
The Current Landscape of Targeted Clinical Trials in Non-WNT/Non-SHH Medulloblastoma


1
Hopp Children’s Cancer Center Heidelberg (KiTZ), 69120 Heidelberg, Germany
2
Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Consortium for Translational Cancer Research (DKTK), 69120 Heidelberg, Germany
3
Department of Pediatric Oncology, Hematology and Immunology, Heidelberg University Hospital, 69120 Heidelberg, Germany
4
Pediatric Oncology and Hematology, Pediatrics III, University Hospital of Essen, 45147 Essen, Germany
5
Clinical Cooperation Unit Pediatric Oncology, German Cancer Research Center (DKFZ) and German Consortium for Translational Cancer Research (DKTK), 69120 Heidelberg, Germany
*
Authors to whom correspondence should be addressed.
Cancers 2022, 14(3), 679; https://doi.org/10.3390/cancers14030679
Submission received: 15 December 2021
/
Revised: 23 January 2022
/
Accepted: 24 January 2022
/
Published: 28 January 2022
(This article belongs to the Special Issue Medulloblastoma Systemic Therapy Clinical Trials and Their Foundations)
Abstract
:Simple Summary
Medulloblastoma is a form of malignant brain tumor that arises predominantly in infants and young children and can be divided into different groups based on molecular markers. The group of non-WNT/non-SHH medulloblastoma includes a spectrum of heterogeneous subgroups that differ in their biological characteristics, genetic underpinnings, and clinical course of disease. Non-WNT/non-SHH medulloblastoma is currently treated with surgery, chemotherapy, and radiotherapy; however, new drugs are needed to treat patients who are not yet curable and to reduce treatment-related toxicity and side effects. We here review which new treatment options for non-WNT/non-SHH medulloblastoma are currently clinically tested. Furthermore, we illustrate the challenges that have to be overcome to reach a new therapeutic standard for non-WNT/non-SHH medulloblastoma, for instance the current lack of good preclinical models, and the necessity to conduct trials in a comparably small patient collective.
Abstract
Medulloblastoma is an embryonal pediatric brain tumor and can be divided into at least four molecularly defined groups. The category non-WNT/non-SHH medulloblastoma summarizes medulloblastoma groups 3 and 4 and is characterized by considerable genetic and clinical heterogeneity. New therapeutic strategies are needed to increase survival rates and to reduce treatment-related toxicity. We performed a noncomprehensive targeted review of the current clinical trial landscape and literature to summarize innovative treatment options for non-WNT/non-SHH medulloblastoma. A multitude of new drugs is currently evaluated in trials for which non-WNT/non-SHH patients are eligible, for instance immunotherapy, kinase inhibitors, and drugs targeting the epigenome. However, the majority of these trials is not restricted to medulloblastoma and lacks molecular classification. Whereas many new molecular targets have been identified in the last decade, which are currently tested in clinical trials, several challenges remain on the way to reach a new therapeutic strategy for non-WNT/non-SHH medulloblastoma. These include the severe lack of faithful preclinical models and predictive biomarkers, the question on how to stratify patients for clinical trials, and the relative lack of studies that recruit large, homogeneous patient collectives. Innovative trial designs and international collaboration will be a key to eventually overcome these obstacles.
1. Introduction
Medulloblastoma (MB) is the most common malignant central nervous system (CNS) tumor of infancy as well as early childhood and accounts for a significant share of both cancer-related morbidity and mortality in this age group [1,2]. Historically, MB has been diagnosed based on histomorphological features and stratified into four respective types: Classic, Large Cell/Anaplastic, Desmoplastic/Nodular, and MB with Extensive Nodularity [3]. However, a growing body of work on the (epi-)genetic background of MB resulted in a first consensus in 2012 that established four molecular groups: WNT, SHH, Group 3, and Group 4 (Gr. 3/Gr. 4) [4]. These developments have culminated in the recognition of a molecularly defined MB classification system in the WHO classification of CNS tumors [5]. The recently published fifth edition includes the molecular MB diagnoses WNT-activated, SHH-activated/TP53-mutated, SHH-activated/TP53-wildtype, and non-WNT-/non-SHH alongside one category for histologically defined MB [6]. DNA methylation-based classification has developed into an accepted tool to confidently stratify MB into its molecular groups [7,8]. Other methods, such as nanoString-based RNA-assays and PCR-arrays, are also applied for subgrouping [9,10].
WNT MB accounts for about 10% of all patients. These tumors are defined by mutations related to the respective pathway, such as CTNNB1 or APC, and are generally associated with excellent survival rates [8,10,11,12,13,14]. Notably, germline APC mutations, known to cause familial polyposis syndromes, also predispose to WNT MB [8,15,16]. Within the SHH group, which comprises about 25–30% of all cases, mutations and copy number variations (CNVs) of SHH-pathway members (PTCH1, SUFU, SMO, GLI1/2, MYCN) and alterations of TP53 and TERT are characteristic genetic hallmarks [4,8,10,12,13,17]. Furthermore, this group also accounts for the majority of patients suffering from cancer predisposing germline mutations. Cancer predisposition syndromes that predispose to SHH MB include, amongst others, Li–Fraumeni syndrome and Gorlin–Goltz syndrome [8,15,16,18,19]. The prognosis of SHH MB varies strongly and ranges from favorable to dismal, for instance, depending on the presence of a TP53-mutation [20,21,22,23]. Both WNT- and SHH-activated MB can be further subdivided into two and four subgroups, respectively, and represent comparably well understood and clearly defined molecular groups [6,12,24].
In contrast, the situation concerning Gr. 3 and Gr. 4 MB is more complex. Together, both groups form the spectrum for non-WNT/non-SHH MB, which accounts for roughly 60% of all cases and remains the genetically most heterogeneous and least understood fraction of MB cases [25]. Gr. 3 MB shows the worst outcome across all MB groups and frequently harbors amplification or overexpression of the MYC-gene [10,26,27,28]. Gr. 4 MB is mostly associated with intermediate risk and is enriched for somatic alterations of genes involved in chromatin remodeling and histone modification, such as KDM6A, and notably MYCN amplification in roughly 6% of cases [8,13,29]. However, in contrast to the WNT and SHH groups, no single somatically mutated gene is present in more than 5–10% of either Gr. 3 or Gr. 4 MB patients, constituting a significant challenge in the development of innovative treatment strategies for these children [13]. Furthermore, recent molecular studies with so far unprecedented sample sizes have shown that both molecular groups do not only show significant group-specific heterogeneity, but also partly overlap in the sense that a number of cases were not unambiguously assignable to either of the two groups [13,24,30,31]. These findings are underlined by the fact that Gr. 3/4 MB share several genetic alterations, such as structural whole-chromosome abnormalities or enhancer hijacking-mediated overexpression of GFI1 and GFI1B, which has been shown to drive tumor formation in vivo in combination with MYC-overexpression [10,32]. Furthermore, a recent single cell sequencing study reported the existence of intermediate tumors that share features of both Gr. 3 and Gr. 4 transcriptional programs in roughly 20% of all Gr. 3/4 cases [33].
A consensus study in 2019 established a subdivision of Gr. 3/4 MB into eight molecular subgroups including shared ones that differ in methylation profiles, genetic alterations, epidemiological, and clinical features [34]. The increased level of complexity concerning stratifying non-WNT/non-SHH MB is taken into account in the current 2021 version of the WHO classification of CNS tumors by recommending a layered, integrated diagnostic approach that accounts for both molecular group and subgroup alongside histological appearance [6]. Recently, a randomized clinical trial which tested an intensified therapeutic regime that included carboplatin and isotretinoin resulted in significantly improved outcomes for children suffering from Gr. 3 MB, illustrating the potential of molecular-guided patient stratification [35].
Throughout the last decade, the combined efforts of the MB community have led to an increased understanding of the biological underpinnings of this heterogeneous disease and a refined molecular classification that lays the foundation for new ways of treating MB in the future. In this noncomprehensive review, we describe the current landscape of precision-medicine trials for which non-WNT/non-SHH MB patients are eligible, with a strong focus on preclinically validated molecular targets and treatment strategies that are or have recently been explored in the clinic.
2. Molecular Targets and Treatment Strategies in Non-WNT/Non-SHH MB
2.1. Kinase Inhibitors
Kinase inhibitors have developed into one of the standard drug classes in the repertoire of personalized oncology [36,37], and a multitude of kinase inhibiting compounds has been tested preclinically in MB, most of them targeting enzymes that are involved in cell cycle regulation. However, activating mutations in receptor tyrosine kinases, which are frequently detected in other CNS malignancies such as glioblastoma, are rare in MB [8]. While cycline-dependent kinase (CDK) inhibitors are discussed separately, this paragraph gives a short overview on other kinase inhibitors that are currently tested in (non-WNT/non-SHH) MB (Figure 1). WEE1 is a tyrosine kinase that is involved in G2 checkpoint regulation and expressed in non-WNT/non-SHH MB, and the WEE1-inhibitor Adavosertib has been tested preclinically in Gr. 3/4 model systems [38,39]. In both studies, Adavosertib showed synergistic effects with the chemotherapeutic agents cisplatin and gemcitabine in suppressing MB tumor growth. This finding may be based on the fact that WEE1-inhibition is more effective in the presence of replication stress and DNA damage [40]. The clinical use of Adavosertib for pediatric cancer patients has been investigated in combination with Irinotecan; however, only one MB patient was enrolled in the respective trial (NCT02095132), which showed an acceptable safety profile with a maximum tolerable dose (MTD) of 85 mg/m2/day [41,42]. Importantly, studies in glioblastoma showed heterogeneous and partly limited distribution of Adavosertib across the blood–brain barrier [43,44,45,46]. Similar to WEE1, the protein kinase CHK1 is involved in cell cycle regulation and has been proposed as a potential vulnerability in Gr. 3 MB in vitro and in vivo [47,48,49]. Preclinical evidence from an in vivo Gr. 3 MB model suggests that therapeutically meaningful concentrations of Prexasertib can reach the brain [48,50]. A first-in-pediatrics trial recently illustrated that Prexasertib monotherapy was well tolerated in a pediatric mixed solid tumor population (NCT02808650/ADVL1515) using a dose of 150 mg/m2 administered i.v. on days 1 and 15 of a 28-day cycle, although no objective responses were reported [51]. Currently, one phase I trial investigates the use of Prexasertib in combination with established DNA-damaging agents used in medulloblastoma to evaluate tolerance and pharmacokinetics in recurrent or refractory non-WNT/non-SHH (NCT04023669) (Table 1). Due to their mechanism of action, which allows cells with high levels of DNA damage to enter the cell cycle, both Adavosertib and Prexasertib may be dependent on synergistic chemotherapy that induces high levels of genomic damage to unfold their full efficacy [39,40,47]. It should be noted that for both WEE1 and CHK1, preclinical efficacy is based on overexpression and not on somatically altered genes, with the potential downside that the mere overexpression of tyrosine kinases alone might not represent a long-lasting target in the context of personalized therapy [52].
2.2. CDK Inhibitors
CDKs are centrally involved in the positive regulation of cell cycle activity and their activity is frequently dysregulated in numerous forms of cancer [53,54]. The FDA and EMA approval of the three CDK4/6 inhibitors Palbociclib, Ribociclib, and Abemaciclib for the treatment of certain forms of breast cancer have also sparked great interest in the concept of CDK inhibition in other tumor types, including MB. However, potential pitfalls, for example, resistance mechanisms such as loss of function mutations in the Rb gene, need to be taken into account when testing CDK inhibitors in pediatric tumors. Amongst many other biological mechanisms, CDKs stabilize MYC-family proteins, which make CDK inhibition an interesting therapeutic option, especially for non-WNT/non-SHH MB [55]. Indeed, several CDKs are altered in a subset of non-WNT/non-SHH MB, for instance, in the form of CDK6-amplification in Gr. 4 MB [8,10,34]. Throughout the last years, preclinical evidence that CDK inhibitors could provide new therapeutic opportunities for non-WNT/non-SHH MB has grown, especially for MYC-amplified MB and in combination with other drugs, such as BET bromodomain-inhibition [56,57,58]. Several trials are currently testing CDK inhibitors for CNS tumors in children (Table 1). A recently published phase I trial determined pharmacokinetic properties and the MTD of Palbociclib monotherapy in both mild and heavily pretreated children with progressive brain tumors, including four MB patients, albeit without molecular information (NCT02255461/PBTC-042) [59]. As anticipated from trials in adults, one of the main dose-limiting side effects was neutropenia. No patient showed an objective response to Palbociclib treatment, suggesting that future trials may need to focus on combinatorial approaches. Currently, one treatment arm of the pediatric MATCH basket trial (NCT03155620) investigates the use of Palbociclib as a monotherapy (NCT03526250), whereas two industry-sponsored trials are combining CDK inhibitors Palbociclib and Abemaciclib with different chemotherapy regimens for relapsed or refractory pediatric solid tumors (NCT03709680/NCT04238819). Further, the currently running SJDAWN trial includes one study arm in which patients with recurrent/relapsed non-WNT/non-SHH MB and ependymoma will be treated with gemcitabine and Ribociclib. Results of this study may allow first estimates for duration of objective response and progression-free survival in a biologically homogeneous group of non-WNT/non-SHH MB (NCT03434262).
2.3. HDAC Inhibitors
Histone deacetylases (HDACs) are important epigenetic regulators of transcription and can induce gene repression via chromatin remodeling and altered transcription factor binding [60]. Throughout the last decade, a large body of work has established HDAC inhibition as a promising therapeutic avenue in MB, with a special focus on MYC-amplified non-WNT/non-SHH MB [61,62,63,64,65,66,67]. A recent study demonstrated that HDAC inhibitors stabilize the MYC protein and thus lead to reduced DNA binding and ultimately lower expression of MYC target genes [63]. Furthermore, a combined phase I/II dose escalation trial showed overall manageable toxicity and partial responses in a fraction of patients (including eight MB patients) for the HDAC inhibitor Vorinostat [68]. This is in line with another study that showed a good safety profile for the combination of Vorinostat and Temozolomide in a small cohort of children with relapsed CNS malignancies, including two MB patients (NCT01076530) [69]. Currently, several trials are testing different HDAC inhibitors and therapeutic strategies (Table 1): One early phase I trial is investigating the infusion of Panobinostat into the fourth ventricle of recurrent MB patients (NCT04315064). A second study is testing the pan-HDAC and PI3K inhibitor Fimepinostat in a cohort of pediatric brain tumor patients, including MB (NCT03893487/PNOC016). Vorinostat in combination with Isotretinoin and chemotherapy was investigated in infants with MB and other embryonal brain tumors; however, the results are not yet published (NCT00867178/PBTC-026). HDAC inhibition modifies T-cell regulation and can augment response to check-point inhibition by reducing the number of myeloid-derived suppressor cells [70,71,72,73], thereby creating an immunogenic tumor microenvironment including the induction of major histocompatibility complexes and neoantigens. Therefore, combination of HDAC inhibitors with immune checkpoint inhibitors as in the biomarker-driven INFORM2 NivEnt trial (Nivolumab and Entinostat) represents an exciting approach for non-WNT/non-SHH MB (NCT03838042) [52,74]. While none of these studies is restricted to non-WNT/non-SHH MB, it seems likely that a significant percentage of enrolled patients will be relapsed or progressive Gr. 3/4 MB patients.
2.4. Antiangiogenic Therapy
Antiangiogenic drugs belong to the first successes of personalized medicine and are currently used in a wide range of oncological entities and disease settings. Several studies reported that genes, such as VEGFA and HIF1A, which are important for malignant neovascularization, are expressed in MB [75,76,77]. One publication showed that as compared to other MB groups, VEGFA-levels are elevated in Gr. 3 MB, which corresponded to vessel density and correlated with survival [78]. These findings indicate a potential role of antiangiogenic therapy for non-WNT/non-SHH MB. Axitinib, a multikinase inhibitor that also targets neovascularization, has shown efficacy in vitro and in vivo in c-MYC amplified MB models [79,80]. Furthermore, a number of case reports and small retrospective studies have indicated that antiangiogenic drugs, such as Bevacizumab, can achieve objective response in some MB patients, especially in combination with chemotherapy [81,82,83,84,85,86,87]. A prospective, randomized phase II trial by the Children’s Oncology Group (ACNS082/NCT01217437) showed significantly reduced risk of death as the primary endpoint in children suffering from relapsed MB when treated with temozolomide/irinotecan and bevacizumab as compared to temozolomide/irinotecan alone [88]. For 36/105 patients, molecular data were reported, and 27/36 cases were assigned to the non-WNT/non-SHH group. Additional evidence for such therapeutic strategies may be derived from the metronomic MEMMAT trial that evaluates the activity of a multidrug antiangiogenic approach enrolling relapsed MB (NCT01356290) (Table 1).
2.5. Radiotherapeutics
MB has long been known to be sensitive to radiotherapy, and the idea to use radioactive isotopes that are conjugated to antibodies that are selectively targeting cancer cells in the CNS is more than thirty years old [89]. One phase II trial showed encouraging results and a good safety profile for the radioconjugated antibody 131I-3F8 in a MB cohort of n = 42, including several long-term survivors in a heavily pretreated patient collective (NCT00445965) [90]. 131I-3F8 targets GD2, a cell-surface disialoganglioside that is expressed on a variety of cancers, including MB. Unfortunately, no information on molecular diagnoses was reported, therefore the potential of 131I-3F8 for non-WNT/non-SHH MB cannot yet be judged. Furthermore, two industry-led trials testing radiotherapeutics for which MB patients are eligible are currently ongoing (Table 1): One phase I trial with the radioconjugate CLR131 for solid pediatric tumors including CNS malignancies (NCT03478462), and another phase I/II study that specifically addresses MB patients and tests the B7-H3 targeting radioconjugate 177Lu-DTPA-omburtamab. Interestingly, this study offers a cohort expansion phase for which SHH, Gr. 3 and Gr. 4 MB patients are eligible only (NCT04167618). Additionally, one study will combine Omburtamab-I131 with Irinotecan/Temozolomide and Bevacizumab for the treatment of recurrent MB and ependymoma (NCT04743661).
2.6. Metabolic Therapies
Compared to epigenetics, transcriptomics, and genomics, MB metabolomics is a field still in its infancy. Thus, only few therapeutic concepts exist to target the metabolism of MB in general and non-WNT/non-SHH MB specifically. One approach is the inhibition of IDO1, an enzyme that is centrally involved in tryptophane catabolism and has been identified as a key player in creating an immunosuppressive microenvironment [91]. A small study reported IDO1 expression in a cohort of 27 MB samples, including 16 non-WNT/non-SHH patients [92]. A phase I trial that used the IDO1 inhibitor Indoximod in combination with Temozolomide in a pediatric population suffering from recurrent/refractory CNS malignancies, was completed last year and enrolled 81 patients (NCT02502708). Although results from the trial are not yet published, a phase II study applying the combination of Indoximod with radiochemotherapy is currently open for participation (NCT04049669) (Table 1). Besides tryptophane, polyamine metabolism has been identified as a potential vulnerability in several cancers, including SHH MB, since polyamines are involved in the regulation of a number of cellular processes [93,94]. A possibility to interfere with polyamine synthesis is the inhibition of the enzyme ornithine decarboxylase (ODC) using the drug Difluoromethylornithine (DFMO/Eflornithine), a potent inhibitor of ODC, which has been tested for various types of cancer, including neuroblastoma [94]. DFMO is already approved for the treatment of sleeping sickness and hirsutism, making it an interesting repurposing candidate. However, its use remains debated due to toxicity concerns and pharmacokinetic challenges [94]. While preclinical studies are lacking, DFMO is currently tested in non-WNT/non-SHH MB patients in an expanded use setting (NCT03581240) and in a phase II trial exploring it as maintenance therapy for high-risk MB (NCT04696029). Taken together, as in other malignancies targeting metabolomic vulnerabilities in non-WNT/non-SHH MB may represent a valuable approach to enhance therapeutic mainstays. To date, scarce preclinical and clinical evidence does not allow for any conclusion or outlook in this molecular subgroup.
2.7. Epigenetic Therapies, Chromatin Remodeling, and Superenhancers
Chromatin remodeling and other epigenetic mechanisms represent key drivers in non-WNT/non-SHH MB. [8,12] A significant percentage of genomic alterations in these tumors is detected in genes that are involved in chromatin remodeling, histone modification, enhancer hijacking, and other epigenetic regulatory mechanisms, especially in Gr. 4 MB [8,10,13,32]. Thus, the underlying pathways have long been identified as potential vulnerabilities in non-WNT/non-SHH MB. However, the biological function of the involved genes and complexes, such as the SWI/SNF or PRC2 complex, is often context-specific and extremely complicated, and their roles in MB formation and proliferation are not yet fully understood [95,96,97,98,99]. For instance EZH2, the catalytic subunit of the PRC2 complex, has been reported to be overexpressed in non-WNT/non-SHH MB, and the same study showed that inhibition of EZH2 in two SHH MB cell lines led to decreased proliferation and induced apoptosis [100]. Conflicting with these results, another more recent study reported that inactivation of EZH2 accelerated tumorigenesis in a MYC-driven Gr. 3 MB mouse model, illustrating the potential caveats of applying EZH2 inhibitors in the clinic [101]. Two other studies demonstrated a modest benefit in overall survival for the EZH2 inhibitor Tazemetostat in MB xenografts and antitumor effects in in vitro models of Gr. 3 MB [102,103]. Notably, there is also growing evidence from large scale diagnostic studies that overexpression of target genes in absence of other alterations is not predictive for response to a targeted treatment [52]. Tazemetostat was available for progressive or recurrent non-WNT/non-SHH MB patients with confirmed SMARCA4-loss of function mutations (~9% of Gr. 3 MB [8]) in a currently active, but not recruiting MATCH phase II trial (NCT03213665) (Table 1). Thus, it will be interesting to learn if respective patients were enrolled and what objective response or progression-free survival rates resulted from treatment.
Roughly 20–25% of all non-WNT/non-SHH MB harbor alterations in either MYC or MYCN. Whereas these genes have long been identified as two of the most important oncogenes, directly targeting them has proven challenging [104]. Thus, indirectly inhibiting the effect of MYC family genes has attracted considerable interest in oncological research. The BET/bromodomain family (BRD/BET) consists of several genes that are key players in regulating the expression of oncogenes and in the organization of superenhancers [105]. Amongst many other functions, BRD/BET bromodomains recognize histone acetylation and subsequently activate gene expression mechanisms. Targeting BRD/BET proteins interferes with MYC-dependent transcription and was shown to be a promising strategy in a number of MYC(N)-driven cancers, including MB and neuroblastoma [56,106,107,108,109,110,111,112,113]. Currently, a number of BRD/BET inhibitors are developed, with promising preclinical results and importantly also in a pediatric setting [112]. Additionally, the first pediatric phase I trial testing the BRD/BET inhibitor BMS-986158 was started in 2019 and is open for participation of pediatric cancer patients with MYC(N)-amplification or high copy number gain, thus potentially offering a new therapeutic option for the most aggressive form of non-WNT/non-SHH MB (NCT03936465) (Table 1).
2.8. Immunotherapy
2.8.1. Immune Checkpoint Inhibitors
Immune checkpoint inhibitors that interfere with the PD1/PDL1- and CTLA4-mediated immunosuppressive crosstalk between malignant and immune effector cells are celebrated as one of the most important developments in oncology during the last decade [114]. However, these strategies rely on the presence of tumor-infiltrating lymphocytes and especially the blockade of the PD1/PDL1-axis is at least partly dependent on the expression of PDL1 on the respective tumor cells. A series of studies used immunohistochemistry and bioinformatic deconvolution to assess immune infiltration and PDL1-expression in MB [115,116,117,118,119,120,121,122,123]. Although most studies only analyzed small case series or cohorts of MB patients, together they suggest that MB is an immunologically “cold” tumor with only sparse immune infiltration. Furthermore, apart from one notable exception [118], all studies showed negligible or no PDL1-expression at all, especially for non-WNT/non-SHH MB [115,116,118,119,121,122]. Additionally, studies investigating intratumoral heterogeneity using single cell RNA-sequencing reported only minor infiltration of tumor-specific lymphocytes and a diverse spectrum of myeloid cells and microglia, which potentially contribute to an immunosuppressive tumor microenvironment [33,124,125]. These findings indicate that it may be challenging to implement immune checkpoint inhibition as part of future treatment strategies for these patients, at least in form of a monotherapy.
However, limited preclinical findings still warrant further validation of immune checkpoint blockade as a therapeutic concept for non-WNT/non-SHH MB (Figure 2), e.g., as part of a combination therapy that induces a “hotter” tumor microenvironment such as the abovementioned INFORM2 NivEnt trial [74,117,122]. Currently, several clinical trials that offer immune checkpoint inhibitors as a monotherapy are also open to patients with recurrent or relapsed MB (NCT02359565: Pembrolizumab and NCT03173950: Nivolumab) (Table 1). However, for the latter trial only patients >18 years of age are eligible, and these patients most often do not harbor non-WNT/non-SHH MB. Another phase II trial that tested either Nivolumab as a monotherapy or in combination with Ipilimumab is in the stage of finalization (NCT03130959). Lastly, a currently running industrial trial tests the combination of Nivolumab with the immunostimulant Bempegaldesleukin, a recombinant form of IL-2, in children and young adults with treatment-resistant cancer (NCT04730349). It should be noted though that none of these trials is exclusively recruiting non-WNT-/non-SHH or even generally MB-patients, and it remains to be seen if enough patients will be enrolled to allow for subgroup-specific analysis.
2.8.2. Cellular Immunotherapy
The success of cellular immunotherapy as a new treatment for hematologic malignancies has sparked intensive research activities that aim at translating these novel therapeutic concepts into the clinic for CNS- and other solid tumors. Currently, two main concepts of cellular immunotherapy are tested in MB patients: chimeric antigen receptor (CAR) T-cells and natural killer (NK) cell therapy [126]. CAR T-cells are produced by isolating the patient’s own cytotoxic T-cells, which are then equipped with a CAR that can recognize any form of surface marker and subsequently activates the T-cell to mount an immunological attack against the target cell [127]. It is of crucial importance to choose target antigens that are highly expressed on cancer cells, but not or only negligibly on normal tissue to avoid severe on target/off tumor-toxicity. Currently, several promising targets are investigated for MB-directed CAR T-cell therapy: HER2/Neu, B7-H3 (also called CD276), EPHA2, IL-13Rα2, and PRAME [128,129,130,131,132,133]. HER2/Neu plays an important role as an oncogenic antigen in a number of solid tumors, including breast cancer and glioblastoma, and has also been identified as a possible target for MB-directed cellular therapy both in vitro and in vivo [129,130,134]. Currently, one phase I trial testing HER2/Neu-specific CAR T-cells is recruiting and open to MB patients (NCT03500991), and a recently published interim analysis of the first three enrolled patients (anaplastic astrocytoma, ependymoma) showed no dose-limiting toxicity and presented evidence of immune activation (Table 1) [135]. B7-H3/CD276 is a pancancer antigen that is strongly expressed by MB [128]. Similar to HER2/Neu, B7-H3-specific CAR T-cells have shown preclinical activity against non-WNT/non-SHH MB-models both in vitro and in vivo as well as in a number of other (pediatric) cancers, including atypical teratoid/rhabdoid tumor (AT/RT), another aggressive CNS-tumor of early childhood [128,136,137,138]. These preclinical findings provide a strong rationale to test B7-H3 targeting CAR T-cells in the clinic, which is currently undertaken in one phase I study that enrolls children with B7-H3 positive CNS-malignancies, including MB (NCT04185038). While HER2/Neu and B7-H3 targeting CARs have already been translated into the clinic, several other targets may be of interest based on preclinical data: one study showed strong expression of EPHA2 and IL-13α2 in human Gr.3 MB and subsequently tested both monovalent EPHA2- and trivalent EPHA2-/HER2/Neu-/IL-13α2-targeting CAR T-cells in a Gr. 3 MB-mouse model, with promising results [132]. An IL-13α2 directed CAR T-cell trial is recruiting adult patients with leptomeningeal metastases, including MB, and the results may be of interest to inform future trials in pediatric populations (NCT04661384). Furthermore, another study provided evidence in vitro that PRAME might represent a promising target for CAR T-cell therapy in MB [133]. Additionally, two more phase I CAR-T cell trials are currently open to pediatric patients with CNS-malignancies, including MB. However, the respective target antigens EGFR806 and GD2 have not been tested preclinically in MB patients (NCT03638167 and NCT04099797). Lastly, it should be noted that the delivery of CAR T-cells to the brain poses challenges due to the role of the blood–brain barrier, which could lower the effectiveness of intravenously applied cellular immunotherapies [129]. To date, the best application route for CAR T-cell in neurooncology has not yet been determined. However, the majority (4/5) of currently running CAR T-cell trials for which MB patients are eligible will use intraventricular/intracavital cell delivery, which circumvents the blood–brain barrier.
An exciting alternative to CAR T-cells is the use of NK cells that may offer certain advantages in comparison, such as lower side effects and increased resistance to immune evasion strategies of the tumor [139]. Several preclinical studies have shown that in principle, NK cells are able to recognize and eliminate MB cells; however, additional stimulation may be needed to arrive at clinically meaningful cytotoxic activity levels [140,141,142,143,144]. Interestingly, one study showed a higher and more consistent sensitivity to NK cells in vitro for non-WNT/non-SHH as compared to SHH MB cell lines [141]. In contrast, another study reported significantly higher expression levels of CD1d, an antigen recognized by NK cells, on SHH as compared to Gr. 4 MB [142]. Clearly, further studies are needed to arrive at a conclusive answer concerning which MB groups are the most promising target for NK cell therapy. The safety and feasibility of NK cell therapy for pediatric brain cancer has recently been shown by a phase I trial that also enrolled five MB patients, although without reporting molecular diagnoses (NCT02271711) [145].
2.8.3. Tumor Vaccinations
Cancer vaccine approaches harness the ability of off-the-shelf or patient-specific antigens to induce an antitumoral immune reaction [146]. Several different strategies have been developed, for instance using antigen pulsed dendritic cells, peptides, and nucleic acid vectors. Tumor vaccines have been studied for a long time; however, several early phase trials have recently shown great potential in adult glioblastoma patients [147,148,149]. One study tested dendritic cells that were pulsed with tumor lysate-derived antigens. As compared to AT/RT and high grade glioma, the five included MB patients showed only modest therapy response, albeit no molecular information is available [150]. The results of another phase I trial that used RNA-pulsed dendritic cells and also enrolled patients with recurrent MB and glioma are pending (NCT03615404). An additional trial applies another strategy that uses a peptide-based approach (NCT03299309). Lastly, MB patients are currently also eligible for a first-in-pediatrics trial that investigates a long peptide vaccine that targets the apoptosis inhibitor survivin (NCT04978727) (Table 1). Similar to most of the treatment strategies presented so far, none of these trials is restricted to non-WNT/non-SHH MB. Furthermore, so far, no definitive conclusions can be drawn from the reported data concerning differences in the efficacy of vaccination therapies for MB molecular subgroups.
2.8.4. Other Immunotherapeutic Approaches
Apart from checkpoint inhibition, increasingly more immunomodulatory approaches are entering the clinical stage. Two studies for a mixed pediatric population with CNS malignancies are currently testing immunostimulatory agents (Table 1): firstly, the antibody APX005M that targets CD40 (NCT03389802) and secondly the immune modulator WP1066, which inhibits the transcription factor STAT3 and therefore interferes with the JAK2/STAT3-pathway (NCT04334863). Additionally, oncolytic viruses, which have recently shown promising results for pediatric high grade glioma, have been proposed as a therapeutic option for MB [151,152]. Preclinical studies suggest that Gr. 3 MB might be a potential candidate for oncolytic virus therapy, and several different viral vectors have been tested throughout the last decade [153,154,155,156,157,158,159,160,161]. Oncolytic virus studies that are currently recruiting MB patients include the investigation of a modified measles vaccine (NCT02962167), Herpes Simplex Virus (NCT03911388), and one open phase IB trial that will assess possible agonistic effects between mesenchymal allogenic cells and an adenovirus-based virotherapy (NCT04758533) (Table 1). Two additional early phase studies that are open to MB patients are currently active, but not recruiting; these are testing reovirus in combination with Sagramostim and a modified poliovirus (NCT02444546, NCT03043391).
2.9. Other Molecular Therapeutic Approaches
In addition to the treatment approaches discussed thus far, several other concepts should be mentioned briefly (Table 1). Firstly, the inhibition of placental growth factor (PGF) has been proposed as a promising strategy across all MB subgroups and was tested in a phase I trial using the monoclonal antibody TB-403 (NCT02748135) [162]. The results have been presented at the AACR annual meeting 2021 and were encouraging both in terms of safety and efficacy according to the producing company; however, a peer-reviewed publication is not available to date [163]. Another interesting treatment approach represents the inhibition of DNA damage response pathways using poly (ADP-ribose) polymerase (PARP) inhibitors, a relatively new drug class that has received numerous approvals for breast and ovarian cancer patients with germline BRCA1/2 mutations in the last years [164]. This could be especially relevant to a subset Gr. 3/4 MB, since these are the molecular groups that are enriched for (germline) BRCA2- and PALB2-mutations [13,16]. Thus, it will be interesting to see whether non-WNT/non-SHH patients will be included in currently running trials using PARP inhibitors (NCT03233204 and NCT04236414). Lastly, several preclinical studies with different compounds indicate that the concept of sensitizing cancer cells to radiotherapy using small molecules could be potentially interesting for non-WNT/non-SHH MB [165,166,167]
3. Discussion
In this review, we aimed at summarizing the current landscape of targeted clinical trials and most important molecular targets for non-WNT/non-SHH MB. Although a multitude of promising, molecularly informed new treatment options is tested to date, several significant challenges remain:
(i) Preclinical in vitro and in vivo models for non-WNT/non-SHH MB are almost entirely restricted to MYC-driven Gr. 3 MB and, thus, do not reflect the true heterogeneity of these malignancies. This is especially true for Gr. 4 MB, for which faithful models are still broadly lacking despite it being the most frequent form of MB [8,134,168].
(ii) The considerable heterogeneity of genetic driver events in non-WNT/non-SHH MB makes it more challenging to systematically test therapeutic approaches in reasonably sized patient cohorts. Although there was significant progress in the last years, the biological underpinnings of non-WNT/non-SHH MB are not yet fully understood. Thus far, no targetable alteration has been identified that would be present in more than a small share of non-WNT/non-SHH MB cases. Additionally, many theoretically targetable alterations are not predictive of response [52].
(iii) The rise of molecular diagnostics has further subdivided MB. Whereas these developments offer exciting chances to arrive at more homogeneous, biologically defined risk groups, they also pose a considerable challenge to clinical research, since it becomes increasingly more difficult to collect a significantly large number of patients for clinical trials. Additionally, different competing stratification strategies are conceivable. For instance, non-WNT/non-SHH MB could be stratified into eight molecular subgroups based on DNA methylation patterns. However, this approach would collide with dividing patients according to genetic alterations, such as MYC-amplification, which is not restricted to just one of the abovementioned molecular subgroups [34]. Furthermore, the number of available patients will be too small to test several drug candidates for the same molecular target by different companies, making a prioritization of most promising drug candidates based upon evaluated and consensus criteria necessary [169,170]. Again, this already challenging situation is complicated by the fact that any monotherapy is likely to be of only modest success, and combinatorial approaches will probably have to be tested to arrive at clinically meaningful results. These could include combinations between conventional chemotherapeutic and targeted agents, as currently tested by the MEMMAT (NCT01356290) or SJDAWN-trials (NCT03434262). However, therapy regimens consisting of several immunotherapeutic or targeted drugs, such as the INFORM2 NivENT study, also hold promise to arrive at new options to treat children suffering from non-WNT/non-SHH MB [74]. Recently, an international consensus statement defined the minimum preclinical testing requirements that should be met before translating a drug into the early phase clinical setting [169]. Notably, while these recommendations strongly advocate for the use of several in vivo models (PDX, genetically engineered mice models, etc.) for any new compound before advancing to a phase I trial, this kind of solid proof-of-concept evidence is broadly missing in the field of non-WNT/non-SHH MB. As highlighted earlier, this issue is largely rooted in the lack of faithful preclinical models and has thus been to a certain extent unavoidable in the past. However, as soon as more and better in vivo models are available, any given drug (or combination of drugs) should be tested rigorously in a standardized set of preclinical models before entering the clinic, as pointed out by Vassal et al. [169].
(iv) Throughout the last decade, the vast majority of phase I/II trials in which MB patients were enrolled did not report molecular information; thus, for a large share of the studies conducted so far that tested new treatment modalities for MB, information on the efficacy in non-WNT/non-SHH MB is lacking.
(v) Due to the rarity of pediatric cancer in general, most of the past and currently open trials are not restricted to MB, let alone non-WNT/non-SHH MB. The small number of non-WNT/non-SHH MB patients per trial will be a considerable challenge to generate the level of high-quality evidence that is necessary to change the current therapeutic standard. Conducting classic randomized phase III therapy optimization trials for MB will become increasingly more challenging, especially when it comes to recurrent disease. The basis to overcome these challenges lies in extensive international cooperation and a shared strategy between academia and industry. A blueprint on how research activities can be coordinated and hastened to benefit non-WNT/non-SHH MB patients represents the international ACCELERATE platform, which, for instance, recently published a roadmap for the development of BRT/BET inhibitors [112]. Further efforts are necessary to standardize preclinical models and to determine criteria to choose the most promising amongst several drug candidates with the same target as this, for example, is realized by the pediatric preclinical proof-of-concept platform ITCC-P4 (https://www.itccp4.eu, accessed on 12 December 2021).
Innovative and creative solutions to arrive at solid evidence levels for new drugs will be of paramount importance. New trial designs, for instance, based on Bayesian statistics, “pick the winner” concepts or in the form of molecular basket and umbrella trials, promise to overcome these issues eventually; however, they also pose methodological challenges [171]. Several comprehensive pediatric precision oncology programs such as pediatric MATCH, MOSCATO-01, the ZERO Childhood Cancer Program, and the INFORM platform (Individualized Therapy for Relapsed Malignancies in Childhood) have been established over the recent years to allow for assignment of patients to matching trials [52,172,173,174,175]. These registries could serve as a tool to stratify non-WNT/non-SHH MB patients into molecular-guided early phase I clinical trials. These will be necessary even in the presence of new drugs that are developed solely for non-WNT/non-SHH MB, since the large heterogeneity within this subgroup will probably prevent the design of a randomized trial in which all non-WNT/non-SHH MB patients can be included. In order to extract as much information as possible from every single case, conducting molecular diagnostics for patients who were enrolled on past trials should be pursued wherever archived material is still available for retrospective testing. Additionally, every future trial should comprehensively assess molecular characteristics of non-WNT/non-SHH MB to determine the role of biomarkers and to identify the most promising drug combinations for given targets. Although a multitude of innovative treatment modalities is theoretically available for non-WNT/non-SHH MB already and even newer concepts are currently developed, such as refined ways of targeting epigenetic alterations in cancer or strategies to directly target MYC-family genes [104], none of these concepts currently are standing out.
4. Conclusions
Non-WNT/non-SHH MB belongs to the most heterogeneous and complex forms of embryonal brain tumors in children. Throughout the last decade, our understanding of its biological underpinning has grown considerably, and these findings are slowly translating from bench to bedside. Whereas a multitude of therapeutic options, ranging from kinase inhibitors to immunotherapy and strategies to target the epigenome, is available to non-WNT/non-SHH MB patients in theory, the vast majority of these trials is still in phase I and not restricted to this patient group. A refined preclinical pipeline with new model systems, coordinated international efforts, and innovative trial and research strategies will be necessary to arrive at new therapeutic options and eventually improved clinical results for non-WNT/non-SHH MB.
Author Contributions
Conceptualization, D.R.G. and K.W.P.; methodology, D.R.G.; writing—review and editing, D.R.G., G.F., T.M. and K.W.P.; visualization, D.R.G.; supervision, project administration, and funding acquisition, K.W.P. All authors have read and agreed to the published version of the manuscript.
Funding
D.R.G. and K.W.P. are supported by the Ein Kiwi gegen Krebs-Foundation. D.R.G. received a fellowship of the German Academic Scholarship Foundation.
Acknowledgments
All illustrations were created using and adapting graphics from the free resource bioicons.com (Simon Dürr) provided by the users Servier, DBCLS, and Marcel Tisch, and licensed under the CC 0, CC-BY 3.0 (https://creativecommons.org/licenses/by/3.0/) and CC-BY 4.0 (https://creativecommons.org/licenses/by/4.0/) Unported licenses, respectively.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Erdmann, F.; Kaatsch, P.; Grabow, D.; Spix, C. German Childhood Cancer Registry—Annual Report 2019 (1980–2018); Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz: Mainz, Germany, 2020. [Google Scholar]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncology 2020, 22, iv1–iv96. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.W. Childhood medulloblastoma: Novel approaches to the classification of a heterogeneous disease. Acta Neuropathol. 2010, 120, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Northcott, P.A.; Robinson, G.W.; Kratz, C.P.; Mabbott, D.J.; Pomeroy, S.L.; Clifford, S.C.; Rutkowski, S.; Ellison, D.W.; Malkin, D.; Taylor, M.D.; et al. Medulloblastoma. Nat. Rev. Dis. Primers 2019, 5, 11. [Google Scholar] [CrossRef]
- Cruzeiro, G.A.V.; Salomão, K.B.; de Biagi, C.A.O., Jr.; Baumgartner, M.; Sturm, D.; Lira, R.C.P.; de Almeida Magalhães, T.; Baroni Milan, M.; da Silva Silveira, V.; Saggioro, F.P.; et al. A simplified approach using Taqman low-density array for medulloblastoma subgrouping. Acta Neuropathol. Commun. 2019, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Shih, D.J.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stutz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef]
- Ellison, D.W.; Onilude, O.E.; Lindsey, J.C.; Lusher, M.E.; Weston, C.L.; Taylor, R.E.; Pearson, A.D.; Clifford, S.C. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: The United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J. Clin. Oncol. 2005, 23, 7951–7957. [Google Scholar] [CrossRef]
- Hovestadt, V.; Ayrault, O.; Swartling, F.J.; Robinson, G.W.; Pfister, S.M.; Northcott, P.A. Medulloblastomics revisited: Biological and clinical insights from thousands of patients. Nat. Rev. Cancer 2020, 20, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Grobner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratz, C.P.; Jongmans, M.C.; Cavé, H.; Wimmer, K.; Behjati, S.; Guerrini-Rousseau, L.; Milde, T.; Pajtler, K.W.; Golmard, L.; Gauthier-Villars, M.; et al. Predisposition to cancer in children and adolescents. Lancet Child Adolesc. Health 2021, 5, 142–154. [Google Scholar] [CrossRef]
- Waszak, S.M.; Northcott, P.A.; Buchhalter, I.; Robinson, G.W.; Sutter, C.; Groebner, S.; Grund, K.B.; Brugieres, L.; Jones, D.T.W.; Pajtler, K.W.; et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018, 19, 785–798. [Google Scholar] [CrossRef]
- Garcia-Lopez, J.; Kumar, R.; Smith, K.S.; Northcott, P.A. Deconstructing Sonic Hedgehog Medulloblastoma: Molecular Subtypes, Drivers, and Beyond. Trends Genet. 2021, 37, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Begemann, M.; Waszak, S.M.; Robinson, G.W.; Jager, N.; Sharma, T.; Knopp, C.; Kraft, F.; Moser, O.; Mynarek, M.; Guerrini-Rousseau, L.; et al. Germline GPR161 Mutations Predispose to Pediatric Medulloblastoma. J. Clin. Oncol. 2020, 38, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Waszak, S.M.; Robinson, G.W.; Gudenas, B.L.; Smith, K.S.; Forget, A.; Kojic, M.; Garcia-Lopez, J.; Hadley, J.; Hamilton, K.V.; Indersie, E.; et al. Germline Elongator mutations in Sonic Hedgehog medulloblastoma. Nature 2020, 580, 396–401. [Google Scholar] [CrossRef]
- Pfaff, E.; Remke, M.; Sturm, D.; Benner, A.; Witt, H.; Milde, T.; von Bueren, A.O.; Wittmann, A.; Schottler, A.; Jorch, N.; et al. TP53 mutation is frequently associated with CTNNB1 mutation or MYCN amplification and is compatible with long-term survival in medulloblastoma. J. Clin. Oncol. 2010, 28, 5188–5196. [Google Scholar] [CrossRef]
- Tabori, U.; Baskin, B.; Shago, M.; Alon, N.; Taylor, M.D.; Ray, P.N.; Bouffet, E.; Malkin, D.; Hawkins, C. Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J. Clin. Oncol. 2010, 28, 1345–1350. [Google Scholar] [CrossRef]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, G.W.; Rudneva, V.A.; Buchhalter, I.; Billups, C.A.; Waszak, S.M.; Smith, K.S.; Bowers, D.C.; Bendel, A.; Fisher, P.G.; Partap, S.; et al. Risk-adapted therapy for young children with medulloblastoma (SJYC07): Therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol. 2018, 19, 768–784. [Google Scholar] [CrossRef]
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Jones, D.T.; Kool, M.; Robinson, G.W.; Gilbertson, R.J.; Cho, Y.J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 2012, 12, 818–834. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.J.; Tsherniak, A.; Tamayo, P.; Santagata, S.; Ligon, A.; Greulich, H.; Berhoukim, R.; Amani, V.; Goumnerova, L.; Eberhart, C.G.; et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J. Clin. Oncol. 2011, 29, 1424–1430. [Google Scholar] [CrossRef]
- Dufour, C.; Foulon, S.; Geoffray, A.; Masliah-Planchon, J.; Figarella-Branger, D.; Bernier-Chastagner, V.; Padovani, L.; Guerrini-Rousseau, L.; Faure-Conter, C.; Icher, C.; et al. Prognostic relevance of clinical and molecular risk factors in children with high-risk medulloblastoma treated in the phase II trial PNET HR+5. Neuro-Oncology 2021, 23, 1163–1172. [Google Scholar] [CrossRef]
- Archer, T.C.; Ehrenberger, T.; Mundt, F.; Gold, M.P.; Krug, K.; Mah, C.K.; Mahoney, E.L.; Daniel, C.J.; LeNail, A.; Ramamoorthy, D.; et al. Proteomics, Post-translational Modifications, and Integrative Analyses Reveal Molecular Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2018, 34, 396–410.e8. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Jager, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.J.; Pugh, T.J.; Hovestadt, V.; Stutz, A.M.; et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012, 488, 100–105. [Google Scholar] [CrossRef]
- Schwalbe, E.C.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet Oncol. 2017, 18, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Menyhárt, O.; Giangaspero, F.; Győrffy, B. Molecular markers and potential therapeutic targets in non-WNT/non-SHH (group 3 and group 4) medulloblastomas. J. Hematol. Oncol. 2019, 12, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Lee, C.; Zichner, T.; Stutz, A.M.; Erkek, S.; Kawauchi, D.; Shih, D.J.; Hovestadt, V.; Zapatka, M.; Sturm, D.; et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 2014, 511, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Hovestadt, V.; Smith, K.S.; Bihannic, L.; Filbin, M.G.; Shaw, M.L.; Baumgartner, A.; DeWitt, J.C.; Groves, A.; Mayr, L.; Weisman, H.R.; et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature 2019, 572, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.; Schwalbe, E.C.; Williamson, D.; Sill, M.; Hovestadt, V.; Mynarek, M.; Rutkowski, S.; Robinson, G.W.; Gajjar, A.; Cavalli, F.; et al. Second-generation molecular subgrouping of medulloblastoma: An international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol. 2019, 138, 309–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leary, S.E.S.; Packer, R.J.; Li, Y.; Billups, C.A.; Smith, K.S.; Jaju, A.; Heier, L.; Burger, P.; Walsh, K.; Han, Y.; et al. Efficacy of Carboplatin and Isotretinoin in Children With High-risk Medulloblastoma: A Randomized Clinical Trial From the Children’s Oncology Group. JAMA Oncol. 2021, 7, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Bellantoni, A.J.; Wagner, L.M. Pursuing Precision: Receptor Tyrosine Kinase Inhibitors for Treatment of Pediatric Solid Tumors. Cancers 2021, 13, 3531. [Google Scholar] [CrossRef]
- Burdach, S.E.G.; Westhoff, M.A.; Steinhauser, M.F.; Debatin, K.M. Precision medicine in pediatric oncology. Mol. Cell. Pediatr. 2018, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, P.S.; Venkataraman, S.; Alimova, I.; Birks, D.K.; Balakrishnan, I.; Cristiano, B.; Donson, A.M.; Dubuc, A.M.; Taylor, M.D.; Foreman, N.K.; et al. Integrated genomic analysis identifies the mitotic checkpoint kinase WEE1 as a novel therapeutic target in medulloblastoma. Mol. Cancer 2014, 13, 72. [Google Scholar] [CrossRef] [Green Version]
- Moreira, D.C.; Venkataraman, S.; Subramanian, A.; Desisto, J.; Balakrishnan, I.; Prince, E.; Pierce, A.; Griesinger, A.; Green, A.; Eberhardt, C.G.; et al. Targeting MYC-driven replication stress in medulloblastoma with AZD1775 and gemcitabine. J. NeuroOncol. 2020, 147, 531–545. [Google Scholar] [CrossRef]
- Geenen, J.J.J.; Schellens, J.H.M. Molecular Pathways: Targeting the Protein Kinase Wee1 in Cancer. Clin. Cancer Res. 2017, 23, 4540–4544. [Google Scholar] [CrossRef] [Green Version]
- Cole, K.A.; Ijaz, H.; Surrey, L.; Santi-vicini, M.; Liu, X.; Minard, C.G.; Maris, J.M.; Voss, S.; Fox, E.; Weigal, B. Abstract CT029: Pediatric phase 2 trial of the WEE1 inhibitor adavosertib (AZD1775) and irinotecan: A Children’s Oncology Group Study (ADVL1312). Cancer Res. 2021, 81, CT029. [Google Scholar] [CrossRef]
- Cole, K.A.; Pal, S.; Kudgus, R.A.; Ijaz, H.; Liu, X.; Minard, C.G.; Pawel, B.R.; Maris, J.M.; Haas-Kogan, D.A.; Voss, S.D.; et al. Phase I Clinical Trial of the Wee1 Inhibitor Adavosertib (AZD1775) with Irinotecan in Children with Relapsed Solid Tumors: A COG Phase I Consortium Report (ADVL1312). Clin. Cancer Res. 2020, 26, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Romo, C.G.; Alexander, B.M.; Agar, N.; Ahluwalia, M.S.; Desai, A.S.; Dietrich, J.; Kaley, T.J.; Peereboom, D.M.; Takebe, N.; Desideri, S.; et al. Intratumoral drug distribution of adavosertib in patients with glioblastoma: Interim results of phase I study. J. Clin. Oncol. 2020, 38, 2568. [Google Scholar] [CrossRef]
- De Gooijer, M.C.; Buil, L.C.M.; Beijnen, J.H.; van Tellingen, O. ATP-binding cassette transporters limit the brain penetration of Wee1 inhibitors. Investig. New Drugs 2018, 36, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Pokorny, J.L.; Calligaris, D.; Gupta, S.K.; Iyekegbe, D.O., Jr.; Mueller, D.; Bakken, K.K.; Carlson, B.L.; Schroeder, M.A.; Evans, D.L.; Lou, Z.; et al. The Efficacy of the Wee1 Inhibitor MK-1775 Combined with Temozolomide Is Limited by Heterogeneous Distribution across the Blood-Brain Barrier in Glioblastoma. Clin. Cancer Res. 2015, 21, 1916–1924. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wu, J.; Bao, X.; Honea, N.; Xie, Y.; Kim, S.; Sparreboom, A.; Sanai, N. Quantitative and Mechanistic Understanding of AZD1775 Penetration across Human Blood-Brain Barrier in Glioblastoma Patients Using an IVIVE-PBPK Modeling Approach. Clin. Cancer Res. 2017, 23, 7454–7466. [Google Scholar] [CrossRef] [Green Version]
- Endersby, R.; Whitehouse, J.; Pribnow, A.; Kuchibhotla, M.; Hii, H.; Carline, B.; Gande, S.; Stripay, J.; Ancliffe, M.; Howlett, M.; et al. Small-molecule screen reveals synergy of cell cycle checkpoint kinase inhibitors with DNA-damaging chemotherapies in medulloblastoma. Sci. Transl. Med. 2021, 13, eaba7401. [Google Scholar] [CrossRef]
- Campagne, O.; Davis, A.; Maharaj, A.R.; Zhong, B.; Stripay, J.; Farmer, D.; Roussel, M.F.; Stewart, C.F. CNS penetration and pharmacodynamics of the CHK1 inhibitor prexasertib in a mouse Group 3 medulloblastoma model. Eur. J. Pharm. Sci. 2020, 142, 105106. [Google Scholar] [CrossRef]
- Prince, E.W.; Balakrishnan, I.; Shah, M.; Mulcahy Levy, J.M.; Griesinger, A.M.; Alimova, I.; Harris, P.S.; Birks, D.K.; Donson, A.M.; Davidson, N.; et al. Checkpoint kinase 1 expression is an adverse prognostic marker and therapeutic target in MYC-driven medulloblastoma. Oncotarget 2016, 7, 53881–53894. [Google Scholar] [CrossRef] [Green Version]
- Zhong, B.; Maharaj, A.; Davis, A.; Roussel, M.F.; Stewart, C.F. Development and validation of a sensitive LC MS/MS method for the measurement of the checkpoint kinase 1 inhibitor prexasertib and its application in a cerebral microdialysis study. J. Pharm. Biomed. Anal. 2018, 156, 97–103. [Google Scholar] [CrossRef]
- Cash, T.; Fox, E.; Liu, X.; Minard, C.G.; Reid, J.M.; Scheck, A.C.; Weigel, B.J.; Wetmore, C. A phase 1 study of prexasertib (LY2606368), a CHK1/2 inhibitor, in pediatric patients with recurrent or refractory solid tumors, including CNS tumors: A report from the Children’s Oncology Group Pediatric Early Phase Clinical Trials Network (ADVL1515). Pediatr. Blood Cancer 2021, 68, e29065. [Google Scholar] [CrossRef]
- Van Tilburg, C.M.; Pfaff, E.; Pajtler, K.W.; Langenberg, K.P.S.; Fiesel, P.; Jones, B.C.; Balasubramanian, G.P.; Stark, S.; Johann, P.D.; Blattner-Johnson, M.; et al. The pediatric precision oncology INFORM registry: Clinical outcome and benefit for patients with very high-evidence targets. Cancer Discov. 2021, 11, 2764–2779. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goga, A.; Yang, D.; Tward, A.D.; Morgan, D.O.; Bishop, J.M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 2007, 13, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Bolin, S.; Borgenvik, A.; Persson, C.U.; Sundström, A.; Qi, J.; Bradner, J.E.; Weiss, W.A.; Cho, Y.J.; Weishaupt, H.; Swartling, F.J. Combined BET bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Oncogene 2018, 37, 2850–2862. [Google Scholar] [CrossRef] [Green Version]
- Cook Sangar, M.L.; Genovesi, L.A.; Nakamoto, M.W.; Davis, M.J.; Knobluagh, S.E.; Ji, P.; Millar, A.; Wainwright, B.J.; Olson, J.M. Inhibition of CDK4/6 by Palbociclib Significantly Extends Survival in Medulloblastoma Patient-Derived Xenograft Mouse Models. Clin. Cancer Res. 2017, 23, 5802–5813. [Google Scholar] [CrossRef]
- Genovesi, L.A.; Millar, A.; Tolson, E.; Singleton, M.; Hassall, E.; Kojic, M.; Brighi, C.; Girard, E.; Andradas, C.; Kuchibhotla, M.; et al. Systems pharmacogenomics identifies novel targets and clinically actionable therapeutics for medulloblastoma. Genome Med. 2021, 13, 103. [Google Scholar] [CrossRef]
- Van Mater, D.; Gururangan, S.; Becher, O.; Campagne, O.; Leary, S.; Phillips, J.J.; Huang, J.; Lin, T.; Poussaint, T.Y.; Goldman, S.; et al. A phase I trial of the CDK 4/6 inhibitor palbociclib in pediatric patients with progressive brain tumors: A Pediatric Brain Tumor Consortium study (PBTC-042). Pediatr. Blood Cancer 2021, 68, e28879. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Alshawli, A.S.; Wurdak, H.; Wood, I.C.; Ladbury, J.E. Histone deacetylase inhibitors induce medulloblastoma cell death independent of HDACs recruited in REST repression complexes. Mol. Genet. Genom. Med. 2020, 8, e1429. [Google Scholar] [CrossRef]
- Ecker, J.; Oehme, I.; Mazitschek, R.; Korshunov, A.; Kool, M.; Hielscher, T.; Kiss, J.; Selt, F.; Konrad, C.; Lodrini, M.; et al. Targeting class I histone deacetylase 2 in MYC amplified group 3 medulloblastoma. Acta Neuropathol. Commun. 2015, 3, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ecker, J.; Thatikonda, V.; Sigismondo, G.; Selt, F.; Valinciute, G.; Oehme, I.; Müller, C.; Buhl, J.L.; Ridinger, J.; Usta, D.; et al. Reduced chromatin binding of MYC is a key effect of HDAC inhibition in MYC amplified medulloblastoma. Neuro-Oncology 2021, 23, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lindsey, S.; Graves, B.; Yoo, S.; Olson, J.M.; Langhans, S.A. Sonic hedgehog-induced histone deacetylase activation is required for cerebellar granule precursor hyperplasia in medulloblastoma. PLoS ONE 2013, 8, e71455. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Liu, K.W.; Wang, J.; Garancher, A.; Tao, R.; Esparza, L.A.; Maier, D.L.; Udaka, Y.T.; Murad, N.; Morrissy, S.; et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell 2016, 29, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phi, J.H.; Choi, S.A.; Kwak, P.A.; Lee, J.Y.; Wang, K.C.; Hwang, D.W.; Kim, S.K. Panobinostat, a histone deacetylase inhibitor, suppresses leptomeningeal seeding in a medulloblastoma animal model. Oncotarget 2017, 8, 56747–56757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Gong, Z.; Oladimeji, P.O.; Currier, D.G.; Deng, Q.; Liu, M.; Chen, T.; Li, Y. A high-throughput screening identifies histone deacetylase inhibitors as therapeutic agents against medulloblastoma. Exp. Hematol. Oncol. 2019, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- van Tilburg, C.M.; Milde, T.; Witt, R.; Ecker, J.; Hielscher, T.; Seitz, A.; Schenk, J.-P.; Buhl, J.L.; Riehl, D.; Frühwald, M.C.; et al. Phase I/II intra-patient dose escalation study of vorinostat in children with relapsed solid tumor, lymphoma, or leukemia. Clin. Epigenetics 2019, 11, 188. [Google Scholar] [CrossRef] [Green Version]
- Hummel, T.R.; Wagner, L.; Ahern, C.; Fouladi, M.; Reid, J.M.; McGovern, R.M.; Ames, M.M.; Gilbertson, R.J.; Horton, T.; Ingle, A.M.; et al. A pediatric phase 1 trial of vorinostat and temozolomide in relapsed or refractory primary brain or spinal cord tumors: A Children’s Oncology Group phase 1 consortium study. Pediatr. Blood Cancer 2013, 60, 1452–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Ciesielski, M.; Ramakrishnan, S.; Miles, K.M.; Ellis, L.; Sotomayor, P.; Shrikant, P.; Fenstermaker, R.; Pili, R. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PLoS ONE 2012, 7, e30815. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Iclozan, C.; Liu, C.; Xia, C.; Anasetti, C.; Yu, X.Z. LBH589 enhances T cell activation in vivo and accelerates graft-versus-host disease in mice. Biol. Blood Marrow Transplant. 2012, 18, 1182–1190.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Zhao, W.; Yan, C.; Watson, C.C.; Massengill, M.; Xie, M.; Massengill, C.; Noyes, D.R.; Martinez, G.V.; Afzal, R.; et al. HDAC Inhibitors Enhance T-Cell Chemokine Expression and Augment Response to PD-1 Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2016, 22, 4119–4132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A., Jr.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Tilburg, C.M.; Witt, R.; Heiss, M.; Pajtler, K.W.; Plass, C.; Poschke, I.; Platten, M.; Harting, I.; Sedlaczek, O.; Freitag, A.; et al. INFORM2 NivEnt: The first trial of the INFORM2 biomarker driven phase I/II trial series: The combination of nivolumab and entinostat in children and adolescents with refractory high-risk malignancies. BMC Cancer 2020, 20, 523. [Google Scholar] [CrossRef]
- Cruzeiro, G.A.V.; Dos Reis, M.B.; Silveira, V.S.; Lira, R.C.P.; Carlotti, C.G., Jr.; Neder, L.; Oliveira, R.S.; Yunes, J.A.; Brandalise, S.R.; Aguiar, S.; et al. HIF1A is Overexpressed in Medulloblastoma and its Inhibition Reduces Proliferation and Increases EPAS1 and ATG16L1 Methylation. Curr. Cancer Drug Targets 2018, 18, 287–294. [Google Scholar] [CrossRef]
- Slongo, M.L.; Molena, B.; Brunati, A.M.; Frasson, M.; Gardiman, M.; Carli, M.; Perilongo, G.; Rosolen, A.; Onisto, M. Functional VEGF and VEGF receptors are expressed in human medulloblastomas. Neuro-Oncology 2007, 9, 384–392. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Held-Feindt, J.; Buhl, R.; Mehdorn, H.M.; Mentlein, R. Expression of VEGF and its receptors in different brain tumors. Neurol. Res. 2005, 27, 371–377. [Google Scholar] [CrossRef]
- Thompson, E.M.; Keir, S.T.; Venkatraman, T.; Lascola, C.; Yeom, K.W.; Nixon, A.B.; Liu, Y.; Picard, D.; Remke, M.; Bigner, D.D.; et al. The role of angiogenesis in Group 3 medulloblastoma pathogenesis and survival. Neuro-Oncology 2017, 19, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, M.; Craveiro, R.B.; Velz, J.; Olschewski, M.; Casati, A.; Schönberger, S.; Pietsch, T.; Dilloo, D. The FDA approved PI3K inhibitor GDC-0941 enhances in vitro the anti-neoplastic efficacy of Axitinib against c-myc-amplified high-risk medulloblastoma. J. Cell Mol. Med. 2018, 22, 2153–2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwinn, S.; Mokhtari, Z.; Thusek, S.; Schneider, T.; Sirén, A.L.; Tiemeyer, N.; Caruana, I.; Miele, E.; Schlegel, P.G.; Beilhack, A.; et al. Cytotoxic effects and tolerability of gemcitabine and axitinib in a xenograft model for c-myc amplified medulloblastoma. Sci. Rep. 2021, 11, 14062. [Google Scholar] [CrossRef]
- Aguilera, D.; Mazewski, C.; Fangusaro, J.; MacDonald, T.J.; McNall-Knapp, R.Y.; Hayes, L.L.; Kim, S.; Castellino, R.C. Response to bevacizumab, irinotecan, and temozolomide in children with relapsed medulloblastoma: A multi-institutional experience. Childs Nerv. Syst. 2013, 29, 589–596. [Google Scholar] [CrossRef]
- Bonney, P.A.; Santucci, J.A.; Maurer, A.J.; Sughrue, M.E.; McNall-Knapp, R.Y.; Battiste, J.D. Dramatic response to temozolomide, irinotecan, and bevacizumab for recurrent medulloblastoma with widespread osseous metastases. J. Clin. Neurosci. 2016, 26, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Piha-Paul, S.A.; Shin, S.J.; Vats, T.; Guha-Thakurta, N.; Aaron, J.; Rytting, M.; Kleinerman, E.; Kurzrock, R. Pediatric patients with refractory central nervous system tumors: Experiences of a clinical trial combining bevacizumab and temsirolimus. Anticancer Res. 2014, 34, 1939–1945. [Google Scholar] [PubMed]
- Reismüller, B.; Azizi, A.A.; Peyrl, A.; Heinrich, M.; Gruber-Olipitz, M.; Luckner, D.; Rothschild, K.V.; Slavc, I. Feasibility and tolerability of bevacizumab in children with primary CNS tumors. Pediatr. Blood Cancer 2010, 54, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, Q.; Ma, H.Y.; Sun, T.; Xiang, R.L.; Di, F. Therapeutic effect and side effects of Bevacizumab combined with Irinotecan in the treatment of paediatric intracranial tumours: Meta-analysis and Systematic Review. J. Clin. Pharm. Ther. 2020, 45, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Fangusaro, J.; Gururangan, S.; Poussaint, T.Y.; McLendon, R.E.; Onar-Thomas, A.; Warren, K.E.; Wu, S.; Packer, R.J.; Banerjee, A.; Gilbertson, R.J.; et al. Bevacizumab (BVZ)-associated toxicities in children with recurrent central nervous system tumors treated with BVZ and irinotecan (CPT-11): A Pediatric Brain Tumor Consortium Study (PBTC-022). Cancer 2013, 119, 4180–4187. [Google Scholar] [CrossRef] [Green Version]
- Han, K.; Peyret, T.; Quartino, A.; Gosselin, N.H.; Gururangan, S.; Casanova, M.; Merks, J.H.; Massimino, M.; Grill, J.; Daw, N.C.; et al. Bevacizumab dosing strategy in paediatric cancer patients based on population pharmacokinetic analysis with external validation. Br. J. Clin. Pharmacol. 2016, 81, 148–160. [Google Scholar] [CrossRef] [Green Version]
- Levy, A.S.; Krailo, M.; Chi, S.; Villaluna, D.; Springer, L.; Williams-Hughes, C.; Fouladi, M.; Gajjar, A. Temozolomide with irinotecan versus temozolomide, irinotecan plus bevacizumab for recurrent medulloblastoma of childhood: Report of a COG randomized Phase II screening trial. Pediatr. Blood Cancer 2021, 68, e29031. [Google Scholar] [CrossRef]
- Cheung, N.K.; Landmeier, B.; Neely, J.; Nelson, A.D.; Abramowsky, C.; Ellery, S.; Adams, R.B.; Miraldi, F. Complete tumor ablation with iodine 131-radiolabeled disialoganglioside GD2-specific monoclonal antibody against human neuroblastoma xenografted in nude mice. J. Natl. Cancer Inst. 1986, 77, 739–745. [Google Scholar] [CrossRef]
- Kramer, K.; Pandit-Taskar, N.; Humm, J.L.; Zanzonico, P.B.; Haque, S.; Dunkel, I.J.; Wolden, S.L.; Donzelli, M.; Goldman, D.A.; Lewis, J.S.; et al. A phase II study of radioimmunotherapy with intraventricular (131) I-3F8 for medulloblastoma. Pediatr. Blood Cancer 2018, 65, e26754. [Google Scholar] [CrossRef]
- Zhai, L.; Bell, A.; Ladomersky, E.; Lauing, K.L.; Bollu, L.; Sosman, J.A.; Zhang, B.; Wu, J.D.; Miller, S.D.; Meeks, J.J.; et al. Immunosuppressive IDO in Cancer: Mechanisms of Action, Animal Models, and Targeting Strategies. Front. Immunol. 2020, 11, 1185. [Google Scholar] [CrossRef]
- Folgiero, V.; Miele, E.; Carai, A.; Ferretti, E.; Alfano, V.; Po, A.; Bertaina, V.; Goffredo, B.M.; Benedetti, M.C.; Camassei, F.D.; et al. IDO1 involvement in mTOR pathway: A molecular mechanism of resistance to mTOR targeting in medulloblastoma. Oncotarget 2016, 7, 52900–52911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, D.; Antonucci, L.; Di Magno, L.; Coni, S.; Sdruscia, G.; Macone, A.; Miele, E.; Infante, P.; Di Marcotullio, L.; De Smaele, E.; et al. Non-canonical Hedgehog/AMPK-Mediated Control of Polyamine Metabolism Supports Neuronal and Medulloblastoma Cell Growth. Dev. Cell 2015, 35, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casero, R.A., Jr.; Murray Stewart, T.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Veneti, Z.; Gkouskou, K.K.; Eliopoulos, A.G. Polycomb Repressor Complex 2 in Genomic Instability and Cancer. Int. J. Mol. Sci. 2017, 18, 1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracken, A.P.; Brien, G.L.; Verrijzer, C.P. Dangerous liaisons: Interplay between SWI/SNF, NuRD, and Polycomb in chromatin regulation and cancer. Genes Dev. 2019, 33, 936–959. [Google Scholar] [CrossRef] [PubMed]
- Mittal, P.; Roberts, C.W.M. The SWI/SNF complex in cancer—Biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef]
- Yi, J.; Wu, J. Epigenetic regulation in medulloblastoma. Mol. Cell Neurosci. 2017, 87, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Alimova, I.; Venkataraman, S.; Harris, P.; Marquez, V.E.; Northcott, P.A.; Dubuc, A.; Taylor, M.D.; Foreman, N.K.; Vibhakar, R. Targeting the enhancer of zeste homologue 2 in medulloblastoma. Int. J. Cancer 2012, 131, 1800–1809. [Google Scholar] [CrossRef] [Green Version]
- Vo, B.T.; Li, C.; Morgan, M.A.; Theurillat, I.; Finkelstein, D.; Wright, S.; Hyle, J.; Smith, S.M.C.; Fan, Y.; Wang, Y.D.; et al. Inactivation of Ezh2 Upregulates Gfi1 and Drives Aggressive Myc-Driven Group 3 Medulloblastoma. Cell Rep. 2017, 18, 2907–2917. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, D.; Zhang, Z.; Kaluz, S.; Yu, B.; Devi, N.S.; Olson, J.J.; Van Meir, E.G. EZH2 targeting reduces medulloblastoma growth through epigenetic reactivation of the BAI1/p53 tumor suppressor pathway. Oncogene 2020, 39, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, C.; Anderle, M.; Gianesello, M.; Lago, C.; Miele, E.; Cardano, M.; Aiello, G.; Piazza, S.; Caron, D.; Gianno, F.; et al. Modeling medulloblastoma in vivo and with human cerebellar organoids. Nat. Commun. 2020, 11, 583. [Google Scholar] [CrossRef] [PubMed]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhayay, P.; Bergthold, G.; Nguyen, B.; Schubert, S.; Gholamin, S.; Tang, Y.; Bolin, S.; Schumacher, S.E.; Zeid, R.; Masoud, S.; et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin. Cancer Res. 2014, 20, 912–925. [Google Scholar] [CrossRef] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Felgenhauer, J.; Tomino, L.; Selich-Anderson, J.; Bopp, E.; Shah, N. Dual BRD4 and AURKA Inhibition Is Synergistic against MYCN-Amplified and Nonamplified Neuroblastoma. Neoplasia 2018, 20, 965–974. [Google Scholar] [CrossRef]
- Henssen, A.; Althoff, K.; Odersky, A.; Beckers, A.; Koche, R.; Speleman, F.; Schäfers, S.; Bell, E.; Nortmeyer, M.; Westermann, F.; et al. Targeting MYCN-Driven Transcription By BET-Bromodomain Inhibition. Clin. Cancer Res. 2016, 22, 2470–2481. [Google Scholar] [CrossRef] [Green Version]
- Henssen, A.; Thor, T.; Odersky, A.; Heukamp, L.; El-Hindy, N.; Beckers, A.; Speleman, F.; Althoff, K.; Schäfers, S.; Schramm, A.; et al. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget 2013, 4, 2080–2095. [Google Scholar] [CrossRef] [Green Version]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [Green Version]
- Pearson, A.D.; DuBois, S.G.; Buenger, V.; Kieran, M.; Stegmaier, K.; Bandopadhayay, P.; Bennett, K.; Bourdeaut, F.; Brown, P.A.; Chesler, L.; et al. Bromodomain and extra-terminal inhibitors-A consensus prioritisation after the Paediatric Strategy Forum for medicinal product development of epigenetic modifiers in children-ACCELERATE. Eur. J. Cancer 2021, 146, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, S.; Alimova, I.; Balakrishnan, I.; Harris, P.; Birks, D.K.; Griesinger, A.; Amani, V.; Cristiano, B.; Remke, M.; Taylor, M.D.; et al. Inhibition of BRD4 attenuates tumor cell self-renewal and suppresses stem cell signaling in MYC driven medulloblastoma. Oncotarget 2014, 5, 2355–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Hino, M.; Koh, K.; Kyushiki, M.; Kishimoto, H.; Arakawa, Y.; Hanada, R.; Kawashima, H.; Kurihara, J.; Shimojo, N.; et al. Low Frequency of Programmed Death Ligand 1 Expression in Pediatric Cancers. Pediatr. Blood Cancer 2016, 63, 1461–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majzner, R.G.; Simon, J.S.; Grosso, J.F.; Martinez, D.; Pawel, B.R.; Santi, M.; Merchant, M.S.; Geoerger, B.; Hezam, I.; Marty, V.; et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer 2017, 123, 3807–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, C.D.; Flores, C.; Yang, C.; Pinheiro, E.M.; Yearley, J.H.; Sayour, E.J.; Pei, Y.; Moore, C.; McLendon, R.E.; Huang, J.; et al. Differential Immune Microenvironments and Response to Immune Checkpoint Blockade among Molecular Subtypes of Murine Medulloblastoma. Clin. Cancer Res. 2016, 22, 582–595. [Google Scholar] [CrossRef] [Green Version]
- Murata, D.; Mineharu, Y.; Arakawa, Y.; Liu, B.; Tanji, M.; Yamaguchi, M.; Fujimoto, K.I.; Fukui, N.; Terada, Y.; Yokogawa, R.; et al. High programmed cell death 1 ligand-1 expression: Association with CD8+ T-cell infiltration and poor prognosis in human medulloblastoma. J. Neurosurg. 2018, 128, 710–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockmayr, M.; Mohme, M.; Klauschen, F.; Winkler, B.; Budczies, J.; Rutkowski, S.; Schüller, U. Subgroup-specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology 2018, 7, e1462430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maximov, V.; Chen, Z.; Wei, Y.; Robinson, M.H.; Herting, C.J.; Shanmugam, N.S.; Rudneva, V.A.; Goldsmith, K.C.; MacDonald, T.J.; Northcott, P.A.; et al. Tumour-associated macrophages exhibit anti-tumoural properties in Sonic Hedgehog medulloblastoma. Nat. Commun. 2019, 10, 2410. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, J.F.; Van Hecke, W.; Adriaansen, E.J.M.; Jansen, M.K.; Bouma, R.G.; Villacorta Hidalgo, J.; Fisch, P.; Broekhuizen, R.; Spliet, W.G.M.; Kool, M.; et al. Prognostic relevance of tumor-infiltrating lymphocytes and immune checkpoints in pediatric medulloblastoma. Oncoimmunology 2018, 7, e1398877. [Google Scholar] [CrossRef]
- Martin, A.M.; Nirschl, C.J.; Polanczyk, M.J.; Bell, W.R.; Nirschl, T.R.; Harris-Bookman, S.; Phallen, J.; Hicks, J.; Martinez, D.; Ogurtsova, A.; et al. PD-L1 expression in medulloblastoma: An evaluation by subgroup. Oncotarget 2018, 9, 19177–19191. [Google Scholar] [CrossRef] [Green Version]
- Hwang, K.; Koh, E.J.; Choi, E.J.; Kang, T.H.; Han, J.H.; Choe, G.; Park, S.H.; Yearley, J.H.; Annamalai, L.; Blumenschein, W.; et al. PD-1/PD-L1 and immune-related gene expression pattern in pediatric malignant brain tumors: Clinical correlation with survival data in Korean population. J. NeuroOncol. 2018, 139, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Vladoiu, M.C.; El-Hamamy, I.; Donovan, L.K.; Farooq, H.; Holgado, B.L.; Sundaravadanam, Y.; Ramaswamy, V.; Hendrikse, L.D.; Kumar, S.; Mack, S.C.; et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 2019, 572, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Riemondy, K.A.; Venkataraman, S.; Willard, N.; Nellan, A.; Sanford, B.; Griesinger, A.M.; Amani, V.; Mitra, S.; Hankinson, T.C.; Handler, M.H.; et al. Neoplastic and immune single cell transcriptomics define subgroup-specific intra-tumoral heterogeneity of childhood medulloblastoma. Neuro-Oncology 2021. [Google Scholar] [CrossRef] [PubMed]
- Kabir, T.F.; Kunos, C.A.; Villano, J.L.; Chauhan, A. Immunotherapy for Medulloblastoma: Current Perspectives. Immunotargets Ther. 2020, 9, 57–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef] [PubMed]
- Nellan, A.; Rota, C.; Majzner, R.; Lester-McCully, C.M.; Griesinger, A.M.; Mulcahy Levy, J.M.; Foreman, N.K.; Warren, K.E.; Lee, D.W. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J. Immunother. Cancer 2018, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Ratnayake, M.; Savoldo, B.; Perlaky, L.; Dotti, G.; Wels, W.S.; Bhattacharjee, M.B.; Gilbertson, R.J.; Shine, H.D.; Weiss, H.L.; et al. Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res. 2007, 67, 5957–5964. [Google Scholar] [CrossRef] [Green Version]
- Orentas, R.J.; Lee, D.W.; Mackall, C. Immunotherapy targets in pediatric cancer. Front. Oncol. 2012, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Orlando, D.; Miele, E.; De Angelis, B.; Guercio, M.; Boffa, I.; Sinibaldi, M.; Po, A.; Caruana, I.; Abballe, L.; Carai, A.; et al. Adoptive Immunotherapy Using PRAME-Specific T Cells in Medulloblastoma. Cancer Res. 2018, 78, 3337–3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, D.P.; Coyle, B.; Walker, D.A.; Grabowska, A.M. In vitro models of medulloblastoma: Choosing the right tool for the job. J. Biotechnol. 2016, 236, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Vitanza, N.A.; Johnson, A.J.; Wilson, A.L.; Brown, C.; Yokoyama, J.K.; Künkele, A.; Chang, C.A.; Rawlings-Rhea, S.; Huang, W.; Seidel, K.; et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: An interim analysis. Nat. Med. 2021, 27, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Luo, L.; Wang, J.; He, B.; Feng, R.; Xian, N.; Zhang, Q.; Chen, L.; Huang, G. B7-H3 specific T cells with chimeric antigen receptor and decoy PD-1 receptors eradicate established solid human tumors in mouse models. Oncoimmunology 2020, 9, 1684127. [Google Scholar] [CrossRef] [Green Version]
- Theruvath, J.; Sotillo, E.; Mount, C.W.; Graef, C.M.; Delaidelli, A.; Heitzeneder, S.; Labanieh, L.; Dhingra, S.; Leruste, A.; Majzner, R.G.; et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat. Med. 2020, 26, 712–719. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, C.; Liu, Z.; Yang, M.; Tang, X.; Wang, Y.; Zheng, M.; Huang, J.; Zhong, K.; Zhao, S.; et al. B7-H3-Targeted CAR-T Cells Exhibit Potent Antitumor Effects on Hematologic and Solid Tumors. Mol. Ther. Oncolytics 2020, 17, 180–189. [Google Scholar] [CrossRef]
- Kimpo, M.S.; Oh, B.; Lee, S. The Role of Natural Killer Cells as a Platform for Immunotherapy in Pediatric Cancers. Curr. Oncol. Rep. 2019, 21, 93. [Google Scholar] [CrossRef] [Green Version]
- Fernández, L.; Portugal, R.; Valentín, J.; Martín, R.; Maxwell, H.; González-Vicent, M.; Díaz, M.; de Prada, I.; Pérez-Martínez, A. In vitro Natural Killer Cell Immunotherapy for Medulloblastoma. Front. Oncol. 2013, 3, 94. [Google Scholar] [CrossRef] [Green Version]
- Kennis, B.A.; Michel, K.A.; Brugmann, W.B.; Laureano, A.; Tao, R.H.; Somanchi, S.S.; Einstein, S.A.; Bravo-Alegria, J.B.; Maegawa, S.; Wahba, A.; et al. Monitoring of intracerebellarly-administered natural killer cells with fluorine-19 MRI. J. Neuro-Oncology 2019, 142, 395–407. [Google Scholar] [CrossRef]
- Liu, D.; Song, L.; Brawley, V.S.; Robison, N.; Wei, J.; Gao, X.; Tian, G.; Margol, A.; Ahmed, N.; Asgharzadeh, S.; et al. Medulloblastoma expresses CD1d and can be targeted for immunotherapy with NKT cells. Clin. Immunol. 2013, 149, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Martinez, A.; Fernandez, L.; Diaz, M.A. The therapeutic potential of natural killer cells to target medulloblastoma. Expert Rev. Anticancer Ther. 2016, 16, 573–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, A.B.; Yadavilli, S.; Saunders, D.; Van Pelt, S.; Chorvinsky, E.; Burga, R.A.; Albihani, S.; Hanley, P.J.; Xu, Z.; Pei, Y.; et al. Medulloblastoma rendered susceptible to NK-cell attack by TGFβ neutralization. J. Transl. Med. 2019, 17, 321. [Google Scholar] [CrossRef] [PubMed]
- Khatua, S.; Cooper, L.J.N.; Sandberg, D.I.; Ketonen, L.; Johnson, J.M.; Rytting, M.E.; Liu, D.D.; Meador, H.; Trikha, P.; Nakkula, R.J.; et al. Phase I study of intraventricular infusions of autologous ex vivo expanded NK cells in children with recurrent medulloblastoma and ependymoma. Neuro-Oncology 2020, 22, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Olsen, H.E.; Lynn, G.M.; Valdes, P.A.; Cerecedo Lopez, C.D.; Ishizuka, A.S.; Arnaout, O.; Bi, W.L.; Peruzzi, P.P.; Chiocca, E.A.; Friedman, G.K.; et al. Therapeutic cancer vaccines for pediatric malignancies: Advances, challenges, and emerging technologies. Neurooncol. Adv. 2021, 3, vdab027. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanovic, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef]
- Ardon, H.; De Vleeschouwer, S.; Van Calenbergh, F.; Claes, L.; Kramm, C.M.; Rutkowski, S.; Wolff, J.E.; Van Gool, S.W. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatr. Blood Cancer 2010, 54, 519–525. [Google Scholar] [CrossRef]
- Varela-Guruceaga, M.; Tejada-Solís, S.; García-Moure, M.; Fueyo, J.; Gomez-Manzano, C.; Patiño-García, A.; Alonso, M.M. Oncolytic Viruses as Therapeutic Tools for Pediatric Brain Tumors. Cancers 2018, 10, 226. [Google Scholar] [CrossRef] [Green Version]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Carrera, D.; Phillips, J.J.; Weiss, W.A.; Raffel, C. An oncolytic measles virus-sensitive Group 3 medulloblastoma model in immune-competent mice. Neuro-Oncology 2018, 20, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Studebaker, A.W.; Hutzen, B.; Pierson, C.R.; Russell, S.J.; Galanis, E.; Raffel, C. Oncolytic measles virus prolongs survival in a murine model of cerebral spinal fluid-disseminated medulloblastoma. Neuro-Oncology 2012, 14, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Studebaker, A.W.; Kreofsky, C.R.; Pierson, C.R.; Russell, S.J.; Galanis, E.; Raffel, C. Treatment of medulloblastoma with a modified measles virus. Neuro-Oncology 2010, 12, 1034–1042. [Google Scholar] [CrossRef]
- Hutzen, B.; Bid, H.K.; Houghton, P.J.; Pierson, C.R.; Powell, K.; Bratasz, A.; Raffel, C.; Studebaker, A.W. Treatment of medulloblastoma with oncolytic measles viruses expressing the angiogenesis inhibitors endostatin and angiostatin. BMC Cancer 2014, 14, 206. [Google Scholar] [CrossRef]
- Hutzen, B.; Pierson, C.R.; Russell, S.J.; Galanis, E.; Raffel, C.; Studebaker, A.W. Treatment of medulloblastoma using an oncolytic measles virus encoding the thyroidal sodium iodide symporter shows enhanced efficacy with radioiodine. BMC Cancer 2012, 12, 508. [Google Scholar] [CrossRef] [Green Version]
- Friedman, G.K.; Moore, B.P.; Nan, L.; Kelly, V.M.; Etminan, T.; Langford, C.P.; Xu, H.; Han, X.; Markert, J.M.; Beierle, E.A.; et al. Pediatric medulloblastoma xenografts including molecular subgroup 3 and CD133+ and CD15+ cells are sensitive to killing by oncolytic herpes simplex viruses. Neuro-Oncology 2016, 18, 227–235. [Google Scholar] [CrossRef]
- Studebaker, A.W.; Hutzen, B.J.; Pierson, C.R.; Haworth, K.B.; Cripe, T.P.; Jackson, E.M.; Leonard, J.R. Oncolytic Herpes Virus rRp450 Shows Efficacy in Orthotopic Xenograft Group 3/4 Medulloblastomas and Atypical Teratoid/Rhabdoid Tumors. Mol. Ther. Oncolytics 2017, 6, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Baxter, P.A.; Zhao, X.; Liu, Z.; Wadhwa, L.; Zhang, Y.; Su, J.M.; Tan, X.; Yang, J.; Adesina, A.; et al. A single intravenous injection of oncolytic picornavirus SVV-001 eliminates medulloblastomas in primary tumor-based orthotopic xenograft mouse models. Neuro-Oncology 2011, 13, 14–27. [Google Scholar] [CrossRef]
- Lacroix, J.; Schlund, F.; Leuchs, B.; Adolph, K.; Sturm, D.; Bender, S.; Hielscher, T.; Pfister, S.M.; Witt, O.; Rommelaere, J.; et al. Oncolytic effects of parvovirus H-1 in medulloblastoma are associated with repression of master regulators of early neurogenesis. Int. J. Cancer 2014, 134, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Snuderl, M.; Batista, A.; Kirkpatrick, N.D.; Ruiz de Almodovar, C.; Riedemann, L.; Walsh, E.C.; Anolik, R.; Huang, Y.; Martin, J.D.; Kamoun, W.; et al. Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell 2013, 152, 1065–1076. [Google Scholar] [CrossRef] [Green Version]
- Sholler, G.L.S.; Duda, D.G.; Bergendahl, G.; Ebb, D.; Snuderl, M.; Laetsch, T.W.; Michlitsch, J.; Hanson, D.; Isakoff, M.; Bielamowicz, K.; et al. Abstract CT015: A phase 1 dose escalation study of TB-403 in pediatric relapsed or refractory medulloblastoma, neuroblastoma, Ewing sarcoma, or alveolar rhabdomyosarcoma. Cancer Res. 2021, 81, CT015. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Veo, B.; Pierce, A.; Fosmire, S.; Madhavan, K.; Balakrishnan, I.; Donson, A.; Alimova, I.; Sullivan, K.D.; Joshi, M.; et al. A novel PLK1 inhibitor onvansertib effectively sensitizes MYC-driven medulloblastoma to radiotherapy. Neuro-Oncology 2021. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.; Foray, C.; Gatto, A.; Larcher, M.; Heinrich, S.; Lupu, M.; Mispelter, J.; Boussin, F.D.; Pouponnot, C.; Dutreix, M. AsiDNA Is a Radiosensitizer with no Added Toxicity in Medulloblastoma Pediatric Models. Clin. Cancer Res. 2020, 26, 5735–5746. [Google Scholar] [CrossRef]
- Buck, J.; Dyer, P.J.C.; Hii, H.; Carline, B.; Kuchibhotla, M.; Byrne, J.; Howlett, M.; Whitehouse, J.; Ebert, M.A.; McDonald, K.L.; et al. Veliparib Is an Effective Radiosensitizing Agent in a Preclinical Model of Medulloblastoma. Front. Mol. Biosci. 2021, 8, 633344. [Google Scholar] [CrossRef]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Vassal, G.; Houghton, P.J.; Pfister, S.M.; Smith, M.A.; Caron, H.N.; Li, X.N.; Shields, D.J.; Witt, O.; Molenaar, J.J.; Colombetti, S.; et al. International Consensus on Minimum Preclinical Testing Requirements for the Development of Innovative Therapies For Children and Adolescents with Cancer. Mol. Cancer Ther. 2021, 20, 1462–1468. [Google Scholar] [CrossRef]
- Grossman, S.A.; Romo, C.G.; Rudek, M.A.; Supko, J.; Fisher, J.; Nabors, L.B.; Wen, P.Y.; Peereboom, D.M.; Ellingson, B.M.; Elmquist, W.; et al. Baseline requirements for novel agents being considered for phase II/III brain cancer efficacy trials: Conclusions from the Adult Brain Tumor Consortium’s first workshop on CNS drug delivery. Neuro-Oncology 2020, 22, 1422–1424. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Müller, P.; Ji, Y. Innovative trial design in precision oncology. Semin. Cancer Biol. 2020, in press. [Google Scholar] [CrossRef]
- Allen, C.E.; Laetsch, T.W.; Mody, R.; Irwin, M.S.; Lim, M.S.; Adamson, P.C.; Seibel, N.L.; Parsons, D.W.; Cho, Y.J.; Janeway, K.; et al. Target and Agent Prioritization for the Children’s Oncology Group-National Cancer Institute Pediatric MATCH Trial. J. Natl. Cancer Inst. 2017, 109, djw274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harttrampf, A.C.; Lacroix, L.; Deloger, M.; Deschamps, F.; Puget, S.; Auger, N.; Vielh, P.; Varlet, P.; Balogh, Z.; Abbou, S.; et al. Molecular Screening for Cancer Treatment Optimization (MOSCATO-01) in Pediatric Patients: A Single-Institutional Prospective Molecular Stratification Trial. Clin. Cancer Res. 2017, 23, 6101–6112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, M.; Mayoh, C.; Lau, L.M.S.; Khuong-Quang, D.A.; Pinese, M.; Kumar, A.; Barahona, P.; Wilkie, E.E.; Sullivan, P.; Bowen-James, R.; et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat. Med. 2020, 26, 1742–1753. [Google Scholar] [CrossRef] [PubMed]
- Worst, B.C.; van Tilburg, C.M.; Balasubramanian, G.P.; Fiesel, P.; Witt, R.; Freitag, A.; Boudalil, M.; Previti, C.; Wolf, S.; Schmidt, S.; et al. Next-generation personalised medicine for high-risk paediatric cancer patients—The INFORM pilot study. Eur. J. Cancer 2016, 65, 91–101. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Personalized and molecular therapeutic strategies currently tested for non-WNT/non-SHH MB. RTKi = receptor tyrosine kinase inhibitor.
Figure 1.
Personalized and molecular therapeutic strategies currently tested for non-WNT/non-SHH MB. RTKi = receptor tyrosine kinase inhibitor.
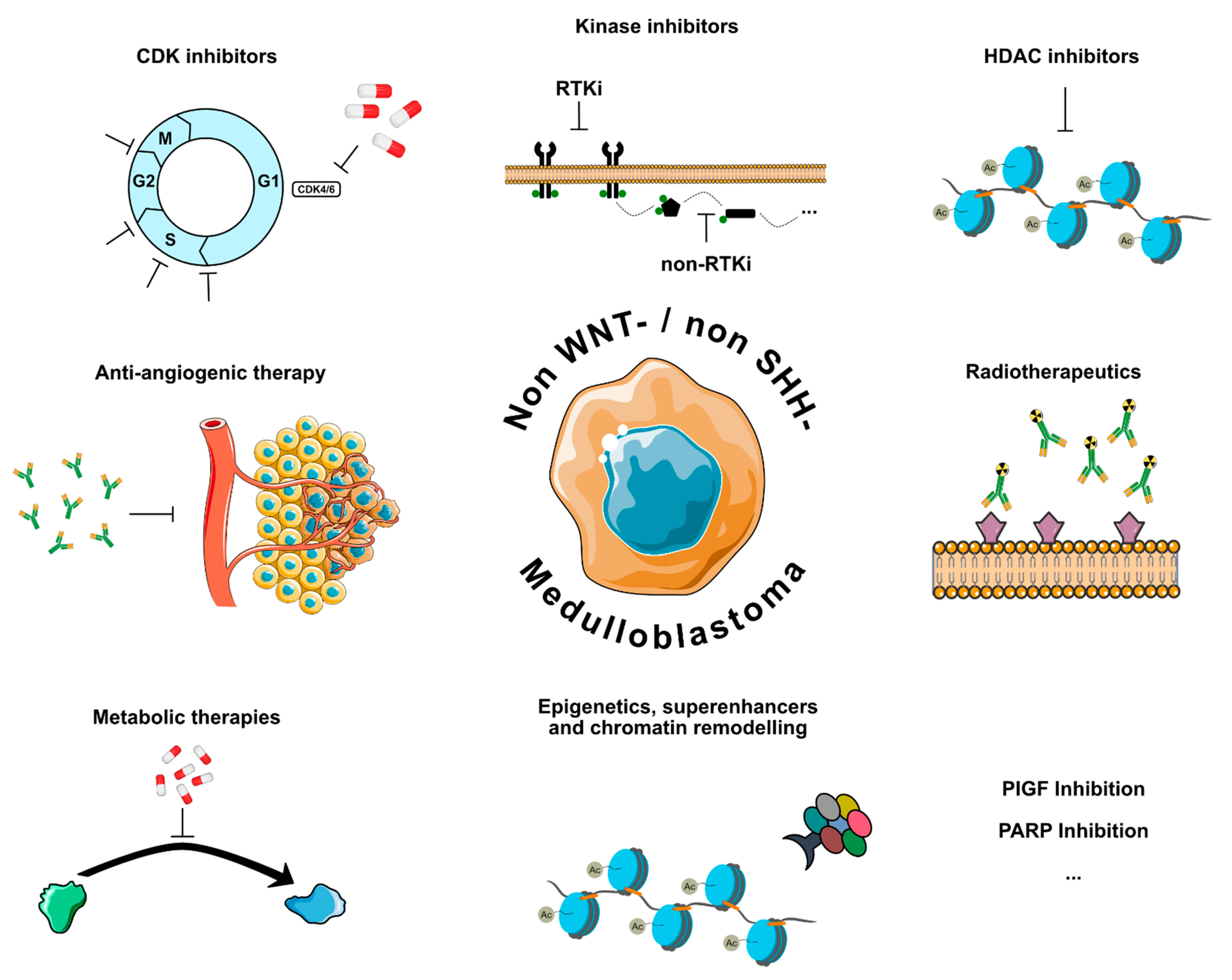
Figure 2.
Immunotherapeutic approaches currently tested for non-WNT/non-SHH MB.
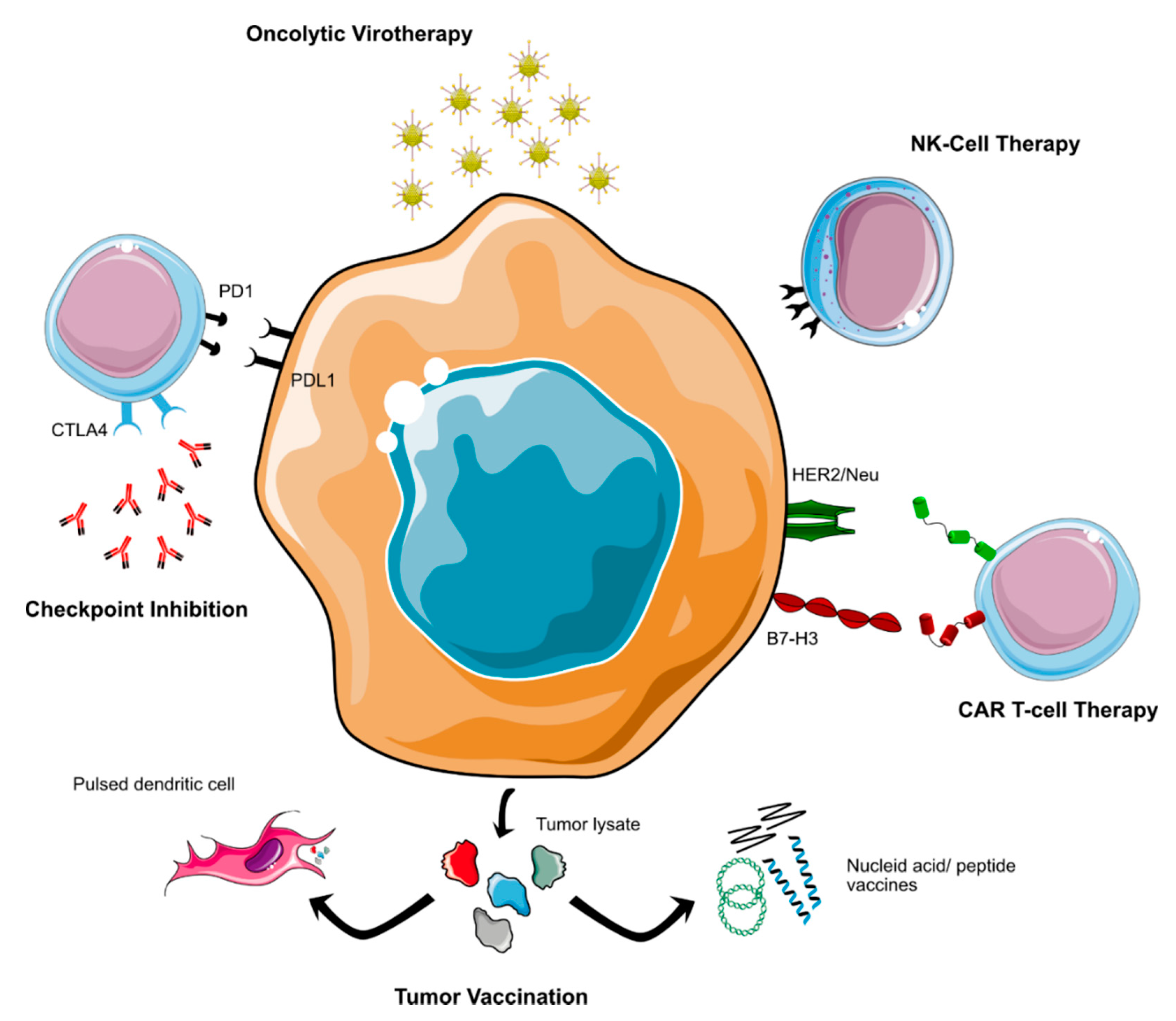

{kind=link}
{kind=link}
Table 1.
Active trials with molecular targets relevant to non-WNT/non-SHH MB. CTx = chemotherapy, RTx = radiotherapy.
Table 1.
Active trials with molecular targets relevant to non-WNT/non-SHH MB. CTx = chemotherapy, RTx = radiotherapy.
| Drug Class | Molecular Target | Drug | Trial Number |
|---|---|---|---|
| Kinase inhibitors | WEE1 | Adavosertib/CTx | NCT02095132 |
| CHK1 | Prexasertib/CTx | NCT04023669 | |
| CDK inhibitors | CDK4/6 | Palbociclib | NCT03526250 |
| CDK4/6 | Palbociclib/CTx | NCT03709680 | |
| CDK4/6 | Abemaciclib/CTx | NCT04238819 | |
| CDK4/6 | Ribociclib/CTx | NCT03434262 | |
| HDAC inhibitors | pan-HDAC | Panobinostat | NCT04315064 |
| pan-HDAC/PI3K | Fimepinostat | NCT03893487 | |
| HDAC class I,II,IV | Vorinostat/CTx | NCT00867178 | |
| HDAC class I,III/PD1 | Entinostat/Nivolumab | NCT03838042 | |
| Anti-angiogenic therapy | VEGF-A | Bevacizumab/CTx | NCT01356290 |
| Radiotherapeutics | - | CLR131 | NCT03478462 |
| B7-H3 | 177Lu-DTPA-omburtamab | NCT04167618 | |
| B7-H3/VEGF-A | Omburtamab-I131/Bevacizumab/CTx | NCT04743661 | |
| Metabolic therapy | IDO1 | Indoximod/CTx/RTx | NCT04049669 |
| ODC | DFMO | NCT03581240 | |
| ODC | DFMO | NCT04696029 | |
| Epigenetic therapy | EZH2 | Tazemetostat | NCT03213665 |
| BRD/BET | BMS-986158 | NCT03936465 | |
| Immune checkpoint Inhibition | PD1 | Pembrolizumab | NCT02359565 |
| PD1 | Nivolumab | NCT03173950 | |
| PD1/CTLA4 | Nivolumab/Ipilimumab | NCT03130959 | |
| PD1/CD122 | Nivolumab/Bempegaldesleukin | NCT04730349 | |
| Cellular immunotherapy | HER2/Neu | CAR T-cells | NCT03500991 |
| B7-H3 | CAR T-cells | NCT04185038 | |
| IL-13α2 | CAR T-cells | NCT04661384 | |
| EGFR806 | CAR T-cells | NCT03638167 | |
| GD2 | CAR T-cells | NCT04099797 | |
| Tumor vaccinations | - | PEP-CMV | NCT03299309 |
| Survivin | SurVaxM | NCT04978727 | |
| Immune modulators | CD40 | APX005M | NCT03389802 |
| STAT3 | WP1066 | NCT04334863 | |
| Oncolytic viruses | - | Modified measles virus | NCT02962167 |
| - | HSV G207/RTx | NCT03911388 | |
| - | AloCELYVIR | NCT04758533 | |
| - | Reovirus/GM-CSF | NCT02444546 | |
| - | PVSRIPO | NCT03043391 | |
| Other approaches | PGF | TB-403 | NCT02748135 |
| PARP | Olaparib | NCT03233204 | |
| PARP | Olaparib | NCT04236414 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ghasemi, D.R.; Fleischhack, G.; Milde, T.; Pajtler, K.W. The Current Landscape of Targeted Clinical Trials in Non-WNT/Non-SHH Medulloblastoma. Cancers 2022, 14, 679. https://doi.org/10.3390/cancers14030679
AMA Style
Ghasemi DR, Fleischhack G, Milde T, Pajtler KW. The Current Landscape of Targeted Clinical Trials in Non-WNT/Non-SHH Medulloblastoma. Cancers. 2022; 14(3):679. https://doi.org/10.3390/cancers14030679
Chicago/Turabian StyleGhasemi, David R., Gudrun Fleischhack, Till Milde, and Kristian W. Pajtler. 2022. "The Current Landscape of Targeted Clinical Trials in Non-WNT/Non-SHH Medulloblastoma" Cancers 14, no. 3: 679. https://doi.org/10.3390/cancers14030679
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.