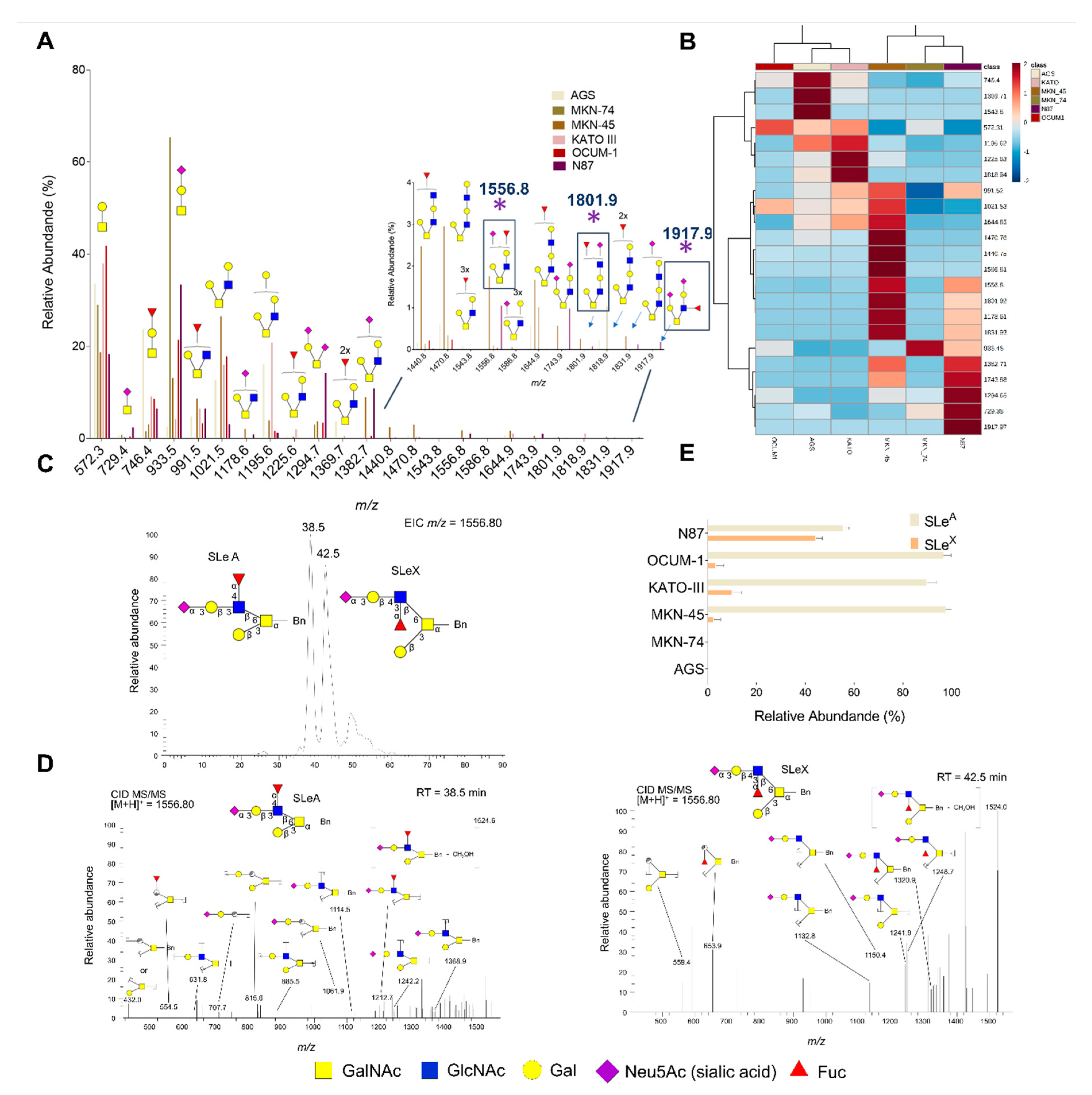
3.1. Glycomics and Affinity for E-selectin
3.2. Targeted Glycoproteomics and Glycobiomarker Discovery
3.3. Nucleolin Expression in Gastric Cancer Cells
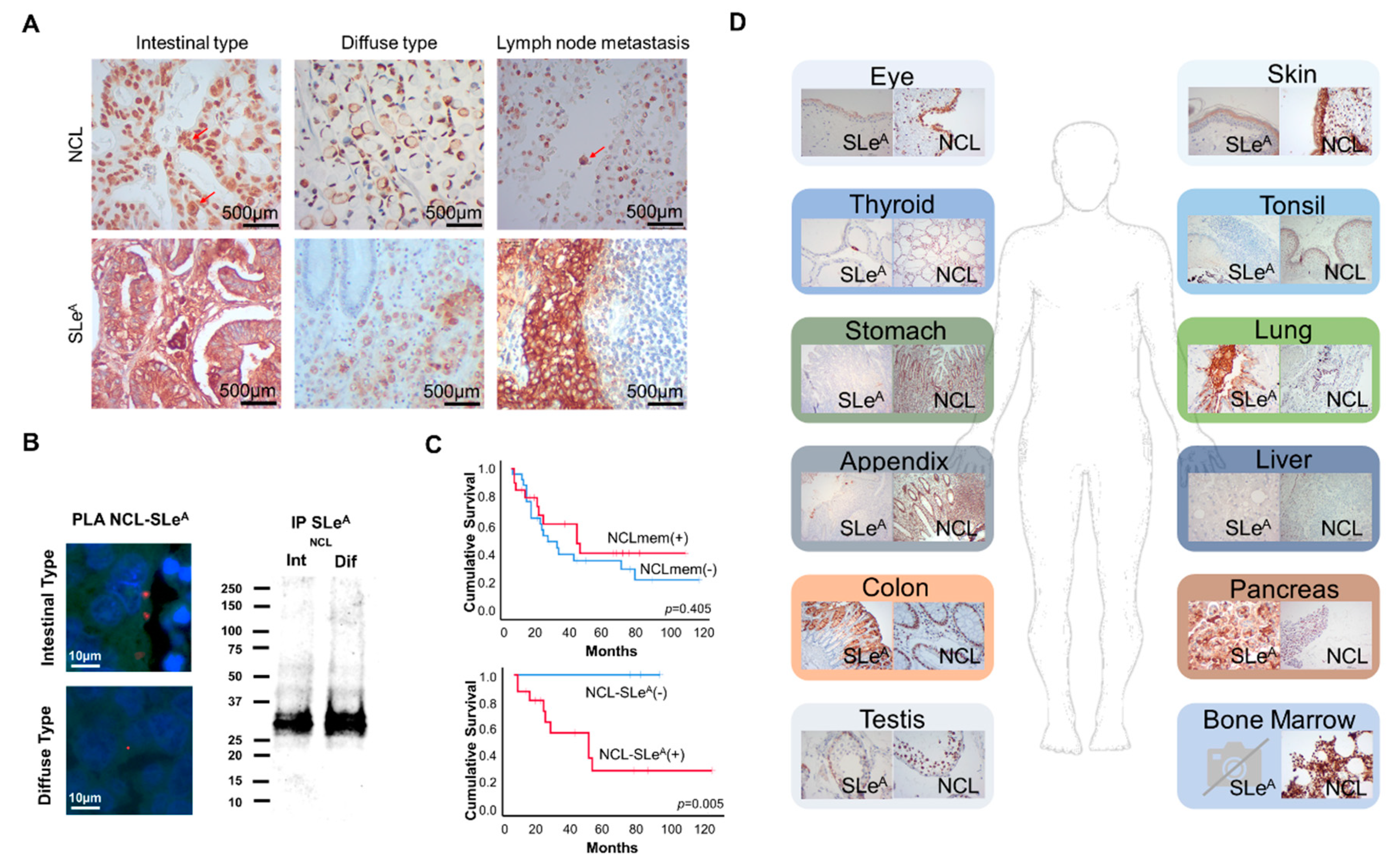
3.4. SLeA and NCL Expression in Gastric Cancer and Healthy Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Catenacci, D.V.T.; Henderson, L.; Xiao, S.-Y.; Penuel, E.; Patel, P.; Yauch, R.L.; Peterson, A.; Salgia, R. Durable Complete Response of Metastatic Gastric Cancer with Anti-Met Therapy Followed by Resistance at Recurrence. Cancer Discov. 2011, 1, 573–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.W.; Lo, S.S.; Shen, K.H.; Hsieh, M.C.; Chen, J.H.; Chiang, J.H.; Lin, H.J.; Li, A.F.; Lui, W.Y. Incidence and Factors Associated with Recurrence Patterns after Intended Curative Surgery for Gastric Cancer. World J. Surg. 2003, 27, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, Y.; Kodera, Y.; Ito, S.; Yamamura, Y.; Kanemitsu, Y.; Shimizu, Y.; Hirai, T.; Yasui, K.; Inada, K.-I.; Kato, T. Treatment and Risk Factors for Recurrence after Curative Resection of Gastrointestinal Stromal Tumors of the Stomach. World J. Surg. 2004, 28, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Youn, H.G.; An, J.Y.; Choi, M.G.; Noh, J.H.; Sohn, T.S.; Kim, S. Recurrence after Curative Resection of Early Gastric Cancer. Ann. Surg. Oncol. 2009, 17, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Lordick, F.; Siewert, J. Recent Advances in Multimodal Treatment for Gastric Cancer: A Review. Gastric Cancer 2005, 8, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastre, J.; García-Sáenz, J.A.; Diaz-Rubio, E. Chemotherapy for Gastric Cancer. World J. Gastroenterol. 2006, 12, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, K.; Kouhuji, K.; Kizaki, J.; Isobe, T.; Hashimoto, K.; Shirouzu, K. Molecular Targeting to Treat Gastric Cancer. World J. Gastroenterol. 2014, 20, 13741–13755. [Google Scholar] [CrossRef]
- Ishii, T.; Kawazoe, A.; Shitara, K. Dawn of Precision Medicine on Gastric Cancer. Int. J. Clin. Oncol. 2019, 24, 779–788. [Google Scholar] [CrossRef]
- Samson, P.; Lockhart, A.C. Biologic Therapy in Esophageal and Gastric Malignancies: Current Therapies and Future Directions. J. Gastrointest. Oncol. 2017, 8, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Kirstein, M.M.; Lange, A.; Prenzler, A.; Manns, M.P.; Kubicka, S.; Vogel, A. Targeted Therapies in Metastatic Colorectal Cancer: A Systematic Review and Assessment of Currently Available Data. Oncologist 2014, 19, 1156–1168. [Google Scholar] [CrossRef] [Green Version]
- Kuo, W.-Y.; Hsu, H.-J.; Wu, C.-Y.; Chen, H.-S.; Chou, Y.-C.; Tsou, Y.-L.; Peng, H.-P.; Jian, J.-W.; Yu, C.-M.; Chiu, Y.-K.; et al. Antibody-Drug Conjugates with Her2-Targeting Antibodies from Synthetic Antibody Libraries Are Highly Potent against Her2-Positive Human Gastric Tumor in Xenograft Models. mAbs 2019, 11, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peixoto, A.; Relvas, M.; Azevedo, R.; Santos, L.L.; Ferreira, J.A. Protein Glycosylation and Tumor Microenvironment Alterations Driving Cancer Hallmarks. Front. Oncol. 2019, 9, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, R.; Peixoto, A.; Gaiteiro, C.; Fernandes, E.; Neves, M.; Lima, L.; Santos, L.L.; Ferreira, J.A. Over Forty Years of Bladder Cancer Glycobiology: Where Do Glycans Stand Facing Precision Oncology? Oncotarget 2017, 8, 91734–91764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinho, S.S.; Reis, C.A. Glycosylation in Cancer: Mechanisms and Clinical Implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Trinchera, M.; Aronica, A.; Dall’Olio, F. Selectin Ligands Sialyl-Lewis a and Sialyl-Lewis x in Gastrointestinal Cancers. Biology 2017, 6, 16. [Google Scholar] [CrossRef]
- Sakamoto, S.; Watanabe, T.; Tokumaru, T.; Takagi, H.; Nakazato, H.; Lloyd, K.O. Expression of Lewisa, Lewisb, Lewisx, Lewisy, Siayl-Lewisa, and Sialyl-Lewisx Blood Group Antigens in Human Gastric Carcinoma and in Normal Gastric Tissue. Cancer Res. 1989, 49, 745–752. [Google Scholar]
- Worrede, A.; Meucci, O.; Fatatis, A.; Worrede-Mahdi, A. Limiting Tumor Seeding as a Therapeutic Approach for Metastatic Disease. Pharmacol. Ther. 2019, 199, 117–128. [Google Scholar] [CrossRef]
- Kannagi, R.; Izawa, M.; Koike, T.; Miyazaki, K.; Kimura, N. Carbohydrate-Mediated Cell Adhesion in Cancer Metastasis and Angiogenesis. Cancer Sci. 2004, 95, 377–384. [Google Scholar] [CrossRef]
- Mannori, G.; Santoro, D.; Carter, L.; Corless, C.; Nelson, R.M.; Bevilacqua, M.P. Inhibition of Colon Carcinoma Cell Lung Colony Formation by a Soluble Form of E-Selectin. Am. J. Pathol. 1997, 151, 233–243. [Google Scholar]
- Krause, T.; Turner, G. Are Selectins Involved in Metastasis? Clin. Exp. Metastasis 1999, 17, 183–192. [Google Scholar] [CrossRef]
- Kannagi, R. Carbohydrate Antigen Sialyl Lewis a—Its Pathophysiological Significance and Induction Mechanism in Cancer Progression. Chang. Gung Med. J. 2007, 30, 189–209. [Google Scholar] [PubMed]
- DeAngelo, D.J.; Erba, H.P.; Jonas, B.A.; O’Dwyer, M.; Marlton, P.; Huls, G.A.; Liesveld, J.; Cooper, B.W.; Bhatnagar, B.; Armstrong, M.; et al. A Phase Iii Trial to Evaluate the Efficacy of Uproleselan (Gmi-1271) with Chemotherapy in Patients with Relapsed/Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2019, 37, TPS7066. [Google Scholar] [CrossRef]
- Song, Y.-X.; Huang, X.-Z.; Gao, P.; Sun, J.-X.; Chen, X.-W.; Yang, Y.-C.; Zhang, C.; Liu, H.-P.; Wang, H.-C.; Wang, Z. Clinicopathologic and Prognostic Value of Serum Carbohydrate Antigen 19-9 in Gastric Cancer: A Meta-Analysis. Dis. Markers 2015, 2015, 549843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, Y.; Mori, M.; Kajiyama, K.; Haraguchi, Y.; Sasaki, O.; Sugimachi, K. Immunohistochemical Expression of Sialyl Tn, Sialyl Lewis a, Sialyl Lewis a-B-, and Sialyl Lewis X in Primary Tumor and Metastatic Lymph Nodes in Human Gastric Cancer. J. Surg. Oncol. 1996, 62, 171–176. [Google Scholar] [CrossRef]
- Fernandes, E.; Ferreira, J.A.; Peixoto, A.; Freitas, R.; Relvas-Santos, M.; Palmeira, C.; Martins, G.; Barros, A.; Santos, L.L.; Sarmento, B.; et al. Glycoengineered Nanoparticles Enhance the Delivery of 5-Fluoroucil and Paclitaxel to Gastric Cancer Cells of High Metastatic Potential. Int. J. Pharm. 2019, 570, 118646. [Google Scholar] [CrossRef]
- Itai, S.; Arii, S.; Tobe, R.; Kitahara, A.; Kim, Y.-C.; Yamabe, H.; Ohtsuki, H.; Kirihara, Y.; Shigeta, K.; Kannagi, R. Significance of 2-3 and 2-6 Sialylation of Lewis a Antigen in Pancreas Cancer. Cancer 1988, 61, 775–787. [Google Scholar] [CrossRef]
- Itai, S.; Nishikata, J.; Yoneda, T.; Ohmori, K.; Yamabe, H.; Arii, S.; Tobe, T.; Kannagi, R. Tissue Distribution of 2-3 and 2-6 Sialyl Lewis a Antigens and Significance of the Ratio of Two Antigens for the Differential Diagnosis of Malignant and Benign Disorders of the Digestive Tract. Cancer 1991, 67, 1576–1587. [Google Scholar] [CrossRef]
- Tuo, X.H.; Itai, S.; Nishikata, J.; Mori, T.; Tanaka, O.; Kannagi, R. Stage-Specific Expression of Cancer-Associated Type 1 and Type 2 Chain Polylactosamine Antigens in the Developing Pancreas of Human Embryos. Cancer Res. 1992, 52, 5744–5751. [Google Scholar]
- Ferreira, J.A.; Magalhães, A.; Gomes, J.; Peixoto, A.; Gaiteiro, C.; Fernandes, E.; Santos, L.L.; Reis, C.A. Protein Glycosylation in Gastric and Colorectal Cancers: Toward Cancer Detection and Targeted Therapeutics. Cancer Lett. 2017, 387, 32–45. [Google Scholar] [CrossRef]
- Kudelka, M.R.; Antonopoulos, A.; Wang, Y.; Duong, D.M.; Song, X.; Seyfried, N.T.; Dell, A.; Haslam, S.M.; Cummings, R.D.; Ju, T. Cellular O-Glycome Reporter/Amplification to Explore O-Glycans of Living Cells. Nat. Methods 2016, 13, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Carrascal, M.; Silva, M.; Ferreira, J.A.; Azevedo, R.; Ferreira, D.; Silva, A.; Ligeiro, D.; Santos, L.; Sackstein, R.; Videira, P.A. A Functional Glycoproteomics Approach Identifies Cd13 as a Novel E-Selectin Ligand in Breast Cancer. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2069–2080. [Google Scholar] [CrossRef] [PubMed]
- Carrascal, M.A.; Silva, M.; Ramalho, J.S.; Pen, C.; Martins, M.; Pascoal, C.; Amaral, C.; Serrano, I.; Oliveira, M.J.; Sackstein, R.; et al. Inhibition of Fucosylation in Human Invasive Ductal Carcinoma Reduces E-Selectin Ligand Expression, Cell Proliferation, and Erk1/2 and P38 Mapk Activation. Mol. Oncol. 2018, 12, 579–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thankamony, S.P.; Sackstein, R. Enforced Hematopoietic Cell E- and L-Selectin Ligand (Hcell) Expression Primes Transendothelial Migration of Human Mesenchymal Stem Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 2258–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotton, S.; Azevedo, R.; Gaiteiro, C.; Ferreira, D.; Lima, L.; Peixoto, A.; Fernandes, E.; Neves, M.; Neves, D.; Amaro, T.; et al. Targeted O-Glycoproteomics Explored Increased Sialylation and Identified Muc16 as a Poor Prognosis Biomarker in Advanced-Stage Bladder Tumours. Mol. Oncol. 2017, 11, 895–912. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.V.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.; Roth, A.; Bork, P.; et al. The String Database in 2017: Quality-Controlled Protein-Protein Association Networks, Made Broadly Accessible. Nucleic Acids Res. 2016, 45, D362–D368. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Kalyana-Sundaram, S.; Mahavisno, V.; Varambally, R.; Yu, J.; Briggs, B.B.; Barrette, T.R.; Anstet, M.J.; Kincead-Beal, C.; Kulkarni, P.; et al. Oncomine 3.0: Genes, Pathways, and Networks in a Collection of 18,000 Cancer Gene Expression Profiles. Neoplasia 2007, 9, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. Cluego: A Cytoscape Plug-in to Decipher Functionally Grouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Galon, J.; Mlecnik, B. Cluepedia Cytoscape Plugin: Pathway Insights Using Integrated Experimental and in Silico Data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Steentoft, C.; Vakhrushev, S.Y.; Joshi, H.; Kong, Y.; Vester-Christensen, M.B.; Schjoldager, K.T.-B.G.; Lavrsen, K.; Dabelsteen, S.; Pedersen, N.B.; Da Silva, L.M.; et al. Precision Mapping of the Human O-Galnac Glycoproteome through Simplecell Technology. EMBO J. 2013, 32, 1478–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. Metaboanalyst 4.0: Towards More Transparent and Integrative Metabolomics Analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, H.; Freitas, D.; Gomes, C.; Gomes, J.; Magalhães, A.; Reis, C.A. Mucin-Type O-Glycosylation in Gastric Carcinogenesis. Biomolecules 2016, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Holgersson, J.; Löfling, J. Glycosyltransferases Involved in Type 1 Chain and Lewis Antigen Biosynthesis Exhibit Glycan and Core Chain Specificity. Glycobiology 2006, 16, 584–593. [Google Scholar] [CrossRef]
- Ozaki, H.; Matsuzaki, H.; Ando, H.; Kaji, H.; Nakanishi, H.; Ikehara, Y.; Narimatsu, H. Enhancement of Metastatic Ability by Ectopic Expression of St6galnaci on a Gastric Cancer Cell Line in a Mouse Model. Clin. Exp. Metastasis 2012, 29, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Freitas, D.; Campos, D.; Gomes, J.; Pinto, F.; Macedo, J.A.; Matos, R.; Mereiter, S.; Pinto, M.; Polónia, A.; Gartner, F.; et al. O-Glycans Truncation Modulates Gastric Cancer Cell Signaling and Transcription Leading to a More Aggressive Phenotype. EBioMedicine 2019, 40, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Domon, B.; Costello, C.E. A Systematic Nomenclature for Carbohydrate Fragmentations in Fab-Ms/Ms Spectra of Glycoconjugates. Glycoconj. J. 1988, 5, 397–409. [Google Scholar] [CrossRef]
- Lee, K.; Byun, K.; Hong, W.; Chuang, H.-Y.; Pack, C.-G.; Bayarsaikhan, E.; Paek, S.H.; Kim, H.; Shin, H.Y.; Ideker, T.; et al. Proteome-Wide Discovery of Mislocated Proteins in Cancer. Genome Res. 2013, 23, 1283–1294. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, S. Protein Mislocalization: Mechanisms, Functions and Clinical Applications in Cancer. Biochim. Biophys. Acta. 2014, 1846, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Fujiki, H.; Watanabe, T.; Suganuma, M. Cell-Surface Nucleolin Acts as a Central Mediator for Carcinogenic, Anti-Carcinogenic, and Disease-Related Ligands. J. Cancer Res. Clin. Oncol. 2014, 140, 689–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaoui, K.; Boudhraa, Z.; Khalifé, P.; Carmona, E.; Provencher, D.; Mes-Masson, A.-M. Ran Promotes Membrane Targeting and Stabilization of Rhoa to Orchestrate Ovarian Cancer Cell Invasion. Nat. Commun. 2019, 10, 2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Li, H.; Liu, N.; Xing, Y.; Zhou, G.; Wu, Y.; Liu, Y.; Chen, W.; Yue, J.; Han, B.; et al. The Implications and Mechanisms of the Extra-Nuclear Nucleolin in the Esophageal Squamous Cell Carcinomas. Med. Oncol. 2015, 32, 45. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.-M.; Zhang, P.; Liu, R.-Y.; Sang, Y.-X.; Zhou, C.; Xu, G.-C.; Yang, J.-L.; Tong, A.; Wang, C.-T. Phosphorylation and Changes in the Distribution of Nucleolin Promote Tumor Metastasis Via the Pi3k/Akt Pathway in Colorectal Carcinoma. FEBS Lett. 2014, 588, 1921–1929. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, X.; Tian, B.; Liu, J.; Yang, L.; Zeng, L.; Chen, T.; Hong, A.; Wang, X. Nucleolin-targeted Extracellular Vesicles as a Versatile Platform for Biologics Delivery to Breast Cancer. Theranostics 2017, 7, 1360–1372. [Google Scholar] [CrossRef]
- Monferran, S.; Paupert, J.; Dauvillier, S.; Salles, B.; Muller, C. The Membrane Form of the DNA Repair Protein Ku Interacts at the Cell Surface with Metalloproteinase 9. EMBO J. 2004, 23, 3758–3768. [Google Scholar] [CrossRef] [Green Version]
- Keller, S.; Kneissl, J.; Grabher-Meier, V.; Heindl, S.; Hasenauer, J.; Maier, D.; Mattes, J.; Winter, P.; Luber, B. Evaluation of Epidermal Growth Factor Receptor Signaling Effects in Gastric Cancer Cell Lines by Detailed Motility-Focused Phenotypic Characterization Linked with Molecular Analysis. BMC Cancer 2017, 17, 845. [Google Scholar] [CrossRef] [Green Version]
- Galizia, G.; Lieto, E.; Orditura, M.; Castellano, P.; La Mura, A.; Imperatore, V.; Pinto, M.; Zamboli, A.; De Vita, F.; Ferraraccio, F. Epidermal Growth Factor Receptor (EGFR) Expression is Associated With a Worse Prognosis in Gastric Cancer Patients Undergoing Curative Surgery. World J. Surg. 2007, 31, 1458–1468. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.-M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular Analysis of Gastric Cancer Identifies Subtypes Associated with Distinct Clinical Outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- Kannagi, R. Carbohydrate-Mediated Cell Adhesion Involved in Hematogenous Metastasis of Cancer. Glycoconj. J. 1997, 14, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Maeda, M.; Watanabe, G.; Imamura, M. High Serum Soluble E-Selectin Levels Are Associated with Postoperative Haematogenic Recurrence in Esophageal Squamous Cell Carcinoma Patients. Oncol. Rep. 2003, 10, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Nakashio, T.; Narita, T.; Sato, M.; Akiyama, S.; Kasai, Y.; Fujiwara, M.; Ito, K.; Takagi, H.; Kannagi, R. The Association of Metastasis with the Expression of Adhesion Molecules in Cell Lines Derived from Human Gastric Cancer. Anticancer Res. 1997, 17, 293–299. [Google Scholar] [PubMed]
- Natoni, A.; Smith, T.A.G.; Keane, N.; McEllistrim, C.; Connolly, C.; Jha, A.; Andrulis, M.; Ellert, E.; Raab, M.S.; Glavey, S.V.; et al. E-Selectin Ligands Recognised by Heca452 Induce Drug Resistance in Myeloma, Which Is Overcome by the E-Selectin Antagonist, Gmi-1271. Leukemia 2017, 31, 2642–2651. [Google Scholar] [CrossRef]
- Catarina, G.; Almeida, A.; Barreira, A.; Calheiros, J.; Pinto, F.; Abrantes, R.; Costa, A.; Polonia, A.; Campos, D.; Osório, H.; et al. Carcinoembryonic Antigen Carrying SLeX as a New Biomarker of More Aggressive Gastric Carcinomas. Theranostics 2019, 9, 7431–7446. [Google Scholar]
- Rho, J.-H.; Mead, J.R.; Wright, W.S.; Brenner, D.E.; Stave, J.W.; Gildersleeve, J.C.; Lampe, P.D. Discovery of Sialyl Lewis a and Lewis X Modified Protein Cancer Biomarkers Using High Density Antibody Arrays. J. Proteom. 2013, 96, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Alley, S.C.; O’Meara, M.; Gardai, S.J.; Okeley, N.M. Abstract Ddt02-02: Sgn-2ff: A Novel Small Molecule Inhibitor of Fucosylation with Preclinical Antitumor Activity through Multiple Immune Mechanisms. Cancer Res. 2017, 77, DDT02-02. [Google Scholar]
- Berger, C.M.; Gaume, X.; Bouvet, P. The Roles of Nucleolin Subcellular Localization in Cancer. Biochimie 2015, 113, 78–85. [Google Scholar] [CrossRef]
- Semenkovich, C.F.; Ostlund, R.E.; Olson, M.O.J.; Yang, J.W. A Protein Partially Expressed on the Surface of Hepg2 Cells That Binds Lipoproteins Specifically Is Nucleolin. Biochemistry 1990, 29, 9708–9713. [Google Scholar] [CrossRef]
- Suganuma, M.; Watanabe, T.; Yamaguchi, K.; Takahashi, A.; Fujiki, H. Human Gastric Cancer Development with Tnf-Alpha-Inducing Protein Secreted from Helicobacter Pylori. Cancer Lett. 2012, 322, 133–138. [Google Scholar] [CrossRef]
- Watanabe, T.; Tsuge, H.; Imagawa, T.; Kise, D.; Hirano, K.; Beppu, M.; Takahashi, A.; Yamaguchi, K.; Fujiki, H.; Suganuma, M. Nucleolin as Cell Surface Receptor for Tumor Necrosis Factor-Alpha Inducing Protein: A Carcinogenic Factor of Helicobacter Pylori. J. Cancer Res. Clin. Oncol. 2010, 136, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, B.; Schlüter, L.; Lange, P.; Mercanoglu, B.; Ewald, F.; Fölster, A.; Picksak, A.-S.; Harder, S.; El Gammal, A.T.; Grupp, K.; et al. Cosmc Knockdown Mediated Aberrant O-Glycosylation Promotes Oncogenic Properties in Pancreatic Cancer. Mol. Cancer 2015, 14, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losfeld, M.-E.; Leroy, A.; Coddeville, B.; Carpentier, M.; Mazurier, J.; Legrand, D. N-Glycosylation Influences the Structure and Self-Association Abilities of Recombinant Nucleolin. FEBS J. 2011, 278, 2552–2564. [Google Scholar] [CrossRef] [PubMed]
- Hoja-Łukowicz, D.; Kedracka-Krok, S.; Duda, W.; Litynska, A. The Lectin-Binding Pattern of Nucleolin and Its Interaction with Endogenous Galectin-3. Cell. Mol. Boil. Lett. 2014, 19, 461–482. [Google Scholar] [CrossRef]
- Watanabe, T.; Takahashi, A.; Suzuki, K.; Kurusu-Kanno, M.; Yamaguchi, K.; Fujiki, H.; Suganuma, M. Epithelial-Mesenchymal Transition in Human Gastric Cancer Cell Lines Induced by Tnf-Alpha-Inducing Protein of Helicobacter Pylori. Int. J. Cancer 2014, 134, 2373–2382. [Google Scholar] [CrossRef]
- Nair, N.; Shoaib, M.; Sørensen, C.S. Chromatin Dynamics in Genome Stability: Roles in Suppressing Endogenous DNA Damage and Facilitating DNA Repair. Int. J. Mol. Sci. 2017, 18, 1486. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.; Zhou, F.; Zhang, Q.; Sun, X.; Shi, X.; Liang, Y.; Wang, X.; Yue, L. Overexpression of Nucleolin and Different Expression Sites Both Related to the Prognosis of Gastric Cancer. APMIS 2013, 121, 919–925. [Google Scholar] [CrossRef]
- Farin, K.; Schokoroy, S.; Haklai, R.; Cohen-Or, I.; Elad-Sfadia, G.; Reyes-Reyes, E.; Bates, P.J.; Cox, A.D.; Kloog, Y.; Pinkas-Kramarski, R. Oncogenic Synergism between ErbB1, Nucleolin, and Mutant Ras. Cancer Res. 2011, 71, 2140–2151. [Google Scholar] [CrossRef] [Green Version]
- Schokoroy, S.; Juster, D.; Kloog, Y.; Pinkas-Kramarski, R. Disrupting the Oncogenic Synergism between Nucleolin and Ras Results in Cell Growth Inhibition and Cell Death. PLoS ONE 2013, 8, e75269. [Google Scholar] [CrossRef]
- Carrascal, M.A.; Talina, C.; Borralho, P.; Mineiro, A.G.; Henriques, A.R.; Pen, C.; Martins, M.; Braga, S.; Sackstein, R.; Videira, P.A. Staining of E-Selectin Ligands on Paraffin-Embedded Sections of Tumor Tissue. BMC Cancer 2018, 18, 495. [Google Scholar] [CrossRef]
- Goldson, T.M.; Turner, K.L.; Huang, Y.; Carlson, G.E.; Caggiano, E.G.; Oberhauser, A.F.; Fennewald, S.M.; Burdick, M.; Resto, V.A. Nucleolin Mediates the Binding of Cancer Cells to L-Selectin under Conditions of Lymphodynamic Shear Stress. Am. J. Physiol. Cell Physiol. 2020, 318, C83–C93. [Google Scholar] [CrossRef] [PubMed]
- De Bruijne-Admiraal, L.G.; Modderman, P.W.; Borne, A.E.V.D.; Sonnenberg, A. P-Selectin Mediates Ca(2+)-Dependent Adhesion of Activated Platelets to Many Different Types of Leukocytes: Detection by Flow Cytometry. Blood 1992, 80, 134–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Manufacture | Reference | Clonality | Clone | Host | Application | Experimental Conditions |
|---|---|---|---|---|---|---|---|
| Anti-TPR | Novus Biologicals, Centennial, CO, USA | NB100-2866 | Polyclonal | - | Rabbit | WB | 1:1000, 4 h RT |
| Anti-ß-Actin | Sigma-Aldrich, St. Louis, MO, USA | A1978 | Monoclonal | AC-15 | Mouse | WB | 1.4µg/mL, 1 h RT |
| Anti-B2M | Abcam, Cambridge, UK | ab75853 | Monoclonal | EP2978Y | Rabbit | WB | 1:100, 1 h, RT |
| Anti-CA19.9 (SLeA) | Abcam, Cambridge, UK | ab116024 | Monoclonal | CA19.9-9-203 | Mouse | IP, WB, IF, PLA, IHC, FC | Flow/IF—1:100, 1 h RT WB—1:1000, 1 h RT IHC—1:100, 4 °C/12 h |
| Anti-NCL | Abcam, Cambridge, UK | ab22758 | Polyclonal | - | Rabbit | IP, WB | WB—1:1000, 1 h RT |
| Anti-NCL | Abcam, Cambridge, UK | ab129200 | Monoclonal | EPR7952 | Rabbit | PLA, IHC | 1:250, 4 °C 12 h |
| Alexa Fluor 647 Anti-NCL | Abcam, Cambridge, UK | ab202709 | Monoclonal | EPR7952 | Rabbit | Flow, IF | 1:100, 1 h RT |
| Rabbit IgG isotype | Thermo Fisher Scientific, Waltham, MA, USA | LTI-02-6102 | - | - | Rabbit | IP, PLA, FC | The same as for anti-NCL |
| Mouse IgG1 isotype | Abcam, Cambridge, UK | ab18443 | - | - | Mouse | IP, PLA, FC | The same as for anti-CA19-9 |
| Anti-Rabbit IgG HRP | Thermo Fisher Scientific, Waltham, MA, USA | 656120 | Polyclonal | - | Goat | WB | 1:60,000, 30 min. RT |
| Anti-Mouse IgG HRP | Jackson ImmunoResearch, West Grove, PA, USA | 115-035-205 | Polyclonal | - | Goat | WB | 1:70,000, 30 min. RT |
| Alexa Fluor 594 Anti-Rabbit IgG | Thermo Fisher Scientific, Waltham, MA, USA | A11012 | Polyclonal | - | Goat | IF | 1:300, 30 min. RT |
| Alexa Fluor 488 Anti-Mouse IgG | Thermo Fisher Scientific, Waltham, MA, USA | A11001 | Polyclonal | - | Goat | IF, FC | IF—1:300, 30 min. RT Flow—1:300, 15 min. RT |
| FITC Anti-human IgG | Agilent Technologies, Santa Clara, CA, USA | F0202 | Polyclonal | - | Rabbit | IF | 1:100, 30 min. RT |
| Alexa Fluor 594 Anti-mouse IgG | Thermo Fisher Scientific, Waltham, MA, USA | A11005 | Polyclonal | - | Goat | IF | 1:300, 30 min. RT |
| Recombinant Mouse E-Selectin Ig Chimera Protein | R&D Systems, Minneapolis, MN, USA | 575-ES-100 | - | - | Mouse | IF | 1 µg/mL, 2 h RT |
| Title | n for SLeA (%) | n for NCL (%) |
|---|---|---|
| Stage | ||
| I | 18 (9%) | 2 (4%) |
| II | 60 (30%) | 12 (26%) |
| III | 89 (44%) | 25 (53%) |
| IV | 35 (17%) | 8 (17%) |
| Tumor (T) | ||
| T1 | 12 (6%) | 0 (0%) |
| T2 | 17 (8%) | 0 (0%) |
| T3 | 105 (52%) | 26 (55%) |
| T4 | 64 (32%) | 21 (45%) |
| Missing Information | 4 (2%) | 0 (0%) |
| Lymph node metastasis (N) | ||
| N0 | 49 (24%) | 10 (21%) |
| N1 | 40 (20%) | 8 (17%) |
| N2 | 32 (16%) | 7 (15%) |
| N3 | 76 (37%) | 21 (45%) |
| Missing Information | 5 (3%) | 1 (2%) |
| Distant metastasis (M) | ||
| M0 | 151 (75%) | 36 (77%) |
| M1 | 36 (19%) | 8 (17%) |
| Missing Information | 15 (7%) | 3 (6%) |
| Lauren Classification | ||
| Intestinal subtype | 124 (61%) | 23 (49%) |
| Mixed | 22 (11%) | 0 (0%) |
| Diffuse | 51 (25%) | 24 (51%) |
| Missing Information | 5 (2%) | 0 (0%) |
| Borrmann Classification | ||
| Type I | 9 (5%) | 1 (2%) |
| Type II | 76 (38%) | 18 (38%) |
| Type III | 42 (21%) | 7 (15%) |
| Type IV | 58 (29%) | 18 (38%) |
| Missing Information | 17 (8%) | 3 (6%) |
| Overall Survival (OS) | ||
| Median (months±SD) | 26 ± 34 | 28 ± 32 |
| Mean (months±SD) | 40 ± 34 | 40 ± 32 |
| Age | 64 ± 13 | 66 ± 12 |
| Gender | ||
| Male | 109 (54%) | 23 (49%) |
| Female | 93 (46%) | 24 (51%) |
| Clinicopathological Variables | SLeA Expression n (%) | p*-Value | NCLmem n (%) | p*-Value | NCLmem+SLeA n (%) | p*-Value |
|---|---|---|---|---|---|---|
| Stage | ||||||
| I | 8 (44%) | 0.020 | 2 (100%) | 0.337 | 0 (0%) | 0.772 |
| II | 37 (62%) | 4 (33%) | 4 (33%) | |||
| III | 50 (57%) | 12 (48%) | 9 (36%) | |||
| IV | 29 (83%) | 3 (38%) | 3 (38%) | |||
| Tumor size/extension (T) | ||||||
| T1 | 6 (50%) | 0.633 | - | 0.716 | - | 0.927 |
| T2 | 11 (65%) | - | - | |||
| T3 | 68 (65%) | 11 (42%) | 9 (35%) | |||
| T4 | 36 (63%) | 10 (48%) | 7 (33%) | |||
| Lymph node metastasis (N) | ||||||
| N0 | 28 (57%) | 0.651 | 4 (40%) | 0.862 | 2 (20%) | 0.551 |
| N1 | 24 (60%) | 3 (38%) | 2 (25%) | |||
| N2 | 18 (56%) | 4 (57%) | 3 (43%) | |||
| N3 | 50 (67%) | 10 (48%) | 9 (43%) | |||
| Distant metastasis (M) | ||||||
| M0 | 84 (56%) | 0.005 | 18 (50%) | 0.522 | 13 (33%) | 0.941 |
| M1 | 29 (81%) | 3 (38%) | 3 (38%) | |||
| Lauren Classification | ||||||
| Intestinal subtype | 77 (62%) | 0.191 | 11 (48%) | 0.671 | 8 (35%) | 0.917 |
| Mixed | 10 (46%) | - | - | |||
| Diffuse | 35 (69%) | 10 (42%) | 8 (33%) | |||
| Borrmann Classification | ||||||
| Type I | 4 (44%) | 0.744 | 1 (100%) | 0.641 | 1 (100%) | 0.228 |
| Type II | 46 (61%) | 7 (39%) | 5 (28%) | |||
| Type III | 27 (64%) | 3 (43%) | 1 (14%) | |||
| Type IV | 35 (61%) | 9 (50%) | 9 (50%) | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, E.; Freitas, R.; Ferreira, D.; Soares, J.; Azevedo, R.; Gaiteiro, C.; Peixoto, A.; Oliveira, S.; Cotton, S.; Relvas-Santos, M.; et al. Nucleolin-Sle A Glycoforms as E-Selectin Ligands and Potentially Targetable Biomarkers at the Cell Surface of Gastric Cancer Cells. Cancers 2020, 12, 861. https://doi.org/10.3390/cancers12040861
Fernandes E, Freitas R, Ferreira D, Soares J, Azevedo R, Gaiteiro C, Peixoto A, Oliveira S, Cotton S, Relvas-Santos M, et al. Nucleolin-Sle A Glycoforms as E-Selectin Ligands and Potentially Targetable Biomarkers at the Cell Surface of Gastric Cancer Cells. Cancers. 2020; 12(4):861. https://doi.org/10.3390/cancers12040861
Chicago/Turabian StyleFernandes, Elisabete, Rui Freitas, Dylan Ferreira, Janine Soares, Rita Azevedo, Cristiana Gaiteiro, Andreia Peixoto, Sara Oliveira, Sofia Cotton, Marta Relvas-Santos, and et al. 2020. "Nucleolin-Sle A Glycoforms as E-Selectin Ligands and Potentially Targetable Biomarkers at the Cell Surface of Gastric Cancer Cells" Cancers 12, no. 4: 861. https://doi.org/10.3390/cancers12040861