
Pre-Clinical Evaluation of the Hypomethylating Agent Decitabine for the Treatment of T-Cell Lymphoblastic Lymphoma
 , , , , , , , , add
Show full author list
, , , , , , , , add
Show full author list
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Patient Characterization
2.2. In Vivo Treatment of Xenografts
2.3. DNA Methylation Profiling
2.4. Gene Expression Profiling
2.5. Data Analysis
2.6. HiC Analysis
3. Results
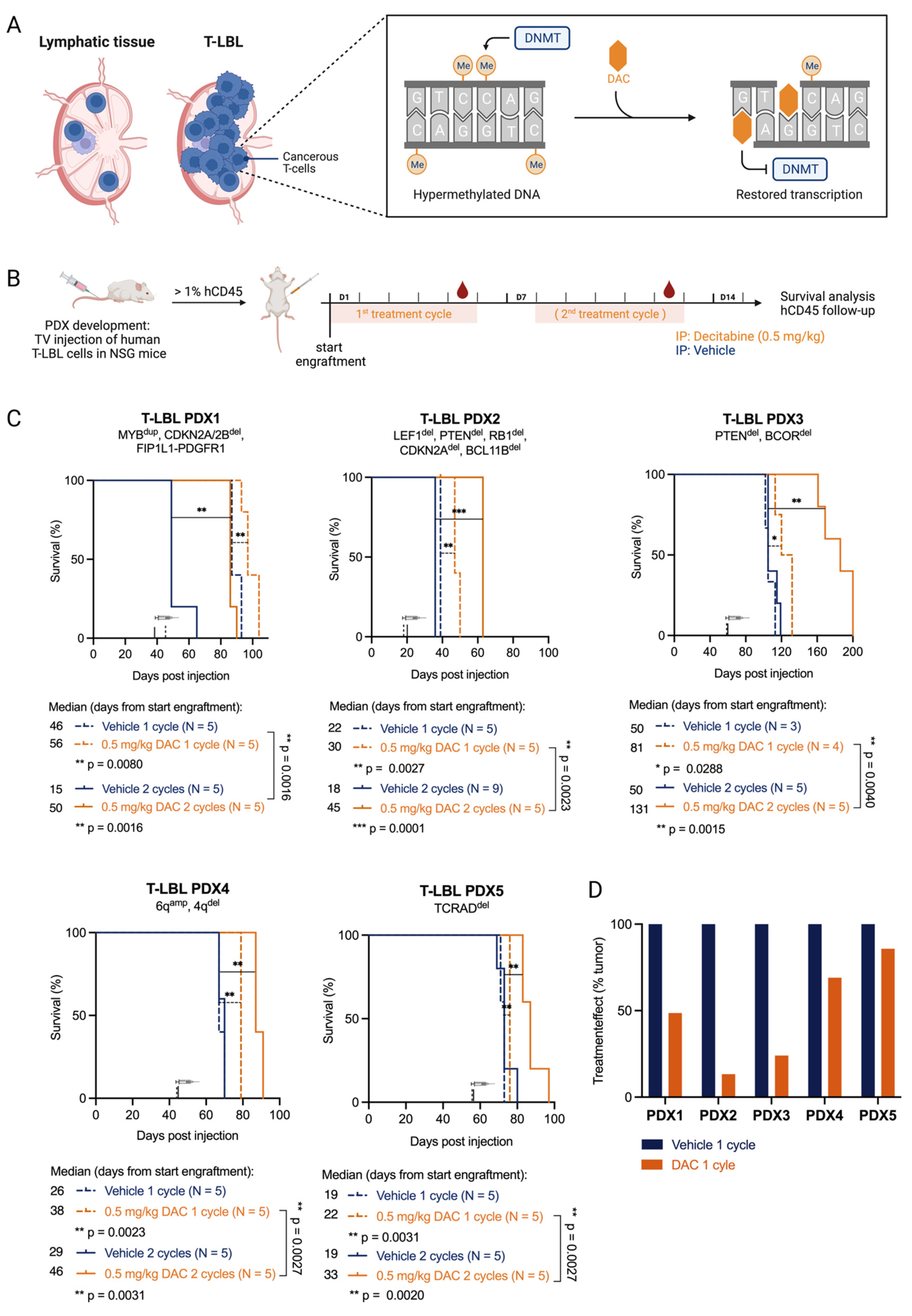
3.1. T-LBL PDX Models Are Sensitive to the FDA-Approved Hypomethylating Agent Decitabine
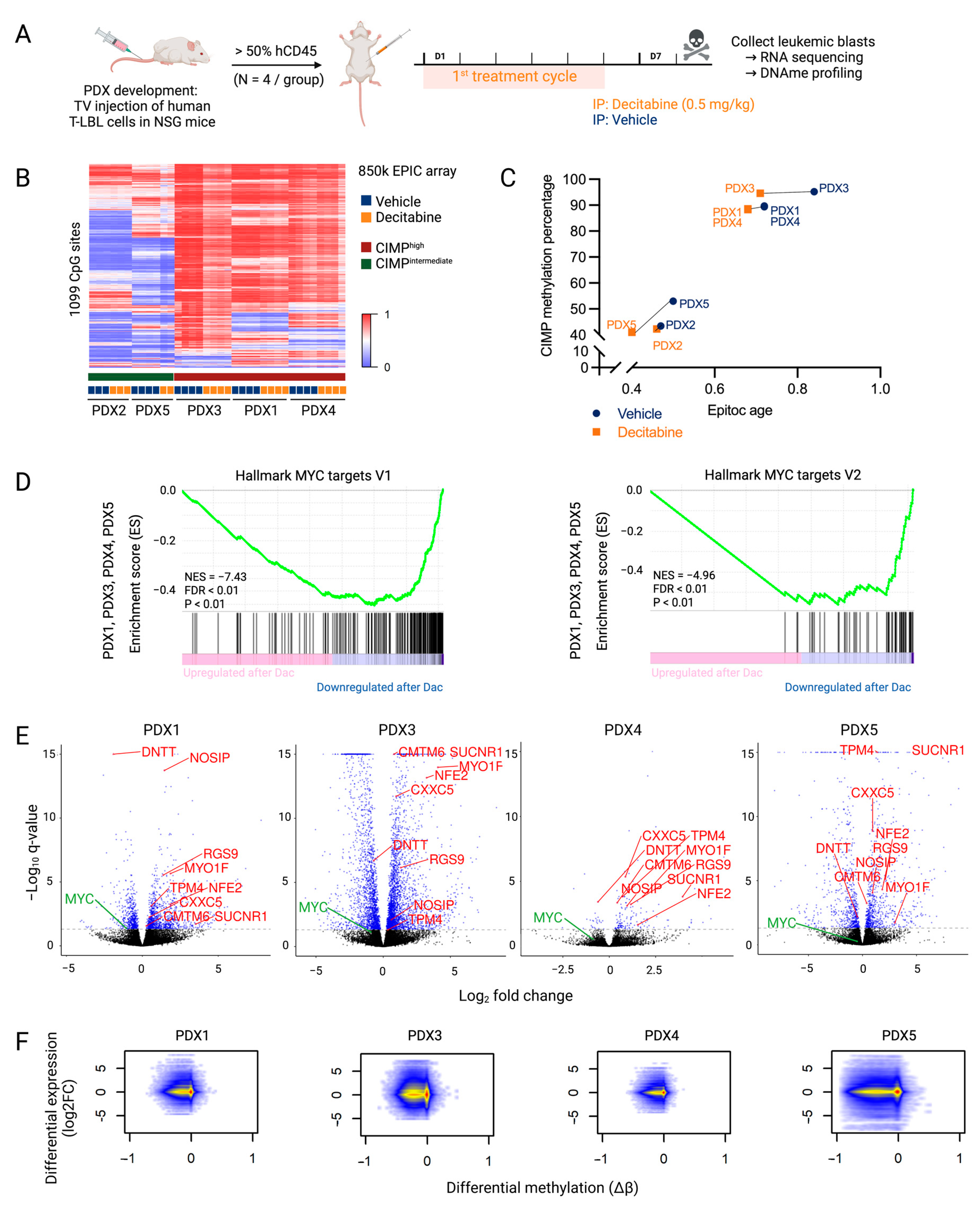
3.2. Overall Anti-Lymphoma Effects of Decitabine Treatment Are Mediated by Downregulation of the MYC Pathway
3.3. Higher Sensitivity to Decitabine Is Mediated by Downregulation of Genes Involved in the Cell Cycle and DNA Replication
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lepretre, S.; Graux, C.; Touzart, A.; Macintyre, E.; Boissel, N. Adult T-type lymphoblastic lymphoma: Treatment advances and prognostic indicators. Exp. Hematol. 2017, 51, 7–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroeze, E.; Loeffen, J.L.C.; Poort, V.M.; Meijerink, J.P.P. T-cell lymphoblastic lymphoma and leukemia: Different diseases from a common premalignant progenitor? Blood Adv. 2020, 4, 3466–3473. [Google Scholar] [CrossRef] [PubMed]
- Michalak, E.; Burr, M.; Bannister, A.J.; Dawson, M.A. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Haider, Z.; Landfors, M.; Golovleva, I.; Erlanson, M.; Schmiegelow, K.; Flægstad, T.; Kanerva, J.; Norén-Nyström, U.; Hultdin, M.; Degerman, S. DNA methylation and copy number variation profiling of T-cell lymphoblastic leukemia and lymphoma. Blood Cancer J. 2020, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Roels, J.; Thénoz, M.; Szarzyńska, B.; Landfors, M.; De Coninck, S.; Demoen, L.; Provez, L.; Kuchmiy, A.; Strubbe, S.; Reunes, L.; et al. Aging of Preleukemic Thymocytes Drives CpG Island Hypermethylation in T-cell Acute Lymphoblastic Leukemia. Blood Cancer Discov. 2020, 1, 274–289. [Google Scholar] [CrossRef]
- Stomper, J.; Rotondo, J.C.; Greve, G.; Lübbert, M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: Mechanisms of resistance and novel HMA-based therapies. Leukemia 2021, 35, 1873–1889. [Google Scholar] [CrossRef]
- Diesch, J.; Zwick, A.; Garz, A.-K.; Palau, A.; Buschbeck, M.; Götze, K.S. A clinical-molecular update on azanucleoside-based therapy for the treatment of hematologic cancers. Clin. Epigenet. 2016, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.-H.; Kang, K.-W.; Jeon, M.J.; Yu, E.S.; Kim, D.S.; Choi, H.; Lee, S.R.; Sung, H.J.; Kim, B.S.; Choi, C.W.; et al. Comparison between 5-day decitabine and 7-day azacitidine for lower-risk myelodysplastic syndromes with poor prognostic features: A retrospective multicentre cohort study. Sci. Rep. 2020, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zhou, Z.; Chen, L.; Wang, X. Comparison of Azacitidine and Decitabine in Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Network Meta-analysis. Clin. Lymphoma Myeloma Leuk. 2021, 21, e530–e544. [Google Scholar] [CrossRef]
- Ma, J.; Ge, Z. Comparison Between Decitabine and Azacitidine for Patients with Acute Myeloid Leukemia and Higher-Risk Myelodysplastic Syndrome: A Systematic Review and Network Meta-Analysis. Front. Pharmacol. 2021, 12, 701690. [Google Scholar] [CrossRef]
- Touzart, A.; Mayakonda, A.; Smith, C.; Hey, J.; Toth, R.; Cieslak, A.; Andrieu, G.P.; Quang, C.T.; Latiri, M.; Ghysdael, J.; et al. Epigenetic analysis of patients with T-ALL identifies poor outcomes and a hypomethylating agent-responsive subgroup. Sci. Transl. Med. 2021, 13, eabc4834. [Google Scholar] [CrossRef]
- Yang, Y.; Yao, S.; Zhang, J.; Yan, Z.; Chu, J.; Wang, H.; Yao, Z.; Zhang, F.; Xia, Q.; Liu, Y. Decitabine-containing G-CSF priming regimen overcomes resistance of primary mediastinal neoplasm from early T-cell precursors to conventional chemotherapy: A case report. Onco Targets Ther. 2019, 12, 7039–7044. [Google Scholar] [CrossRef] [Green Version]
- Van Dongen, J.J.M.; Lhermitte, L.; Böttcher, S.; Almeida, J.R.S.; Van Der Velden, V.H.J.; Flores-Montero, J.; Rawstron, A.; Asnafi, V.; Lécrevisse, Q.; Lucio, P.; et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia 2012, 26, 1908–1975. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Laird, P.W.; Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2016, 45, e22. [Google Scholar] [CrossRef]
- Gaunt, T.R.; Shihab, H.A.; Hemani, G.; Min, J.L.; Woodward, G.; Lyttleton, O.; Zheng, J.; Duggirala, A.; McArdle, W.L.; Ho, K.; et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016, 17, 61. [Google Scholar] [CrossRef] [Green Version]
- Richter-Pechańska, P.; Kunz, J.B.; Bornhauser, B.; von Knebel Doeberitz, C.; Rausch, T.; Erarslan-Uysal, B.; Assenov, Y.; Frismantas, V.; Marovca, B.; Waszak, S.M.; et al. PDX models recapitulate the genetic and epigenetic landscape of pediatric T-cell leukemia. EMBO Mol. Med. 2018, 10, e9443. [Google Scholar] [CrossRef]
- Borssén, M.; Haider, Z.; Landfors, M.; Norén-Nyström, U.; Schmiegelow, K.; Åsberg, A.E.; Kanerva, J.; Madsen, H.O.; Marquart, H.; Heyman, M.; et al. DNA Methylation Adds Prognostic Value to Minimal Residual Disease Status in Pediatric T-Cell Acute Lymphoblastic Leukemia. Pediatr. Blood Cancer 2016, 63, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wong, A.; Kuh, D.; Paul, D.S.; Rakyan, V.K.; Leslie, R.D.; Zheng, S.C.; Widschwendter, M.; Beck, S.; Teschendorff, A.E. Correlation of an epigenetic mitotic clock with cancer risk. Genome Biol. 2016, 17, 205. [Google Scholar] [CrossRef]
- Kloetgen, A.; Thandapani, P.; Ntziachristos, P.; Ghebrechristos, Y.; Nomikou, S.; Lazaris, C.; Chen, X.; Hu, H.; Bakogianni, S.; Wang, J.; et al. Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat. Genet. 2020, 52, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Zhang, M.; Zhu, J.; Zhang, M.; Ci, C.; Shang, S.; Wei, Y.; Liu, H.; Li, X.; Zhang, Y. Exploration of drug-response mechanism by integrating genetics and epigenetics across cancers. Epigenomics 2018, 10, 993–1010. [Google Scholar] [CrossRef] [PubMed]
- Murcia, O.; Juárez, M.; Rodríguez-Soler, M.; Hernández-Illán, E.; Giner-Calabuig, M.; Alustiza, M.; Egoavil, C.; Castillejo, A.; Alenda, C.; Barberá, V.; et al. Colorectal cancer molecular classification using BRAF, KRAS, microsatellite instability and CIMP status: Prognostic implications and response to chemotherapy. PLoS ONE 2018, 13, e0203051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjan, N.; Pandey, V.; Panigrahi, M.; Klumpp, L.; Naumann, U.; Babu, P. The Tumor Suppressor MTUS1/ATIP1 Modulates Tumor Promotion in Glioma: Association with Epigenetics and DNA Repair. Cancers 2021, 13, 1245. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Jia, Y.; Wang, Y.; Han, X.; Duan, Y.; Lv, W.; Ma, M.; Liu, L. Methylation decreases the Bin1 tumor suppressor in ESCC and restoration by decitabine inhibits the epithelial mesenchymal transition. Oncotarget 2017, 8, 19661–19673. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Chen, Y.; Pei, X.; Zang, Y.; Han, S. Effects of Decitabine on the proliferation of K562 cells and the expression of DR4 gene. Saudi J. Biol. Sci. 2018, 25, 242–247. [Google Scholar] [CrossRef]
- Zhu, H.; Gu, S.; Yin, M.; Shi, M.; Xin, C.; Zhu, J.; Wang, J.; Huang, S.; Xie, C.; Ma, J.; et al. Analysis of infantile fibrosarcoma reveals extensive T-cell responses within tumors: Implications for immunotherapy. Pediatr. Blood Cancer 2017, 65, e26813. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Y.-C.; Chen, X.-Y.; Shen, Z.-Y.; Cao, H.; Zhang, Y.-J.; Yu, J.; Zhu, J.-D.; Lu, Y.-Y.; Fang, J.-Y. CTHRC1 is upregulated by promoter demethylation and transforming growth factor-β1 and may be associated with metastasis in human gastric cancer. Cancer Sci. 2012, 103, 1327–1333. [Google Scholar] [CrossRef]
- Rawłuszko-Wieczorek, A.A.; Horbacka, K.; Krokowicz, P.; Misztal, M.; Jagodziński, P.P. Prognostic Potential of DNA Methylation and Transcript Levels of HIF1A and EPAS1 in Colorectal Cancer. Mol. Cancer Res. 2014, 12, 1112–1127. [Google Scholar] [CrossRef]
- Dudzik, P.; Trojan, S.E.; Ostrowska, B.; Zemanek, G.; Dulińska-Litewka, J.; Laidler, P.; Kocemba-Pilarczyk, K.A. The Epigenetic Modifier 5-Aza-2-deoxycytidine Triggers the Expression of CD146 Gene in Prostate Cancer Cells. Anticancer Res. 2019, 39, 2395–2403. [Google Scholar] [CrossRef]
- Lucarini, V.; Buccione, C.; Ziccheddu, G.; Peschiaroli, F.; Sestili, P.; Puglisi, R.; Mattia, G.; Zanetti, C.; Parolini, I.; Bracci, L.; et al. Combining Type I Interferons and 5-Aza-2′-Deoxycitidine to Improve Anti-Tumor Response against Melanoma. J. Investig. Dermatol. 2017, 137, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.; Huang, S.-F.; Cao, J.; Wen, Y.-A.; Zhang, L.-P.; Ren, G.-S. Silencing of PCDH10 in hepatocellular carcinoma via de novo DNA methylation independent of HBV infection or HBX expression. Clin. Exp. Med. 2013, 13, 127–134. [Google Scholar] [CrossRef]
- Nakamura, N.; Takenaga, K. Hypomethylation of the metastasis-associated S100A4 gene correlates with gene activation in human colon adenocarcinoma cell lines. Clin. Exp. Metastasis 1998, 16, 471–479. [Google Scholar] [CrossRef]
- Bartling, B.; Rehbein, G.; Simm, A.; Silber, R.-E.; Hofmann, H.-S. Porcupine expression is associated with the expression of S100P and other cancer-related molecules in non-small cell lung carcinoma. Int. J. Oncol. 2010, 36, 1015–1021. [Google Scholar] [CrossRef] [Green Version]
- Miasaki, F.Y.; Vivaldi, A.; Ciampi, R.; Agate, L.; Collecchi, P.; Capodanno, A.; Pinchera, A.; Elisei, R. Retinoic acid receptor β2 re-expression and growth inhibition in thyroid carcinoma cell lines after 5-aza-2′-deoxycytidine treatment. J. Endocrinol. Investig. 2008, 31, 724–730. [Google Scholar] [CrossRef]
- Henrique, R.; Costa, V.L.; Cerveira, N.; Carvalho, A.L.; Hoque, M.O.; Ribeiro, F.R.; Oliveira, J.; Teixeira, M.R.; Sidransky, D.; Jerónimo, C. Hypermethylation of Cyclin D2 is associated with loss of mRNA expression and tumor development in prostate cancer. J. Mol. Med. 2006, 84, 911–918. [Google Scholar] [CrossRef]
- Oshimo, Y.; Nakayama, H.; Ito, R.; Kitadai, Y.; Yoshida, K.; Chayama, K.; Yasui, W. Promoter methylation of cyclin D2 gene in gastric carcinoma. Int. J. Oncol. 2003, 23, 1663–1670. [Google Scholar] [CrossRef]
- Sekita, N.; Suzuki, H.; Ichikawa, T.; Kito, H.; Akakura, K.; Igarashi, T.; Nakayama, T.; Watanabe, M.; Shiraishi, T.; Toyota, M.; et al. Epigenetic Regulation of the KAI1 Metastasis Suppressor Gene in Human Prostate Cancer Cell Lines. Jpn. J. Cancer Res. 2001, 92, 947–951. [Google Scholar] [CrossRef]
- Schayek, H.; Bentov, I.; Sun, S.; Plymate, S.R.; Werner, H. Progression to metastatic stage in a cellular model of prostate cancer is associated with methylation of the androgen receptor gene and transcriptional suppression of the insulin-like growth factor-I receptor gene. Exp. Cell Res. 2010, 316, 1479–1488. [Google Scholar] [CrossRef]
- Di Tullio, A.; Passaro, D.; Rouault-Pierre, K.; Purewal, S.; Bonnet, D. Nuclear Factor Erythroid 2 Regulates Human HSC Self-Renewal and T Cell Differentiation by Preventing NOTCH1 Activation. Stem Cell Rep. 2017, 9, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeken, J.C.; Jutzi, J.S.; Wehrle, J.; Koellerer, C.; Staehle, H.F.; Becker, H.; Schoenwandt, E.; Seeger, T.S.; Schanne, D.H.; Gothwal, M.; et al. Epigenetic regulation of NFE2 overexpression in myeloproliferative neoplasms. Blood 2018, 131, 2065–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borssén, M.; Palmqvist, L.; Karrman, K.; Abrahamsson, J.; Behrendtz, M.; Heldrup, J.; Forestier, E.; Roos, G.; Degerman, S. Promoter DNA Methylation Pattern Identifies Prognostic Subgroups in Childhood T-Cell Acute Lymphoblastic Leukemia. PLoS ONE 2013, 8, e65373. [Google Scholar] [CrossRef]
- Tomar, T.; de Jong, S.; Alkema, N.G.; Hoekman, R.L.; Meersma, G.J.; Klip, H.G.; van der Zee, A.G.; Wisman, G.B.A. Genome-wide methylation profiling of ovarian cancer patient-derived xenografts treated with the demethylating agent decitabine identifies novel epigenetically regulated genes and pathways. Genome Med. 2016, 8, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, K.; Das, V.; Vyas, P.; Hajdúch, M. Nucleosidic DNA demethylating epigenetic drugs—A comprehensive review from discovery to clinic. Pharmacol. Ther. 2018, 188, 45–79. [Google Scholar] [CrossRef]
- Pappalardi, M.B.; Keenan, K.; Cockerill, M.; Kellner, W.A.; Stowell, A.; Sherk, C.; Wong, K.; Pathuri, S.; Briand, J.; Steidel, M.; et al. Discovery of a first-in-class reversible DNMT1-selective inhibitor with improved tolerability and efficacy in acute myeloid leukemia. Nat. Cancer 2021, 2, 1002–1017. [Google Scholar] [CrossRef]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Maes, K.; De Smedt, E.; Lemaire, M.; De Raeve, H.; Menu, E.; Van Valckenborgh, E.; McClue, S.; Vanderkerken, K.; De Bruyne, E. The role of DNA damage and repair in decitabine-mediated apoptosis in multiple myeloma. Oncotarget 2014, 5, 3115–3129. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Liu, Y.; Shang, L.; Wei, W.; Shen, Y.; Gu, Q.; Xie, X.; Dong, W.; Lin, Y.; Yue, Y.; et al. Decitabine and all-trans retinoic acid synergistically exhibit cytotoxicity against elderly AML patients via miR-34a/MYCN axis. Biomed. Pharmacother. 2020, 125, 109878. [Google Scholar] [CrossRef]
- Guan, H.; Xie, L.; Klapproth, K.; Weitzer, C.D.; Wirth, T.; Ushmorov, A. Decitabine represses translocated MYC oncogene in Burkitt lymphoma. J. Pathol. 2013, 229, 775–783. [Google Scholar] [CrossRef]
- Poole, C.J.; Zheng, W.; Lodh, A.; Yevtodiyenko, A.; Liefwalker, D.; Li, H.; Felsher, D.W.; van Riggelen, J. DNMT3B overexpression contributes to aberrant DNA methylation and MYC-driven tumor maintenance in T-ALL and Burkitt’s lymphoma. Oncotarget 2017, 8, 76898–76920. [Google Scholar] [CrossRef] [Green Version]
- Bogenberger, J.; Kornblau, S.M.; Pierceall, W.E.; Lena, R.; Chow, D.; Shi, C.-X.; Mantei, J.; Ahmann, G.J.; Gonzales, I.M.; Choudhary, A.; et al. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia 2014, 28, 1657–1665. [Google Scholar] [CrossRef] [Green Version]
- Dinardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2018, 133, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Gibson, A.; Trabal, A.; McCall, D.; Khazal, S.; Toepfer, L.; Bell, D.H.; Roth, M.; Mahadeo, K.M.; Nunez, C.; Short, N.J.; et al. Venetoclax for Children and Adolescents with Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancers 2021, 14, 150. [Google Scholar] [CrossRef]
- Peirs, S.; Matthijssens, F.; Goossens, S.; Van de Walle, I.; Ruggero, K.; de Bock, C.E.; Degryse, S.; Canté-Barrett, K.; Briot, D.; Clappier, E.; et al. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood 2014, 124, 3738–3747. [Google Scholar] [CrossRef]
- Baig, M.U.; Rytting, M.; Roth, M.; Morani, A.C.; Nunez, C.; Lin, P.; Cuglievan, B. Venetoclax and Decitabine in Pediatric Refractory T-cell Lymphoblastic Lymphoma. J. Pediatr. Hematol. Oncol. 2021, 43, e991–e996. [Google Scholar] [CrossRef]
- Farhadfar, N.; Li, Y.; May, W.S.; Adams, C.B. Venetoclax and decitabine for treatment of relapsed T-cell acute lymphoblastic leukemia: A case report and review of literature. Hematol. Stem Cell Ther. 2021, 14, 246–251. [Google Scholar] [CrossRef]
- Yau, H.L.; Bell, E.; Ettayebi, I.; de Almeida, F.C.; Boukhaled, G.M.; Shen, S.Y.; Allard, D.; Morancho, B.; Marhon, S.A.; Ishak, C.A.; et al. DNA hypomethylating agents increase activation and cytolytic activity of CD8+ T cells. Mol. Cell 2021, 81, 1469–1483. [Google Scholar] [CrossRef]
- Goossens, S.; Cauwels, A.; Pieters, T.; De Smedt, R.; T’Sas, S.; Almeida, A.; Daneels, W.; Van Vlierberghe, P.; Tavernier, J. Direct and indirect anti-leukemic properties of activity-on-target interferons for the treatment of T-cell acute lymphoblastic leukemia. Haematologica 2022, 107, 1448–1453. [Google Scholar] [CrossRef]
- Van Thillo, Q.; De Bie, J.; Seneviratne, J.A.; Demeyer, S.; Omari, S.; Balachandran, A.; Zhai, V.; Tam, W.L.; Sweron, B.; Geerdens, E.; et al. Oncogenic cooperation between TCF7-SPI1 and NRAS(G12D) requires β-catenin activity to drive T-cell acute lymphoblastic leukemia. Nat. Commun. 2021, 12, 4164. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Qi, Y.; Wang, Y.; Ma, X.; Xu, Y.; Wang, J.; Zhang, X.; Gao, M.; Cong, B.; Han, S. Overexpression of CD59 inhibits apoptosis of T-acute lymphoblastic leukemia via AKT/Notch1 signaling pathway. Cancer Cell Int. 2019, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.A.; Spolski, R.; Kovanen, P.E.; Suzuki, T.; Bollenbacher, J.; Pise-Masison, C.A.; Radonovich, M.F.; Lee, S.; Jenkins, N.A.; Copeland, N.G.; et al. Stat5 Synergizes with T Cell Receptor/Antigen Stimulation in the Development of Lymphoblastic Lymphoma. J. Exp. Med. 2003, 198, 79–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, W.Z.; Tan, S.H.; Ngoc, P.C.T.; Amanda, S.; Yam, A.W.Y.; Liau, W.-S.; Gong, Z.; Lawton, L.N.; Tenen, D.G.; Sanda, T. ARID5B as a critical downstream target of the TAL1 complex that activates the oncogenic transcriptional program and promotes T-cell leukemogenesis. Genes Dev. 2017, 31, 2343–2360. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Kraszewska, M.D.; Dawidowska, M.; Kosmalska, M.; Sędek, L.; Grzeszczak, W.; Kowalczyk, J.R.; Szczepański, T.; Witt, M. BCL11B, FLT3, NOTCH1 and FBXW7 mutation status in T-cell acute lymphoblastic leukemia patients. Blood Cells Mol. Dis. 2013, 50, 33–38. [Google Scholar] [CrossRef]
- Fantin, V.R.; Loboda, A.; Paweletz, C.P.; Hendrickson, R.C.; Pierce, J.W.; Roth, J.A.; Li, L.; Gooden, F.; Korenchuk, S.; Hou, X.S.; et al. Constitutive Activation of Signal Transducers and Activators of Transcription Predicts Vorinostat Resistance in Cutaneous T-Cell Lymphoma. Cancer Res. 2008, 68, 3785–3794. [Google Scholar] [CrossRef] [Green Version]
- Kok, C.H.; Yeung, D.T.; Lu, L.; Watkins, D.B.; Leclercq, T.M.; Dang, P.; Saunders, V.A.; Reynolds, J.; White, D.L.; Hughes, T.P. Gene expression signature that predicts early molecular response failure in chronic-phase CML patients on frontline imatinib. Blood Adv. 2019, 3, 1610–1621. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Adusumilli, P.S.; Eisenberg, D.P.; Darr, E.; Ghossein, R.A.; Li, S.; Liu, S.; Singh, B.; Shah, J.P.; Fong, Y.; et al. Nectin-1 Expression by Squamous Cell Carcinoma is a Predictor of Herpes Oncolytic Sensitivity. Mol. Ther. 2007, 15, 103–113. [Google Scholar] [CrossRef]
- Cha, K.; Li, Y.; Yi, G.-S. Discovering gene expression signatures responding to tyrosine kinase inhibitor treatment in chronic myeloid leukemia. BMC Med. Genom. 2016, 9 (Suppl. 1), 29. [Google Scholar] [CrossRef]
- Yotova, I.; Hsu, E.; Do, C.; Gaba, A.; Sczabolcs, M.; Dekan, S.; Kenner, L.; Wenzl, R.; Tycko, B. Epigenetic Alterations Affecting Transcription Factors and Signaling Pathways in Stromal Cells of Endometriosis. PLoS ONE 2017, 12, e0170859. [Google Scholar] [CrossRef] [Green Version]
- Raterman, H.G.; Vosslamber, S.; De Ridder, S.; Nurmohamed, M.T.; Lems, W.F.; Boers, M.; Van De Wiel, M.; Dijkmans, B.A.; Verweij, C.L.; Voskuyl, A.E. The interferon type I signature towards prediction of non-response to rituximab in rheumatoid arthritis patients. Arthritis Res. Ther. 2012, 14, R95. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ni, M.; Li, L.; Wang, J.; Tu, Z.; Zhou, H.; Zhang, S. A novel four-gene signature predicts immunotherapy response of patients with different cancers. J. Clin. Lab. Anal. 2022, 36, e24494. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDX1 | PDX3 | PDX4 | PDX5 | |
|---|---|---|---|---|
| Total number of hypomethylated annotated genes (DM-CpG sites) | 31,174 (175,219) | 36,431 (308,103) | 26,265 (106,835) | 38,903 (422,137) |
| Total number of hypermethylated annotated genes (DM-CpG sites) | 51 (44) | 44 (41) | 5 (4) | 95 (75) |
| Total number of annotated genes with increased expression | 1067 | 2728 | 151 | 781 |
| Total number of annotated genes with decreased expression | 576 | 2099 | 26 | 700 |
| Hypomethylated annotated genes with increased expression | 733 | 2326 | 112 | 689 |
| Hypomethylated annotated genes with decreased expresssion | 488 | 1776 | 21 | 619 |
| Gene Name | Protein Function | Previous Association with DAC | Differentially Methylated CpG Sites |
|---|---|---|---|
| CMTM6 | Regulating T cell activation and antitumor responses | / | Gene: 1 |
| CXXC5 | Transcription factor and epigenetic regulator in a.o. myelopoiesis | / | Gene: 5 Promotor: 6 |
| DNTT | DNA polymerase that functions as DNA nucleotidylexotransferase | / | Gene: 2 |
| MYO1F | Immune cell motility | / | Gene: 2 Enhancer: 1 |
| NFE2 | Megakaryocyte production, regulation of HSC self-renewal and T-cell differentiation by preventing NOTCH1 activation [41] | Decitabine reduces NFE2 expression in HEL cells [42] | Promotor: 4 |
| NOSIP | Nitric oxide production | / | Gene: 1 Promotor: 3 |
| RGS9 | Deactivation of G-proteins | / | Gene: 5 Promotor: 1 |
| SUCNR1 | Hematopoietic progenitor cell development, regulation immune cell responses | / | Gene: 1 |
| TPM4 | Actin-binding protein involved in the contractile system, maintains cell-cell adhesions | / | Gene: 3 Enhancer: 1 Promotor: 2 |
| More Sensitive Subgroup | Less Sensitive Subgroup | |
|---|---|---|
| Total number of hypomethylated annotated genes (DM-CpG sites) | 34,078 (235,386) | 35,017 (272,338) |
| Total number of hypermethylated annotated genes (DM-CpG sites) | 11 (8) | 3 (2) |
| Total number of annotated genes with increased expression | 1235 | 181 |
| Total number of annotated genes with decreased expression | 959 | 133 |
| Hypomethylated annotated genes with increased expression | 930 | 149 |
| Hypomethylated annotated genes with decreased expresssion | 808 | 115 |
| Open Chromatin | Closed Chromatin | Relative Risk | |
|---|---|---|---|
| Proportion of CpG sites (number of CpG sites on the Methylation Infinium EPICarray) | 0.82 (237,233) | 0.18 (53,283) | - |
| Proportion of genes (number of RNA sequenced genes) | 0.74 (9213) | 0.26 (3161) | - |
| More sensitive subgroup | |||
| Proportion of DM CpG sites (number of CpG sites) | 0.80 (74,802) | 0.20 (18,385) | 0.98 |
| Proportion of DEG (number of genes) | 0.90 (860) | 0.10 (96) | 1.23 |
| Less sensitive subgroup | |||
| Proportion of DM CpG sites (number of CpG sites) | 0.83 (90,481) | 0.17 (18,132) | 1.03 |
| Proportion of DEG (number of genes) | 0.94 (144) | 0.06 (9) | 1.27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Provez, L.; Putteman, T.; Landfors, M.; Roels, J.; Reunes, L.; T’Sas, S.; Van Loocke, W.; Lintermans, B.; De Coninck, S.; Thenoz, M.; et al. Pre-Clinical Evaluation of the Hypomethylating Agent Decitabine for the Treatment of T-Cell Lymphoblastic Lymphoma. Cancers 2023, 15, 647. https://doi.org/10.3390/cancers15030647
Provez L, Putteman T, Landfors M, Roels J, Reunes L, T’Sas S, Van Loocke W, Lintermans B, De Coninck S, Thenoz M, et al. Pre-Clinical Evaluation of the Hypomethylating Agent Decitabine for the Treatment of T-Cell Lymphoblastic Lymphoma. Cancers. 2023; 15(3):647. https://doi.org/10.3390/cancers15030647
Chicago/Turabian StyleProvez, Lien, Tom Putteman, Mattias Landfors, Juliette Roels, Lindy Reunes, Sara T’Sas, Wouter Van Loocke, Béatrice Lintermans, Stien De Coninck, Morgan Thenoz, and et al. 2023. "Pre-Clinical Evaluation of the Hypomethylating Agent Decitabine for the Treatment of T-Cell Lymphoblastic Lymphoma" Cancers 15, no. 3: 647. https://doi.org/10.3390/cancers15030647