Metabolic Heterogeneity of Cancer Cells: An Interplay between HIF-1, GLUTs, and AMPK



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Hypoxia-Inducible Factors
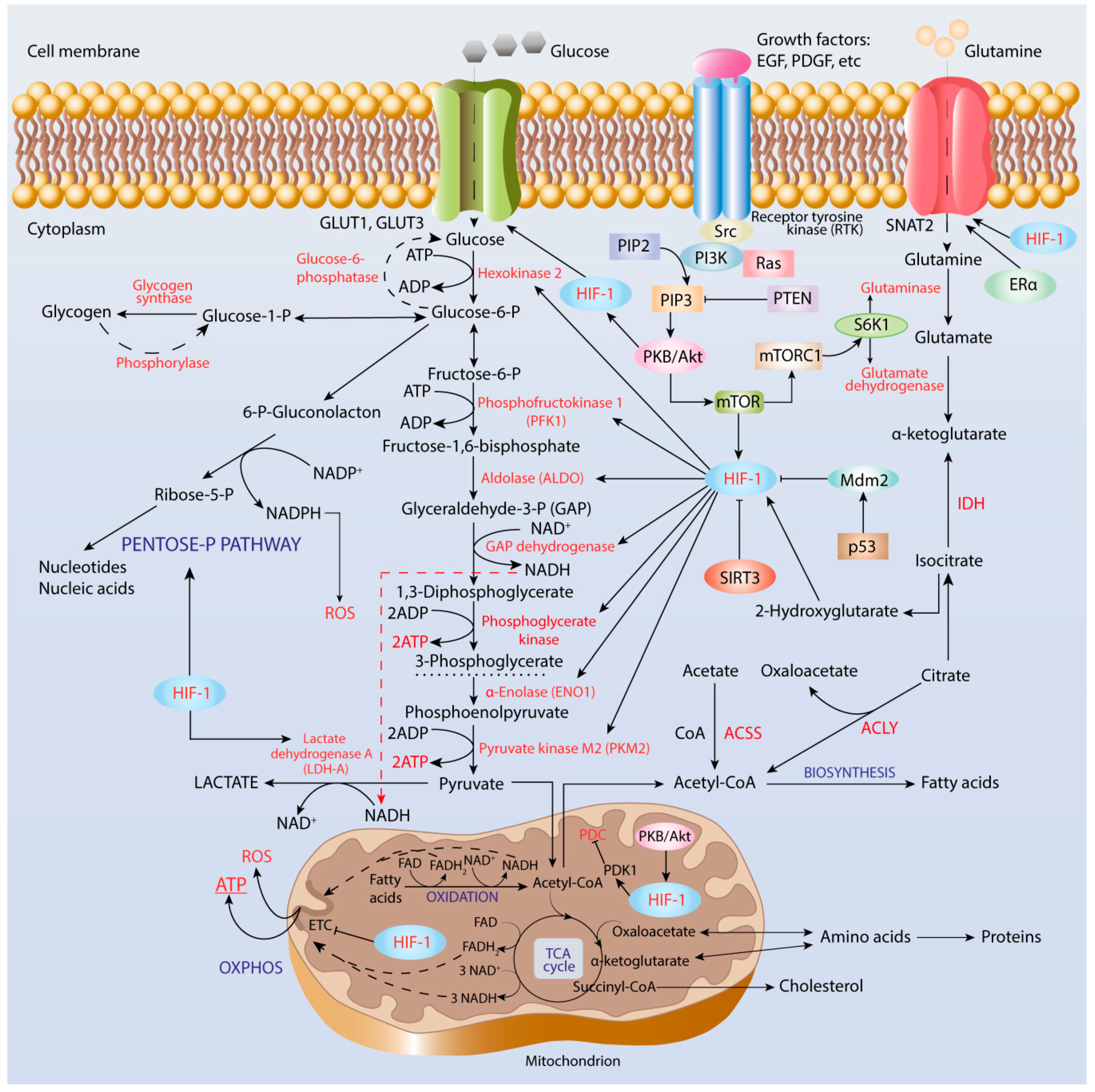
3. Interplay between HIF-1 and Facilitative Glucose Transporters
4. Role of HIF-1 in Metabolic Reprogramming of Cancer Cells
4.1. Enhancement of Glycolysis
4.2. Pentose Phosphate Pathway
4.3. Cancer Acidification and Its Role in Reverse to OXPHOS
4.4. Lipid Biosynthesis
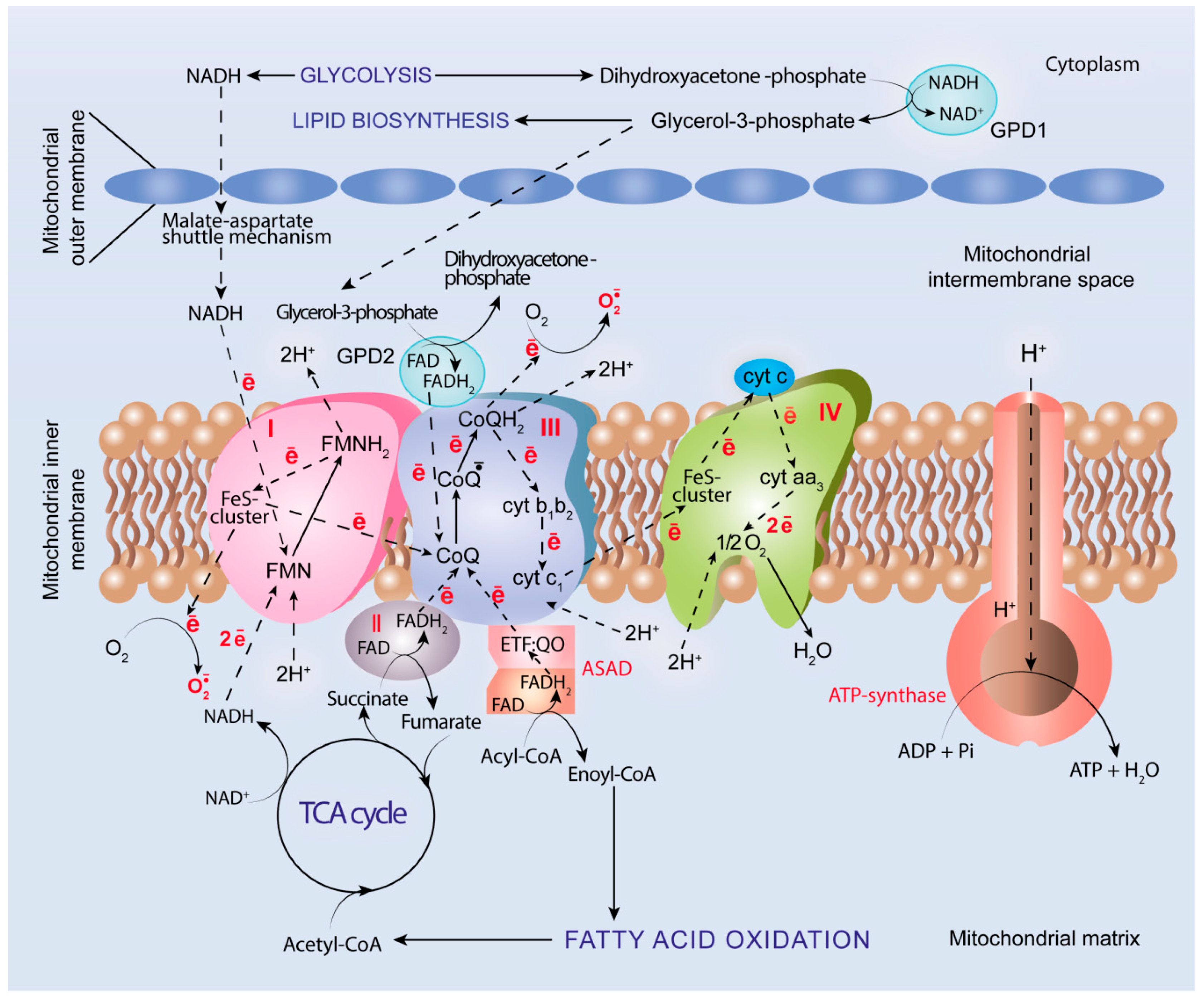
5. Oxidative Metabolism and OXPHOS in Cancer
5.1. PDC and TCA Cycle
5.2. Glutaminolysis
5.3. Fatty Acid β-Oxidation
5.4. ROS Generation
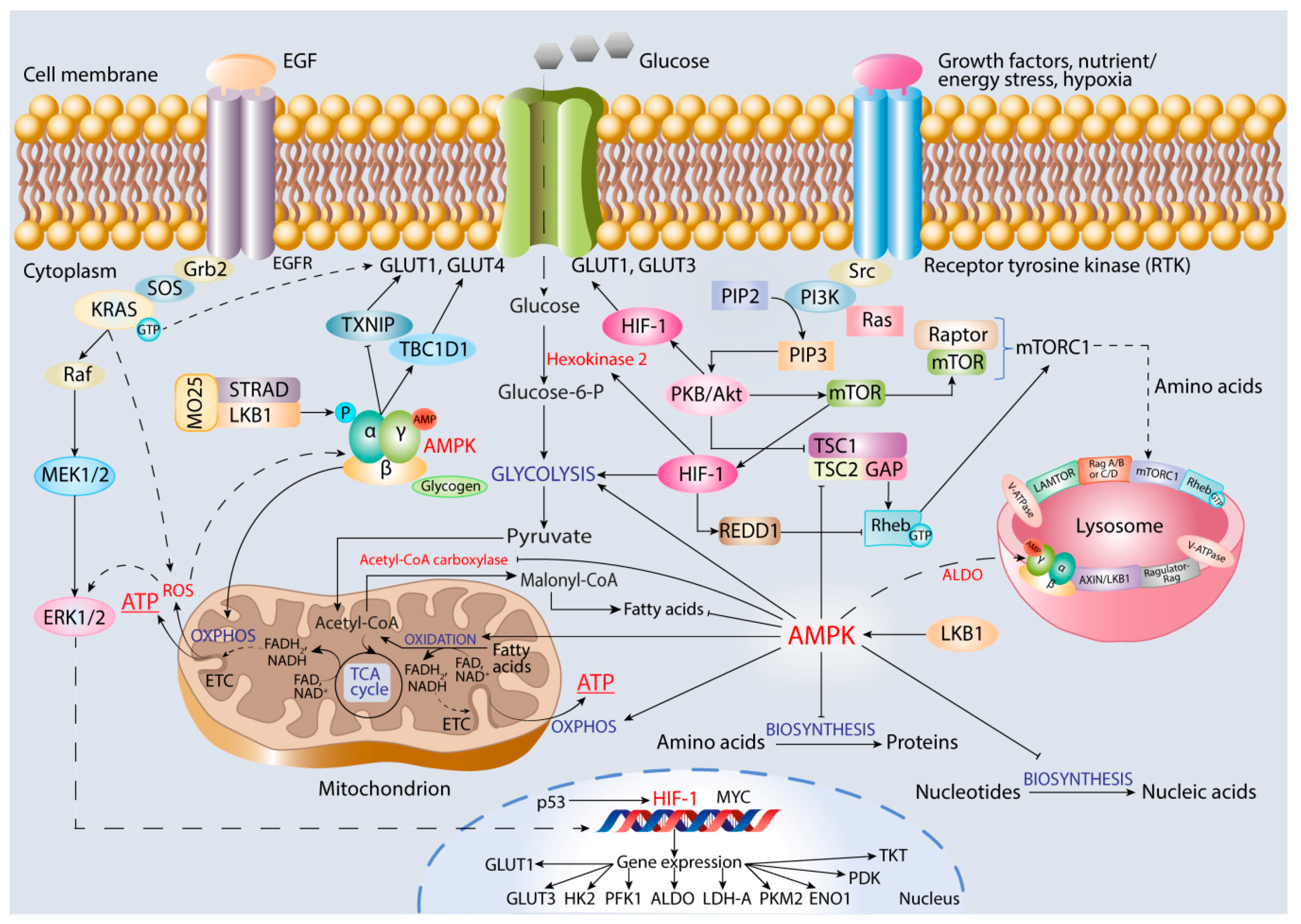
6. Role of AMPK in Promoting Cancer Cell Oxidative Metabolism
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HIF-1 | hypoxia-inducible factor-1 |
| AMPK | AMP-activated protein kinase |
| GLUT | glucose transporter |
| ETC | electron transportation chain |
| OXPHOS | oxidative phosphorylation |
| PDC | pyruvate dehydrogenase complex |
| PDK | pyruvate dehydrogenase complex kinase |
| PFK | phosphofructokinase |
| PK | pyruvate kinase |
| LDH | lactate dehydrogenase |
| TCA | tricarboxylic acid |
| EGF | epidermal growth factor |
| VEGF | vascular endothelial growth factor |
| PDGF | platelet-derived growth factor |
| PKB | protein kinase B |
| mTOR | mechanistic (mammalian) target of rapamycin |
| UPS | ubiquitin-proteasome system |
References
- Smith, K.A.; Waypa, G.B.; Schumacker, P.T. Redox signaling during hypoxia in mammalian cells. Redox Biol. 2017, 13, 228–234. [Google Scholar] [CrossRef]
- Ralph, S.J.; Rodriguez-Enriguez, S.; Neuzil, J.; Saavedra, E.; Moreno-Sanchez, R. The causes of cancer revisited: “Mitochondrial malignancy” and ROS-induced oncogenic transformation—Why mitochondria are targets for cancer therapy. Mol. Asp. Med. 2010, 31, 145–170. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Hypoxia-/HIF-1α-driven factors of the tumor microenvironment impeding antitumor immune response and promoting malignant progression. Adv. Exp. Med. Biol. 2018, 1072, 171–175. [Google Scholar]
- Caino, M.C.; Altieri, D.C. Molecular pathways: Mitochondrial reprogramming in tumor progression and therapy. Clin. Cancer Res. 2016, 22, 540–545. [Google Scholar] [CrossRef]
- Francis, A.; Venkatesh, G.H.; Zaarour, R.F.; Zeinelabdin, N.A.; Nawafleh, H.H.; Prasad, P.; Buart, S.; Terry, S.; Chouaib, S. Tumor hypoxia: A key determinant of microenvironment hostility and major checkpoint during the antitumor response. Crit. Rev. Immunol. 2018, 38, 505–524. [Google Scholar] [CrossRef]
- Gu, Q.; He, Y.; Ji, J.; Yao, Y.; Shen, W.; Luo, J.; Zhu, W.; Cao, H.; Geng, Y.; Zhang, S.; et al. Hypoxia-inducible factor-1α (HIF-1α) and reactive oxygen species (ROS) mediates radiation-induced invasiveness through the SDF-1α/CXCR4 pathway in non-small cell lung carcinoma cells. Oncotarget 2015, 6, 10893–10907. [Google Scholar] [CrossRef]
- Choudhry, H.; Harria, A. Advances in hypoxia-inducible factor biology. Cell Metabol. 2018, 27, 281–298. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Racker, E. Bioenergetics and the problem of tumor growth: An understanding of the mechanism of the generation and control of biological energy may shed light on the problem of tumor growth. Am. Sci. 1972, 60, 56–63. [Google Scholar] [PubMed]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Nanduri, J.; Khan, S.; Semenza, G.L.; Prabhakar, N.R. Induction of HIF-1alpha expression by intermittent hypoxia: Involvement of NADPH oxidase, Ca2þ signaling, prolyl hydroxylases, and mTOR. J. Cell Physiol. 2008, 217, 674–685. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. Metabolic reprogramming by class I and II histone deacetylases. Trends Endocrinol. Metab. 2013, 24, 48–57. [Google Scholar] [CrossRef]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef]
- van der Heijden, M.; Miedema, D.M.; Waclaw, B.; Veenstra, V.L.; Lecca, M.C.; Nijman, L.E.; van Dijk, E.; van Neerven, S.M.; Lodestijin, S.C.; Lenos, K.J.; et al. Spatiotemporal regulation of clonogenicity in colorectal cancer xenografts. Proc. Natl. Acad. Sci. USA 2019, 116, 6140–6145. [Google Scholar] [CrossRef]
- Campbell, P.J.; Pleasance, E.D.; Stephens, P.J.; Dicks, E.; Rance, R.; Goodhead, I.; Follows, G.A.; Green, A.R.; Futreal, P.A.; Stratton, M.R. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc. Natl. Acad. Sci. USA 2008, 105, 13081–13086. [Google Scholar] [CrossRef]
- Russo, M.; Siravegna, G.; Blaszkowsky, L.S.; Corti, G.; Crisafulli, G.; Ahronian, L.G.; Mussolin, B.; Kwak, E.L.; Buscarino, M.; Lazzari, L.; et al. Tumor heterogeneity and lesion-specific response to targeted therapy in colorectal cancer. Cancer Discov. 2016, 6, 147–153. [Google Scholar] [CrossRef]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative metabolic flux profiling of melanoma cell lines: Beyond the Warburg effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef]
- Gentric, G.; Mieulet, V.; Mechta-Grigorou, F. Heterogeneity in cancer metabolism: New concepts in an old field. Antioxid Redox Signal 2017, 26, 462–485. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.; Sotgia, F.; Lisanti, M.P. Tumor microenvironment and metabolic synergy in breast cancers: Critical importance of mitochondrial fuels and function. Semin. Oncol. 2014, 41, 195–216. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signaling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Lutsenko, S.V.; Terentiev, A.A. Reactive oxygen and nitrogen species-induced protein modifications: Implication in carcinogenesis and anti-cancer therapy. Cancer Res. 2018, 78, 6040–6047. [Google Scholar] [CrossRef]
- Hielscher, A.; Gerecht, S. Hypoxia and free radicals: Role in tumor progression and the use of engineering-based platforms to address these relationships. Free Radic. Biol. Med. 2015, 79, 281–291. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Samanta, D.; Semenza, G.L. Maintenance of redox homeostasis by hypoxia-inducible factors. Redox Biol. 2017, 13, 331–335. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix heterodimer that is regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Saito, S.; Lin, Y.-C.; Tsai, M.-H.; Lin, C.-S.; Murayama, Y.; Sato, R.; Yokoyama, K.K. Emerging roles of hypoxia-inducible factors and reactive oxygen species in cancer and pluripotent stem cells. Kaohsiung J. Med. Sci. 2015, 31, 279–286. [Google Scholar] [CrossRef]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta 2010, 1797, 1171–1177. [Google Scholar] [CrossRef]
- Solaini, G.; Sgarbi, G.; Baracca, A. Oxidative phosphorylation in cancer cells. Biochim. Biophys. Acta 2010, 1807, 534–542. [Google Scholar] [CrossRef]
- Berra, E.; Benizri, E.; Ginouves, A.; Volmat, V.; Roux, D.; Pouyssegur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1 alpha in normoxia. EMBO J. 2003, 22, 4082–4090. [Google Scholar] [CrossRef]
- Ginouves, A.; Ilc, K.; Macías, N.; Pouysségur, J.; Berra, E. PHDs overactivation during chronic hypoxia “desensitizes” HIFα and protects cells from necrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 4745–4750. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Activation of the HIF pathway in cancer. Curr. Opin. Genet. Dev. 2001, 11, 293–299. [Google Scholar] [CrossRef]
- Blancher, C.; Moore, J.W.; Robertson, N.; Harris, A.L. Effects of ras and von Hippel-Lindau (VHL) gene mutations on hypoxia-inducible factor (HIF)-1alpha, HIF-2alpha, and vascular endothelial growth factor expression and their regulation by the phosphatidylinositol 3’-kinase/Akt signaling pathway. Cancer Res. 2001, 61, 7349–7355. [Google Scholar] [PubMed]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef]
- Kelly, C.; Smallbone, K.; Brady, M. Tumour glycolysis: The many faces of HIF. J. Theor. Biol. 2008, 254, 508–513. [Google Scholar] [CrossRef]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef]
- Ma, W.; Sung, H.J.; Park, J.Y.; Matoba, S.; Hwang, P.M. A pivotal role for p53: Balancing aerobic respiration and glycolysis. J. Bioenerg. Biomembr. 2007, 39, 243–246. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 2009, 1787, 414–420. [Google Scholar] [CrossRef]
- Sauer, H.; Engel, S.; Milosevic, N.; Sharifpanah, F.; Wartenberg, M. Activation of AMP-kinase by AICAR induces apoptosis of DU-145 prostate cancer cells through generation of reactive oxygen species and activation of c-Jun N-terminal kinase. Int. J. Oncol. 2012, 40, 501–508. [Google Scholar] [CrossRef]
- Stine, Z.P.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, metabolism, and cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Dang, C.V. A time for MYC: Metabolism and therapy. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 79–83. [Google Scholar] [CrossRef]
- Madan, E.; Parker, T.M.; Pelham, C.J.; Palma, A.M.; Peixoto, M.L.; Nagane, M.; Chandaria, A.; Tomás, A.R.; Canas-Marques, R.; Henriques, V.; et al. HIF-transcribed p53 chaperones HIF-1α. Nucleic Acids Res. 2019, 47, 10212–10234. [Google Scholar] [CrossRef]
- Robertson, E.D.; Semenchenko, K.; Wasylyk, B. Crosstalk between Mdm2, p53 and HIF-1α: Distinct responses to oxidative stress and implications for tumour hypoxia. Subcell. Biochem. 2014, 85, 199–214. [Google Scholar] [PubMed]
- Bartoletti-Stella, A.; Mariani, E.; Kurelac, I.; Maresca, A.; Caratozzolo, M.F.; Iommarini, L.; Carelli, V.; Eusebi, L.H.; Guido, A.; Cenacchi, G.; et al. Gamma rays induce a p53-dependent mitochondrial biogenesis that is counter-regulated by HIF-1α. Cell Death Dis. 2013, 4, e663. [Google Scholar] [CrossRef]
- Chun, S.Y.; Johnson, C.; Washburn, J.G.; Cruz-Correa, M.R.; Dang, D.T.; Dang, L.H. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1α and HIF-2α target genes. Mol. Cancer. 2010, 9, 293. [Google Scholar] [CrossRef]
- Ogrunc, R.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gorgoulis, V.G.; d’Adda di Fagagna, F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014, 21, 998–1012. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Pullen, N.; Brennan, P.; Cantrell, D.; Dennis, P.B.; Thomas, G. Regulation of an activated S6 kinase 1 variant reveals a novel mammalian target of rapamycin phosphorylation site. J. Biol. Chem. 2002, 277, 20104–20112. [Google Scholar] [CrossRef]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef]
- Gong, T.; Cui, L.; Wang, H.; Wang, H.; Han, N. Knockdown of KLF5 suppresses hypoxia-induced resistance to cisplatin in NSCLC cells by regulating HIF-1α-dependent glycolysis through inactivation of the PI3K/Akt/mTOR pathway. J. Transl. Med. 2018, 16, 164. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Vaira, V.; Caino, M.C.; Tang, H.Y.; Seo, J.H.; Kossenkov, A.V.; Ottobrini, L.; Martelli, C.; Lucignani, G.; Bertolini, I.; et al. Mitochondrial Akt regulation of hypoxic tumor reprogramming. Cancer Cell 2016, 30, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Kiwai, M.T.; Kitadai, Y.; Tanaka, S.; Onogawa, S.; Matsutani, N.; Kaio, E.; Ito, M.; Chayama, K. Expression of hypoxia-inducible factor-1alpha is associated with tumor vascularization in human colorectal carcinoma. Int. J. Cancer 2003, 105, 176–181. [Google Scholar] [CrossRef]
- Huang, G.W.; Yang, L.Y.; Lu, W.Q. Expression of hypoxia-inducible factor 1alpha and vascular endothelial growth factor in hepatocellular carcinoma: Impact on neovascularization and survival. World J. Gastroenterol. 2005, 11, 1705–1708. [Google Scholar] [CrossRef]
- Naruse, T.; Kawasaki, G.; Yanamoto, S.; Mizuno, A.; Umeda, M. Immunochemical study of VEGF expression in oral squamous cell carcinomas: Correlation with the mTOR-HIF-1α pathway. Anticancer Res. 2011, 31, 4429–4437. [Google Scholar] [PubMed]
- Shao, J.B.; Li, Z.; Zhang, N.; Yang, F.; Gao, W.; Sun, Z.G. Hypoxia-inducible factor-1α in combination with vascular endothelial growth factor could predict the prognosis of postoperative patients with oesophageal squamous cell carcinoma. Pol. J. Pathol. 2019, 70, 84–90. [Google Scholar] [CrossRef]
- Fu, Z.; Chen, D.; Cheng, H.; Wang, F. Hypoxia-inducible factor-1α protects cervical carcinoma cells from apoptosis induced by radiation via modulation of vascular endothelial growth factor and p53 under hypoxia. Med. Sci. Monit. 2015, 21, 318–325. [Google Scholar] [PubMed]
- Zhang, Y.; Chen, C.; Zhang, J. Effects and significance of formononetin on expression levels of HIF-1α and VEGF in mouse cervical cancer tissue. Oncol. Lett. 2019, 18, 2248–2253. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef]
- Zhu, Y.; Yan, Y.; Gius, D.R.; Vassilpoulos, A. Metabolic regulation of sirtuins upon fasting and the implication for cancer. Curr. Opn. Oncol. 2013, 25, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, S.; Liu, X.; Chen, Y.; Denog, H. SIRT3 overexpression inhibits growth of kidney tumor cells and enhances mitochondrial biogenesis. J. Proteome. Res. 2018, 17, 3143–3152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Liu, L.; Cai, M.; Pan, Y.; Fu, J.; Cao, Y.; Yun, J. Low SIRT3 expression correlates with poor differentiation and unfavorable prognosis in primary hepatocellular carcinoma. PLoS ONE 2012, 7, e51703. [Google Scholar] [CrossRef]
- Kweon, K.H.; Lee, C.R.; Jung, S.J.; Ban, E.J.; Kang, S.-W.; Jeong, J.J.; Nam, K.-H.; Jo, Y.S.; Lee, J.; Chung, W.Y. Sirt1 induction confers resistance to etoposide-induced genotoxic apoptosis in thyroid cancers. Int. J. Oncol. 2014, 45, 2065–2075. [Google Scholar] [CrossRef]
- Finley, L.W.; Haigis, M.C. Metabolic regulation by SIRT3: Implications for tumorigenesis. Trends Mol. Med. 2012, 18, 516–523. [Google Scholar] [CrossRef]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef]
- Xiong, Y.; Wang, L.; Wang, S.; Wang, M.; Zhao, J.; Zhang, Z.; Li, X.; Jia, L.; Han, Y. SIRT3 deacetylates and promotes degradation of PTEN-defective non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2018, 144, 189–198. [Google Scholar] [CrossRef]
- Scheepers, A.; Joost, H.-G.; Schurmann, A. The glucose transporter families SGLT and GLUT: Molecular basis of normal and aberrant function. JPEN J. Parenter. Enteral Nutr. 2004, 28, 364–371. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Toyoda, T.; Taylor, E.B.; Yu, H.; Fujii, N.; Hirshman, M.F.; Goodyear, L.J. TBC1D1 regulates insulin- and contraction-induced glucose transport in mouse skeletal muscle. Diabetes 2010, 59, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Mafakheri, S.; Flörke, R.R.; Kanngieber, S.; Hartwig, S.; Espelage, L.; De Wendt, C.; Schönberger, T.; Hamker, N.; Lehr, S.; Chadt, A.; et al. AKT and AMP-activated protein kinase regulate TBC1D1 through phosphorylation and its interaction with the cytosolic tail of insulin-regulated aminopeptidase IRAP. J. Biol. Chem. 2018, 16293, 17853–17862. [Google Scholar] [CrossRef] [PubMed]
- Ancey, P.-B.; Contat, C.; Meylan, E. Glucose transporters in cancer—From tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef] [PubMed]
- Jóźwiak, P.; Krzeŝlak, A.; Pomorski, L.; Lipińka, A. Expression of hypoxia-related glucose transporters GLUT1 and GLUT3 in benign, malignant and non-neoplastic thyroid lesions. Mol. Med. Rep. 2012, 6, 601–606. [Google Scholar] [CrossRef][Green Version]
- Krzeŝlak, A.; Wojcik-Krowiranda, K.; Forma, E.; Jóźwiak, P.; Romanowicz, H.; Bienkiewicz, A.; Brys, M. Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol. Oncol. Res. 2012, 18, 721–728. [Google Scholar] [CrossRef]
- Kuo, C.C.; Ling, H.H.; Chiang, M.C.; Chung, C.H.; Lee, W.Y.; Chu, C.Y.; Wu, Y.C.; Chen, C.H.; Lai, Y.W.; Tsai, I.L.; et al. Metastatic colorectal cancer rewrites metabolic program through a Glut3-YAP-dependent signaling. Theranostics 2019, 9, 2526–2540. [Google Scholar] [CrossRef]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.-H.; Wen, J.; Asara, J.; McGraw, T.E. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef]
- Tanegashiwa, K.; Sato-Miyata, Y.; Funakoshi, M.; Nishito, Y.; Aigaki, T.; Hara, T. Epigenetic regulation of the glucose transporter gene Slc2a1 by the β-hydroxybutyrate underlies preferential glucose supply to the brain of fasted mice. Genes Cells 2017, 22, 71–83. [Google Scholar] [CrossRef]
- Masin, M.; Vazquez, J.; Rossi, S.; Groeneveld, S.; Samson, N.; Schwalie, P.C.; Deplanck, B.; Frawley, L.E.; Gouttenoire, J.; Moradpour, D.; et al. GLUT3 is induced during epithelial-mesenchymaal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metabol. 2014, 2, 11. [Google Scholar] [CrossRef]
- Ali, A.; Levantini, E.; Fhu, C.W.; Teo, J.T.; Clohessy, J.G.; Goggi, J.L.; Wu, C.-S.; Chen, L.; Chin, T.M.; Tenen, D.G. CAV1-GLUT3 signaling is important for cellular energy and can be targeted by atorvastatin in non-small cell lung cancer. Theranostics 2019, 9, 6157–6174. [Google Scholar] [CrossRef]
- Aida, E.; Yasuda, M.; Miyazawa, M.; Fujita, M.; Osamura, R.Y.; Harisawa, T.; Muramatsu, T.; Murakami, M.; Saito, K.; Mikami, M. Hypoxic status in ovarian serous and mucinous tumors: Relationship between histological characteristics and HIF-1alpha/GLUT-1 expression. Arch. Gynecol. Obstet. 2008, 277, 539–546. [Google Scholar] [PubMed]
- Wincewicz, A.; Sulkowska, M.; Koda, M.; Sulkowski, S. Clinicopathological significance and linkage of the distribution of HIF-1 in human primary colorectal cancer. Pathol. Oncol. Res. 2007, 13, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.S.; Liu, Q.; Tian, C.; Zhang, D.X.; Wang, B.; Zhou, D.X.; Li, Z.P.; Yuan, Z.X. Correlation and expression analysis of hypoxia-inducible factor 1α, glucose transporter 1 and lactate dehydrogenase 5 in human gastric cancer. Oncol. Lett. 2019, 18, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.Y.; Hao, L.S.; Guo, D.Z. Expression of hypoxia-inducible factor 1α, glucose transporter 1 and lactate dehydrogenase 5 in colorectal cancer and clinicopathological significance. Zhonghua Bing Li Xue Za Zhi 2017, 46, 93–97. [Google Scholar] [PubMed]
- Bobarykina, A.Y.; Minchenko, D.O.; Opentanova, I.L.; Moenner, M.; Caro, J.; Esumi, H.; Minchenko, O.H. Hypoxic regulation of PFKFB and PFKFB-4 gene expression in gastric and pancreatic cell lines and expression of PFKFB genes in gastric cancers. Acta Biochim. Pol. 2006, 53, 789–799. [Google Scholar] [CrossRef]
- Cori, C.F.; Cori, G.T. The carbohydrate metabolism of tumors: II. Changes in the sugar, lactic acid, and CO2-combining power of blood passing through a tumor. J. Biol. Chem. 1925, 65, 397–405. [Google Scholar]
- Ristow, M. Oxidative metabolism in cancer growth. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 339–345. [Google Scholar] [CrossRef]
- Shoshan, M. On mitochondrial metabolism in tumor biology. Curr. Opin. Oncol. 2017, 29, 48–54. [Google Scholar] [CrossRef]
- Cancemi, P.; Buttacavoli, N.; Roz, E.; Feo, S. Expression of alpha-enolase (ENO1), Myc promoter-binding protein-1 (MBP-1) and matrix metalloproteinases (MMP-2 and MMP-9) reflect the nature and aggressiveness of breast tumors. Int. J. Mol. Sci. 2016, 20, 3952. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, X.; Li, J.; Yang, H.; Lin, X. Aldolase A as a prognostic factor and mediator of progression via inducing epithelial-to-mesenchymal transition in gastric cancer. J. Cell Mol. Med. 2018, 22, 4377–4386. [Google Scholar] [CrossRef]
- Yasuda, A.S.; Mori, A.; Isobe, N.; Yang, W.; Oe, H.; Fujimoto, A.; Yonenaga, Y.; Sakashita, H.; Imamura, M. Hexokinase II and VEGF expression in liver tumors: Correlation with hypoxia-inducible factor 1 alpha and its significance. J. Hepatol. 2004, 40, 117–123. [Google Scholar] [CrossRef]
- Jin, Z.; Gu, J.; Xin, X.; Li, Y.; Wang, H. Expression of hexokinase 2 in epithelial ovarian tumors and its clinical significance in serous ovarian cancer. Eur. J. Gynaecol. Oncol. 2014, 35, 519–524. [Google Scholar] [PubMed]
- Guzman, G.; Chennuri, R.; Chan, A.; Rea, B.; Quintana, A.; Patel, R.; Xu, P.Z.; Xie, H.; Hay, N. Evidence for heightened hexokinase II immunoexpression in hepatocyte dysplasia and hepatocellular carcinoma. Dig. Dis. Sci. 2015, 60, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Bartrons, R.; Simon-Molas, H.; Rodriguez-Garcia, A.; Castano, E.; Navarro-Sabaté, A.; Manzano, A.; Martinez-Outschoom, U.E. Fructose 2.6-bisphosphate in cancer cell metabolism. Front. Oncol. 2018, 8, 331. [Google Scholar] [CrossRef]
- Yang, H.; Shu, Z.; Jiang, Y.; Mao, W.; Pang, L.; Redwood, A.; Jeter-Jones, S.L.; Jennings, N.B.; Ometas, A.; Zhou, J.; et al. 6-Phosphofructo-2-kinase/Fructose-2,6-bisphosphatase-2 regulates TP53-dependent paclitaxel sensitivity in ovarian and breast cancers. Clin. Cancer Res. 2019, 25, 5702–5716. [Google Scholar] [CrossRef]
- Fiorillo, A.; Petrosino, M.; Ilari, A.; Pasquo, A.; Cipollone, A.; Maggi, M.; Chiaraluce, R.; Consalvi, V. The phosphoglycerate kinase 1 variants found in carcinoma cells display different catalytic activity and conformational stability compared to native enzyme. PLoS ONE 2018, 13, e0199191. [Google Scholar] [CrossRef]
- Israelsen, W.J.; Heiden, M.G.V. Pyruvate kinase: Function, regulation and role in cancer. Semin. Cell Dev. Biol. 2015, 43, 43–51. [Google Scholar] [CrossRef]
- Li, H.; Xu, H.; Xing, R.; Pan, Y.; Li, W.; Cui, J.; Lu, Y. Pyruvate kinase M2 contributes to cell growth in gastric cancer via aerobic glycolysis. Pathol. Res. Pract. 2019, 215, 152409. [Google Scholar] [CrossRef]
- Li, S.; Ji, X.; Wang, R.; Miao, Y. Follicle-stimulating hormone promoted pyruvate kinase isozyme type M2-induced glycolysis and proliferation of ovarian cancer cells. Arch. Gynecol. Obstet. 2019, 299, 1443–1451. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Chen, R.; Chang, L.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef]
- Said, H.M.; Polat, B.; Hagemann, C.; Anacker, J.; Fientje, M.; Vordermark, D. Absence of GAPDH regulation in tumor cells of different origin under hypoxic conditions in vitro. BMC Res. Notes 2009, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Sanders, E.; Diehl, S. Analysis and interpretation of transcriptomic data obtained from extended Warburg effect genes in patients with clear cell renal carcinoma. Oncoscience 2015, 2, 151–186. [Google Scholar] [CrossRef] [PubMed]
- Ortega, A.D.; Sanchez-Arago, M.; Giner-Sanchez, D.; Sanchez-Cenizo, L.; Willers, I.; Cuezva, J.M. Glucose avidity of carcinomas. Cancer Lett. 2009, 276, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Opperman, H.; Birkemeyer, C.; Meixensberger, J.; Gaunitz, F. Non-enzymatic reaction of carnosine and glycerol-3-phosphate accompanies metabolic changes in the pentose phosphate pathway. Cell Prolif. 2019, e12702. [Google Scholar] [CrossRef]
- Preuss, J.; Richardson, A.D.; Pinkerton, A.; Hedrick, M.; Sergienko, E.; Rahlfs, S.; Becker, K.; Bode, L. Identification and characterization of novel human glucose-6-phosphate dehydrogenase Inhibitors. J. Biomol. Screen. 2013, 18, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Cai, P.; Xu, C.; Cao, D.; Yu, W.; Zhao, Z.; Huang, M.; Jin, J. Inhibition of glucose-6-phosphate dehydrogenase reverses cisplatin resistance in lung cancer cells via the redox system. Front. Pharmacol. 2018, 9, 43. [Google Scholar] [CrossRef]
- Lucarelli, G.; Galleggiante, V.; Rutigliano, M.; Sanguedolce, F.; Cagiano, S.; Bufo, P.; Lastilla, G.; Maiorano, E.; Ribatti, D.; Giglio, A.; et al. Metabolomic profile of glycolysis and the pentose phosphate pathway identifies the central role of glucose-6-phosphate dehydrogenase in clear cell-renal cell carcinoma. Oncotarget 2015, 6, 13371–13386. [Google Scholar] [CrossRef]
- Zheng, W.; Feng, Q.; Liu, J.; Guo, Y.; Gao, L.; Li, R.; Xu, M.; Yan, G.; Yin, Z.; Zhang, S.; et al. Inhibition of 6-phosphogluconate dehydrogenase reverses cisplatin resistance in ovarian and lung cancer. Front. Pharmacol. 2017, 8, 421. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, H.; Wang, S.; Ni, Z.; Wang, T. 1-Hydroxy-8-methoxy-anthraquinon reverses cisplatin resistance by inhibiting 6PGD in cancer cells. Open Life Sci. 2019, 14, 454–461. [Google Scholar] [CrossRef]
- Diaz-Moralli, S.; Tarrado-Castellarnau, M.; Alenda, C.; Castells, A.; Cascante, M. Transketolase-like 1 expression is modulated during colorectal cancer progression and metastasis formation. PLoS ONE 2011, 6, e25323. [Google Scholar] [CrossRef]
- Benito, A.; Polat, I.H.; Noé, V.; Ciudad, C.J.; Marin, S.; Cascante, M. Glucose-6-phosphate dehydrogenase and transketolase modulate breast cancer cell metabolic reprogramming and correlate with poor patient outcome. Oncotarget 2017, 8, 106693–106706. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.K.; Peng, T.L.; Chuang, W.Y.; Yeh, C.J.; Li, Y.L.; Lu, Y.C.; Cheng, A.J. Transketolase serves a poor prognosticator in esophageal cancer by promoting cell invasion via epithelial-mesenchymal transition. J. Cancer 2016, 7, 1804–1811. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Liu, Y.; Glazer, C.A.; Shao, C.; Bhan, S.; Demokan, S.; Zhao, M.; Rudek, M.A.; Ha, P.K.; Califano, J.A. TKTL1 is activated by promoter hypomethylation and contributes to head and neck squamous cell carcinoma carcinogenesis through increased aerobic glycolysis and HIF1alpha stabilization. Clin. Cancer Res. 2010, 16, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Fritz, P.; Coy, J.F.; Mürdter, T.E.; Ott, G.; Alscher, M.D.; Friedel, G. TKTL-1 expression in lung cancer. Pathol. Res. Pract. 2012, 208, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Mancuso, A.; Wellen, K.E.; Yang, X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013, 493, 689–693. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Keeley, T.P.; Mann, G.E. Defining physiological normoxia for improved translation of cell physiology to animal models and humans. Physiol. Rev. 2019, 99, 161–234. [Google Scholar] [CrossRef]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumors—Implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
- San-Millan, I.; Brooks, G. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Izuka, N.; Tsunedomi, R.; Haamamoto, Y.; Miyamoto, T.; Iida, M.; Tokuhisa, Y.; Sakamoto, K.; Takashima, M.; Tamesa, M.; et al. Glycolysis module activated by hypoxia-inducible factor 1alpha is related to the aggressive phenotype of hepatocellular carcinoma. Int. J. Oncol. 2008, 33, 725–731. [Google Scholar] [PubMed]
- Sanchez-Sanchez, A.M.; Antolin, I.; Puente-Moncada, N.; Suarez, S.; Gomez-Lobo, M.; Rodrigez, C.; Martin, V. Melatonin cytotoxicity is associated to Warburg effect inhibition in Ewing sarcoma cells. PLoS ONE 2015, 10, e0135420. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Sardet, C.; Franchi, A.; L’Allemain, G.; Paris, S. A specific mutation abolishing Na+/H+ antiport activity in hamster fibroblasts precludes growth at neutral and acidic pH. Proc. Natl. Acad. Sci. USA 1984, 81, 4833–4837. [Google Scholar] [CrossRef]
- Park, H.J.; Lyons, J.C.; Ohtsubo, T.; Song, C.W. Acidic environment causes apoptosis by increasing caspase activity. Br. J. Cancer 1999, 80, 1892–1897. [Google Scholar] [CrossRef]
- San-Millán, I.; Julian, C.G.; Matarazzo, C.; Martinez, J.; Brooks, G.A. Is lactate an oncometabolite? Evidence supporting a role for lactate in the regulation of transcriptional activity of cancer-related genes in MCF7 breast cancer cells. Front. Oncol. 2020, 9, 1536. [Google Scholar] [CrossRef]
- Cassim, S.; Pouyssegur, J. Tumor microenvironment: A metabolic player that shapes the immune response. Int. J. Mol. Sci. 2020, 21, 157. [Google Scholar] [CrossRef]
- Ding, J.; Karp, J.E.; Emadi, A. Elevated lactate dehydrogenase (LDH) can be a marker of immune suppression in cancer: Interplay between hematologic and solid neoplastic clones and their microenvironments. Cancer Biomark. 2017, 19, 353–363. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Chiche, J.; Ilc, K.; Laferrière, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef]
- Lobo, R.C.; Hubbard, N.E.; Damonte, P.; Mori, H.; Penzvalto, Z.; Pham, C.; Koehne, A.L.; Go, A.C.; Anderson, S.E.; Cala, P.M.; et al. Glucose uptake and intracellular pH in a mouse model of ductal carcinoma in situ (DCIS) suggests metabolic heterogeneity. Front. Cell Dev. Biol. 2016, 4, 93. [Google Scholar] [CrossRef] [PubMed]
- Parks, S.K.; Chiche, J.; Pouyssegur, J. pH control mechanisms of tumor survival and growth. J. Cell. Physiol. 2011, 226, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Spugnini, E.P.; Sonveaux, P.; Stock, C.; Perez-Sayans, M.; De Milito, A.; Avnet, S.; Garcia, A.G.; Harguindey, S.; Fais, S. Proton channels and exchangers in cancer. Biochim. Biophys. Acta 2015, 1848, 2715–2726. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. Cell-cell and intracellular lactate shuttles. J. Physiol. 2009, 587, 5591–5600. [Google Scholar] [CrossRef]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate metabolism in human lung tumors. Cell 2017, 171, 358–371. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhang, L.; Guo, J.Y.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef]
- Wu, H.; Ying, M.; Hu, X. Lactic acidosis switches cancer cells from aerobic glycolysis back to dominant oxidative phosphorylation. Oncotarget 2016, 7, 40621–40629. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef]
- Qin, C.; Yang, G.; Yang, J.; Ren, B.; Wang, H.; Chen, G.; Zhao, F.; You, L.; Wang, W.; Zhao, Y. Metabolism of pancreatic cancer: Paving the way to better anticancer strategies. Mol. Cancer 2020, 19, 50. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Torres, A.; Henry, R.A.; Trefely, S.; Wallace, M.; Lee, J.V.; Carrer, A.; Sengupta, A.; Campbell, S.L.; Kuo, Y.M.; et al. ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep. 2016, 17, 1037–1052. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, Y.; Furukawa, T.; Saga, T.; Fujibayashi, Y. Acetate/acetyl-CoA metabolism associated with cancer fatty acid synthesis: Overview and application. Cancer Lett. 2015, 356, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Zu, X.Y.; Zhang, Q.H.; Liu, J.H.; Cao, R.X.; Zhong, J.; Yi, G.H.; Quan, Z.H.; Pizzorno, G. ATP citrate lyase inhibitors as novel cancer therapeutic agents. Recent Patents Anticancer. Drug Discov. 2012, 7, 154–167. [Google Scholar] [PubMed]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 22, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gruber, J.J.; Litzenburger, U.M.; Zhou, Y.; Miao, Y.R.; LaGory, E.L.; Li, A.M.; Hu, Z.; Yip, M.; Hart, L.S.; et al. Acetate supplementation restores chromatin accessibility and promotes tumor cell differentiation under hypoxia. Cell Death Dis. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Alpha-ketoglutarate dehydrogenase: A target and generator of oxidative stress. Philos. Trans. R. Soc. Lond. B Biol Sci. 2005, 360, 2335–2345. [Google Scholar] [CrossRef]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef]
- Lien, E.C.; Vander Heiden, M.G. A framework for examining how diet impacts tumour metabolism. Nat. Rev. Cancer 2019, 19, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Peck, B.; Schulze, A. Lipid metabolism at the nexus of diet and tumor microenvironment. Trends Cancer 2019, 5, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid metabolism and lipid droplets in pancreatic cancer and stellate cells. Cancers 2017, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Atoum, M.F.; Alzoughool, F.; Al-Hourani, H. Linkage between obesity. Leptin and breast cancer. Breast Cancer 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, K.; Douglas, I.; Forbes, H.; dos-Santos-Silva, I.; Leon, D.A.; Smeeth, L. Body-mass index and risk of 22 specific cancers: A population-based cohort study of 5·24 million UK adults. Lancet 2014, 384, 755–765. [Google Scholar] [CrossRef]
- Philip, B.; Roland, C.L.; Daniluk, J.; Liu, Y.; Chatterjee, D.; Gomez, S.B.; Ji, B.; Huang, H.; Wang, H.; Fleming, J.B.; et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 2013, 145, 1449–1458. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, Y.; Liu, M.; Wang, D.; Wang, F.; Bi, Y.; Ji, J.; Li, S.; Liu, Y.; Chen, R.; et al. Oncogenic KRAS reduces expression of FGF21 in acinar cells to promote pancreatic tumorigenesis in mice on a high-fat diet. Gastroenterology 2019, 157, 1413–1428. [Google Scholar] [CrossRef]
- Papa, S.; Martino, P.L.; Capitanio, G.; Gaballo, A.; De Rasmo, D.; Signorile, A.; Petruzzella, V. The oxidative phosphorylation system in mammalian mitochondria. Adv. Exp. Med. Biol. 2012, 942, 3–37. [Google Scholar] [PubMed]
- Curry, J.M.; Tuluc, M.; Whitaker-Menezes, D.; Ames, J.A.; Anantharaman, A.; Butera, A.; Leiby, B.; Cognetti, D.M.; Sotgia, F.; Lisanti, M.P.; et al. Cancer metabolism, stemness and tumor recurrence: MCT1 and MCT4 are functional biomarkers of metabolic symbiosis in head and neck cancer. Cell Cycle 2013, 12, 1371–1384. [Google Scholar] [CrossRef]
- De Luca, A.; Fiorillo, M.; Peiris-Pages, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef]
- Mikkilineni, L.; Whitaker-Menezes, D.; Domingo-Vidal, M.; Sprandio, J.; Avena, P.; Cotzia, P.; Dulau-Florea, A.; Gong, J.; Uppal, G.; Zhan, T.; et al. Hodgkin lymphoma: A complex metabolic ecosystem with glycolytic reprogramming of the tumor microenvironment. Semin. Oncol. 2017, 44, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiwicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.W.; Jeong, Y.J.; Park, S.H.; Oh, H.K.; Kang, S. Reverse Warburg effect-related mitochondrial activity and 18F-FDG uptake in invasive ductal carcinoma. Nucl. Med. Mol. Imaging 2019, 53, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [PubMed]
- Vellinga, T.T.; Borovski, T.; de Boer, V.C.; Fatrai, S.; van Schelven, S.; Trumpi, K.; Verheem, A.; Snoeren, N.; Emmink, B.L.; Koster, J.; et al. SIRT1/PGC1α-dependent increase in oxidative phosphorylation supports chemotherapy resistance of colon cancer. Clin. Cancer Res. 2015, 21, 2870–2879. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef]
- Xu, Q.; Biener-Ramanujan, E.; Yang, W.; Ramanujan, V.K. Targeting metabolic plasticity in breast cancer cells via mitochondrial complex I modulation. Breast Cancer Res. Treat. 2015, 150, 43–56. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Ralf, M.; Roberts, K.; Walter, P. Molecular biology of the cell. In The Mitochondrion, 4th ed.; Garland Science: NewYork, NY, USA, 2002. [Google Scholar]
- Fogg, V.C.; Lanning, N.J.; Mackeigan, J.P. Mitochondria in cancer: At the crossroads of life and death. Clin J. Cancer 2011, 30, 526–539. [Google Scholar] [CrossRef]
- Sajnani, K.; Islam, F.; Smith, R.A.; Gopalan, V.; Lam, A.K. Genetic alterations in Krebs cycle and its impact on cancer pathogenesis. Biochimie 2017, 135, 164–172. [Google Scholar] [CrossRef]
- Sudarshan, S.; Shanmugasundaram, K.; Naylor, S.L.; Lin, S.; Livi, C.B.; O’Neill, C.F.; Parekh, D.J.; Yeh, I.T.; Sun, L.Z.; Block, K. Reduced expression of fumarate hydratase in clear cell renal cancer mediates HIF-2α accumulation and promotes migration and invasion. PLoS ONE 2011, 6, e21037. [Google Scholar] [CrossRef]
- Adam, J.; Yang, M.; Soga, T.; Pollard, P.J. Rare insights into cancer biology. Oncogene 2014, 33, 2547–2556. [Google Scholar] [CrossRef] [PubMed]
- Khatami, F.; Aghamir, S.M.K.; Tavangar, S.M. Oncometabolites: A new insight for oncology. Mol. Genet. Genom. Med. 2019, 9, e873. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wahk, D.R. Metabolic abnormalities in glioblastoma and metabolic strategies to overcome treatment resistance. Cancers 2019, 11, 1231. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Cross, J.R.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.A.; Thompson, C.B. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene 2012, 31, 2491–2498. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y. Specific monoclonal antibodies against IDH1/2 mutations as diagnostic tools for gliomas. Brain Tumor Pathol. 2015, 32, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Yang, M.; Soga, T.; Pollard, P.J.; Adam, J. The emerging role of fumarate as an oncometabolite. Front. Oncol. 2012, 31, 85. [Google Scholar] [CrossRef]
- Hoekstra, A.S.; de Graaff, M.A.; Briaire-de Bruijn, I.H.; Ras, C.; Seifar, R.M.; van Minderhout, I.; Cornelisse, C.J.; Hodendoorn, P.C.W.; Breuning, M.H.; Suijker, J.; et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget 2015, 6, 38777–38788. [Google Scholar] [CrossRef]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell. 2018, 9, 216–237. [Google Scholar] [CrossRef]
- Weinberg, F.; Chandel, N.S. Mitochondrial metabolism and cancer. Ann. N. Y. Acad. Sci. 2009, 1177, 66–73. [Google Scholar] [CrossRef]
- Caneba, C.A.; Bellance, N.; Yang, L.; Pabst, L.; Nagrath, D. Pyruvate uptake is increased in highly invasive ovarian cancer cells under anoikis conditions for anaplerosis, mitochondrial function, and migration. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1036–E1052. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed. 2012, 25, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. Glutaminolysis: Supplying carbon or nitrogen or both for cancer cells? Cell Cycle 2010, 9, 3884–3886. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Manusco, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Filipp, F.V.; Ratnikov, B.; De Ingenis, J.; Smith, J.W.; Osterman, A.L.; Scott, D.A. Glutamine-fueled mitochondrial metabolism is decoupled from glycolysis in melanoma. Pigment Cell Melanoma Res. 2012, 25, 732–739. [Google Scholar] [CrossRef]
- Cheng, T.; Sudderth, J.; Yang, C.; Mullen, A.R.; Jin, E.S.; Mates, J.M.; DeBerardinis, R.J. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8674–8679. [Google Scholar] [CrossRef]
- Christen, S.; Lorendeau, D.; Schmieder, R.; Broekaert, D.; Metzger, K.; Veys, K.; Elia, I.; Buescher, J.M.; Orth, M.F.; Davidson, S.M.; et al. Breast cancer-derived lung metastases show increased pyruvate carboxylase-dependent anaplerosis. Cell Rep. 2016, 17, 837–848. [Google Scholar] [CrossRef]
- Mallik, R.; Chowdhury, T.A. Metformin in cancer. Diabetes Res. Clin. Pract. 2018, 143, 409–419. [Google Scholar] [CrossRef]
- Huang, Y.; Zhou, S.; He, C.; Deng, J.; Tao, T.; Su, Q.; Darko, K.O.; Peng, M.; Yang, X. Phenformin alone or combined with gefitinib inhibits bladder cancer via AMPK and EGFR pathways. Cancer Commun. 2018, 38, 50. [Google Scholar] [CrossRef]
- Liu, W.; Le, A.; Hancock, C.; Lane, A.N.; Dang, C.V.; Fan, T.W.; Phang, J.M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c- MYC. Proc. Natl. Acad. Sci. USA 2012, 109, 8983–8988. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Lee, G.; Yoon, S.O.; Tong, H.; Ilter, D.; Elia, I.; Fendt, S.M.; Roberts, T.M.; Blenis, J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. 2014, 24, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Fendt, S.M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Morotti, M.; Bridges, E.; Valli, A.; Choudhry, H.; Sheldon, H.; Wigfield, S.; Gray, N.; Zois, C.E.; Grimm, F.; Jones, D.; et al. Hypoxia-induced switch in SNAT2/ SLC38A2 regulation generates endocrine resistance in breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 12452–12461. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A. Beta oxidation of fatty acids. In Sweet Biochemistry; Academic Press: Cambridge, MA, USA, 2018; pp. 17–19. [Google Scholar]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef]
- Wang, M.D.; Wu, H.; Huang, S.; Zhang, H.L.; Qin, C.J.; Zhao, L.H.; Fu, G.B.; Zhou, X.; Wang, X.M.; Tang, L.; et al. HBx regulates fatty acid oxidation to promote hepatocellular carcinoma survival during metabolic stress. Oncotarget 2016, 7, 6711–6726. [Google Scholar] [CrossRef]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol. 2017, 19, 43–54. [Google Scholar] [CrossRef]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty acid oxidation mediated by acyl-CoA synthetase long chain 3 is required for mutant KRAS lung tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef]
- Shao, H.; Mohamed, E.M.; Xu, G.G.; Waters, M.; Jing, K.; Ma, Y.; Zhang, Y.; Spiegel, S.; Idowu, M.O.; Fang, X. Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer. Oncotarget 2016, 7, 3832–3846. [Google Scholar] [CrossRef]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.-L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Makin, A.; Hullin-Matsuda, F.; Kobayashi, T.; Furihata, M.; Chung, S.; Ashida, S.; Miki, T.; Fujioka, T.; Shuin, T.; et al. Novel lipogenic enzyme ELOVL7 is involved in prostate cancer Growth through saturated long-chain fatty acid metabolism. Cancer Res. 2009, 69, 8133–8140. [Google Scholar] [CrossRef]
- Linher-Melville, K.; Zantinge, S.; Sanli, T.; Gerstein, H.; Tsakiridis, T.; Singh, G. Establishing a relationship between prolactin and altered fatty acid beta-oxidation via carnitine palmitoyl transferase 1 in breast cancer cells. BMC Cancer 2011, 11, 56. [Google Scholar] [CrossRef]
- Tong, L. Acetyl-coenzyme A carboxylase: Crucial metabolic enzyme and attractive target for drug discovery. Cell Mol. Life Sci. 2003, 62, 1784–1803. [Google Scholar] [CrossRef] [PubMed]
- Brownsey, R.W.; Boone, A.N.; Elliott, J.E.; Kulpa, J.E.; Lee, W.M. Regulation of acetyl-CoA carboxylase. Biochem. Soc. Trans. 2006, 34, 223–227. [Google Scholar] [CrossRef]
- German, N.J.; Yoon, H.; Yusuf, R.Z.; Murphy, J.P.; Finley, L.W.; Laurent, G.; Haas, W.; Satterstrom, F.K.; Guarnerio, J.; Zaganjor, E.; et al. PHD3 loss in cancer enables metabolic reliance on fatty acid oxidation via deactivation of ACC2. Mol. Cell 2016, 63, 1006–1020. [Google Scholar] [CrossRef]
- Jiang, L.; Xiao, L.; Sugiura, H.; Huang, X.; Ali, A.; Kuro-o, M.; Deberardinis, R.J.; Boothman, D.A. Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene 2015, 34, 3908–3916. [Google Scholar] [CrossRef]
- Maher, M.; Diesch, J.; Casquero, R.; Buschbeck, M. Epigenetic-transcriptional regulation of fatty acid metabolism and its alterations in leukaemia. Front. Genet. 2018, 9, 405. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Mráček, T.; Holzerová, E.; Drahota, Z.; Kovářová, N.; Vrbacký, M.; Ješina, P.; Houštěk, J. ROS generation and multiple forms of mammalian mitochondrial glycerol-3-phosphate dehydrogenase. Biochim. Biophys. Acta—Bioenerg. 2014, 1837, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Frerman, F.E.; Kim, J.J. Structure of electron transfer flavoprotein-ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool. Proc. Natl. Acad. Sci. USA 2006, 103, 16212–16217. [Google Scholar] [CrossRef]
- Watmough, N.J.; Frerman, F.E. The electron transfer flavoprotein: Ubiquinone reductases. Biochim. Biophys. Acta 2010, 1797, 1910–1916. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modification. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef]
- Porporato, P.E.; Payen, V.L.; Perez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef]
- Oakhill, J.S.; Scott, J.W.; Kemp, B.E. AMPK functions as an adenylate charge-regulated protein kinase. Trends Endocrinol. Metab. 2012, 23, 125–132. [Google Scholar] [CrossRef]
- Carling, D. AMPK signalling in health and disease. Curr. Opin. Cell Biol. 2017, 45, 31–37. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Wang, B.Z.; Yang, J.J.; Zhang, H.; Smith, C.A.; Jiu, K. AMPK signaling regulates the age-related decline of hippocampal neurogenesis. Aging Dis. 2019, 10, 1058–1074. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Boudeau, J.; Baas, A.F.; Deak, M.; Morrice, N.A.; Kieloch, A.; Schutkowski, M.; Prescott, A.R.; Clevers, H.C.; Alessi, D.R. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003, 22, 5102–5114. [Google Scholar] [CrossRef]
- Zhang, C.-S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.-W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef]
- Lin, R.; Elf, S.; Shan, C.; Kang, H.-B.; Ji, Q.; Zhou, L.; Hitosugi, T.; Zhang, L.; Zhang, S.; Seo, J.H.; et al. 6-Phosphogluconate dehydrogenase links Oxidative PPP, pipogenesis and tumour growth by inhibiting LKB1–AMPK signalling. Nat. Cell Biol. 2015, 17, 1484–1496. [Google Scholar] [CrossRef]
- Ducommun, S.; Deak, M.; Sumpton, D.; Ford, R.J.; Nunez Galindo, A.; Kussmann, M.; Viollet, B.; Steinberg, G.R.; Foretz, M.; Davon, L.; et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: Identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal. 2015, 27, 978–988. [Google Scholar] [CrossRef]
- Schaffer, B.E.; Levin, R.S.; Hertz, N.T.; Maures, T.J.; Schoof, M.L.; Hollstein, P.E.; Benayoun, B.A.; Banko, M.R.; Shaw, R.J.; Shokat, K.M.; et al. Identification of AMPK phosphorylation sites reveals a network of proteins involved in cell invasion and facilitates large-scale substrate prediction. Cell Metab. 2015, 22, 907–921. [Google Scholar] [CrossRef]
- Nielsen, J. Systems biology of metabolism. Annu. Rev. Biochem. 2017, 86, 245–275. [Google Scholar] [CrossRef]
- Inoki, K.; Kim, J.; Guan, K.L. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef]
- Shaw, R.J. LKB1 and AMP-activated protein kinase C control of mTOR signaling and growth. Acta Physiol. 2009, 196, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Groenewoud, M.J.; Zwartkruis, F.J. Rheb and Rags come together at the lysosome to activate mTORC1. Biochem. Soc. Trans. 2013, 41, 951–955. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Sabatini, D.M. Ragulator and SLC38A9 activate the Rag GTPases through noncanonical GEF mechanism. Proc. Natl. Acad. Sci. USA 2018, 115, 9445–9550. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.M.; Shin, Y.; Lee, E.J.; Lee, S.; Jeong, S.H.; Kong, H.K.; Park, E.Y.; Kim, H.K.; Han, J.; Chang, M.; et al. Inhibition of aerobic glycolysis represses Akt/mTOR/HIF-1 axis and restores tamoxifen sensitivity in antiestrogen-resistant breast cancer cells. PLoS ONE 2015, 10, e0132285. [Google Scholar] [CrossRef]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the metabolic plasticity of cancer: Mitochondrial reprogramming and hybrid metabolic states. Cells 2018, 7, 21. [Google Scholar] [CrossRef]
- Yu, L.; Lu, M.; Jia, D.; Ma, J.; Ben-Jacob, E.; Levine, H.; Kaipparettu, B.A.; Onuchic, J.N. Modeling of genetic regulation of cancer metabolism: Interplay between glycolysis and oxidative phosphorylation. Cancer Res. 2017, 77, 1564–1574. [Google Scholar] [CrossRef]
- Tyszka-Czochara, M.; Konieczny, P.; Majka, M. Recent advances in the role of AMP-activated protein kinase in metabolic reprogramming of metastatic cancer cells: Targeting cellular bioenergetics and biosynthetic pathways for anti-tumor treatment. J. Physiol. Pharmacol. 2018, 69, 337–349. [Google Scholar] [PubMed]
- Jiang, S.; Wang, Y.; Luo, L.; Shi, F.; Zou, J.; Lin, H.; Ying, Y.; Luo, Y.; Zhan, Z.; Liu, P.; et al. AMP-activated protein kinase regulates cancer cell growth and metabolism via nuclear and mitochondria events. J. Cell Mol. Med. 2019, 23, 3951–3961. [Google Scholar] [CrossRef]
- Lee, M.; Katerelos, M.; Gleich, K.; Galic, S.; Kemp, B.E.; Mount, P.F.; Power, D.A. Phosphorylation of acetyl-CoA carboxylase by AMPK reduces renal fibrosis and is essential for the anti-fibrotic effect of metformin. J. Am. Soc. Nephrol. 2018, 29, 2326–2336. [Google Scholar] [CrossRef]
- Angin, Y.; Beauloye, C.; Horman, S.; Bertrand, L. Regulation of carbohydrate metabolism, lipid metabolism, and protein metabolism by AMPK. Exp. Suppl. 2016, 107, 23–43. [Google Scholar] [PubMed]
- Park, C.; Cha, H.-J.; Choi, E.O.; Lee, H.; Bo, H.H.; Ji, S.Y.; Kim, M.Y.; Kim, S.Y.; Hong, S.H.; Cheong, J.H.; et al. Isorhamnetin induces cell cycle arrest and apoptosis via reactive oxygen species-mediated AMP-activated protein kinase signaling pathway activation in bladder cancer cells. Cancers 2019, 11, 1494. [Google Scholar] [CrossRef]
- Yu, J.; Shi, L.; Shen, X.; Zhao, Y. UCP2 regulates cholangiocarcinoma cell plasticity via mitochondria-to-AMPK signals. Biochem. Pharmacol. 2019, 166, 174–184. [Google Scholar] [CrossRef]
- Zhiang, H.; Wu, S.; Li, H.; Duan, Q.; Zhang, Z.; Shen, Q.; Wang, C.; Yin, T. ROS/KRAS/AMPK signaling contributes to gemcitabine-induced stem-like cell properties in pancreatic cancer. Mol. Ther. Oncolyt. 2019, 14, 299–312. [Google Scholar] [PubMed]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and mitochondrial respiration: A neglected connection. Cancer Res. 2015, 75, 3687–3691. [Google Scholar] [CrossRef]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [PubMed]
- Smolkova, K.; Plecita-Hlavata, L.; Bellance, N.; Benard, G.; Rossignol, R.; Jesek, P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int. J. Biochem. Cell Biol. 2011, 43, 950–968. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Porceli, A.M.; Lenaz, G.; Romeo, G. Relevance of mitochondrial genetics and metabolism in cancer development. Cold Spring Harbor Perspect. Biol. 2013, 5, 011411. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Metabolic Heterogeneity of Cancer Cells: An Interplay between HIF-1, GLUTs, and AMPK. Cancers 2020, 12, 862. https://doi.org/10.3390/cancers12040862
Moldogazieva NT, Mokhosoev IM, Terentiev AA. Metabolic Heterogeneity of Cancer Cells: An Interplay between HIF-1, GLUTs, and AMPK. Cancers. 2020; 12(4):862. https://doi.org/10.3390/cancers12040862
Chicago/Turabian StyleMoldogazieva, Nurbubu T., Innokenty M. Mokhosoev, and Alexander A. Terentiev. 2020. "Metabolic Heterogeneity of Cancer Cells: An Interplay between HIF-1, GLUTs, and AMPK" Cancers 12, no. 4: 862. https://doi.org/10.3390/cancers12040862
APA StyleMoldogazieva, N. T., Mokhosoev, I. M., & Terentiev, A. A. (2020). Metabolic Heterogeneity of Cancer Cells: An Interplay between HIF-1, GLUTs, and AMPK. Cancers, 12(4), 862. https://doi.org/10.3390/cancers12040862